
Abstract
MicroRNAs (miRNAs) play important roles in response of plants to biotic and abiotic stresses. Aluminum (Al) toxicity is a major factor limiting plant growth in acidic soils. However, there has been limited report on the involvement of miRNAs in response of plants to toxic Al3+. To identify Al3+-responsive miRNAs at whole-genome level, high-throughput sequencing technology was used to sequence libraries constructed from root apices of the model legume plant Medicago truncatula treated with and without Al3+. High-throughput sequencing of the control and two Al3+-treated libraries led to generation of 17.1, 14.1 and 17.4 M primary reads, respectively. We identified 326 known miRNAs and 21 new miRNAs. Among the miRNAs, expression of 23 miRNAs was responsive to Al3+, and the majority of Al3+-responsive mRNAs was down-regulated. We further classified the Al3+-responsive miRNAs into three groups based on their expression patterns: rapid-responsive, late-responsive and sustained-responsive miRNAs. The majority of Al3+-responsive miRNAs belonged to the ‘rapid-responsive’ category, i.e. they were responsive to short-term, but not long-term Al3+ treatment. The Al3+-responsive miRNAs were also verified by quantitative real-time PCR. The potential targets of the 21 new miRNAs were predicted to be involved in diverse cellular processes in plants, and their potential roles in Al3+-induced inhibition of root growth were discussed. These findings provide valuable information for functional characterization of miRNAs in Al3+ toxicity and tolerance.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
MicroRNA (miRNA) is one type of endogenous non-coding small RNAs with approximate length of 21 nt, and is a large family of small RNA (sRNA). miRNAs have been found in animals (Lee and Ambros 2001) and plants (Llave et al. 2002). miRNA-like small interfering RNAs (siRNAs) were also reported in fungi (Lee et al. 2010). Similar to siRNAs, miRNAs play an important role in regulation of gene expression. The major difference between miRNA and siRNA lies in their way of generation (Jones-Rhoades et al. 2006). siRNAs are processed from long, double-strand RNAs; whereas miRNAs are generated from single-strand RNAs with stem-loop structure, called pre-miRNAs (Bartel 2004). The generation of miRNA in plants differs from animals. In plants, the primary transcripts of miRNAs (pri-miRNAs) which are translated by RNA polymerase II are digested by DCL1, a dicer like enzyme, leading to the generation of pre-miRNAs, thereafter pre-miRNAs were digested by DCL1 to produce miRNA/miRNA* duplexes (Kurihara and Watanabe 2004). Mature miRNAs which depart from miRNA/miRNA* are loaded into AGO1, an argonaute protein, to assemble RNA-induced silence complex (RISC) (Jones-Rhoades et al. 2006). Multiple proteins participate in this process, including HYPONASTIC LEAVESI (HYL1) (Lu and Fedoroff 2000; Wu et al. 2007), SERRATE (SE) (Lobbes et al. 2006), nuclear cap binding complex (CBC) (Fang and Spector 2007), HUA ENHANCER1 (HEN1) (Park et al. 2002), and DAWDLE (Yu et al. 2008). miRNAs direct RISC to their target mRNAs, and repress the expression of their target genes by cleavage or inhibition of translation (Jones-Rhoades et al. 2006). In a recent study, Wu et al. (2010) reported that miRNAs can also regulate expression of target genes by DNA methylation.
It has been widely reported that miRNAs are involved in the regulation of numerous physiological processes in plants, including seed germination (Reyes and Chua 2007), flower development (Chen 2004), root development (Gutierrez et al. 2009). There is also a growing body of evidence showing that miRNAs play a role in transduction of hormonal signals in plants, such as auxin (Guo et al. 2005; Meng et al. 2009), abscisic acid (Reyes and Chua 2007) and gibberellins (Achard et al. 2004). In addition, the involvement of miRNAs in responses of plants to biotic and abiotic stresses has been reported (for example, Sunkar and Zhu 2004). These include cold (Jian et al. 2010), drought (Li et al. 2008a; Trindade et al. 2009; Wang et al. 2011), salinity (Ding et al. 2009; Liu et al. 2008), nutrition deficiency (Bari et al. 2006; Liang et al. 2010; Pant et al. 2008), heavy metals stress (Zhou et al. 2008) and oxidative stress (Sunkar et al. 2006).
Aluminum (Al) is the most abundant metal in the Earth’s crust and usually occurs in non-phytotoxic forms of aluminosilicate under most conditions. However, it is solubilized to phytotoxic Al3+ species in acidic soils, and becomes a major factor limiting crop production and yield in the acid soils. Inhibition of root elongation is one of the earliest and most distinct symptoms of Al3+ phytotoxicity (Ryan et al. 1993). The root apex in general and the root transition zone in particular have been identified as critical sites for sensing Al3+ toxicity and expressing tolerance to Al3+ (Ryan et al. 1993; Sivaguru and Horst 1998). Many physiological processes have been identified to be associated with Al3+ toxicity and tolerance (Rengel and Zhang 2003; Ryan et al. 2011). However, the primary mechanisms underlying the Al3+ phytotoxicity remain largely unknown and elusive. The involvements of miRNAs in mineral stress have been reported (see review Sunkar et al. 2007), but there has been no report on systemic identification of Al3+-responsive miRNAs and their targets at the global genome level by high-throughput sequencing. Medicago truncatula is an annual legume species distinguished by its small diploid genome and easy transformation, and has been used as a model plant to study functional genomics of legume plants (Trinh et al. 1998). Identification of new miRNAs in plants on a genome-wide scale is one of the essential steps for functional characterization of miRNAs. To understand the role of miRNAs in response of plants to Al3+ toxicity, we identified a number of conserved and non-conserved miRNAs that were responsive to toxic Al3+ by high-throughput sequencing, and their potential role in mediation of Al3+-induced inhibition of root growth and development was discussed.
Materials and methods
Plant materials and growth conditions
Seeds of Medicago truncatula (cv. Jemalong A17, kindly provided by Dr. Carroll Vance, USDA-ARS, Plant Science Research, St. Paul, MN, USA), were soaked in concentrated, anhydrous sulfuric acid for about 5 min to scarify seed coat, and then washed thoroughly with water. After kept at 4°C in 0.8% agar plates for 3 days, the seeds were germinated at 25°C in dark for 2 days. Seedlings were grown hydroponically in aerated nutrient solution. The nutrient solution contained 1 mM NH4NO3, 2.5 mM KNO3, 1 mM KH2PO4, 1 mM MgSO4, 0.25 mM K2SO4, 0.25 mM CaCl2, 100 μM FeNaEDTA, 30 μM H3PO3, 5 μM MnSO4, 1 μM ZnSO4, 1 μM CuSO4, 0.7 μM Na2MoO4, 1 mM NH4NO3, 2.5 mM KNO3 and 50 μM KCl at pH 5.8. Seedlings were grown in a growth chamber under conditions of a 16/8 h light/dark cycle at 25°C for 1 week. During seedling growth, the nutrient solution was changed every 2 days.
Aluminum treatment
Seven-day-old M. truncatula seedlings were exposed to solution containing 0.5 mM CaCl2 with 10 μM AlCl3 (pH 4.5) for varying periods to determine the effect of Al3+ on root growth, while M. truncatula seedlings of the same age incubated in 0.5 mM CaCl2 solution (pH 4.5) were used as controls. Root tips (≈1.5 cm in length) were collected to isolate total RNA for construction of sRNA libraries after exposure to AlCl3 solution for 4 and 24 h, respectively. The corresponding root tips grown in the control solution were used to construct library of sRNA for control.
Construction and sequencing of sRNA libraries
Total RNA was isolated from root tips of the following three groups: control group (CK), and groups treated with Al for 4 h (Al4) and 24 h (Al24), respectively. sRNA libraries were constructed by the methods described previously (Hafner et al. 2008; Wang et al. 2011). Briefly, sRNA with the length of 18–30 nt was separated and purified on a 15% TBE-urea denaturing PAGE gel. The 5′- and 3′-RNA adapters were ligated to sRNA with T4 RNA ligase (TaKaRa). Thereafter, the adapter-ligated sRNAs were transcribed to single-stranded cDNA with superscript II reverse transcriptase (Invitrogen). PCR was conducted using the primer designed according to the adapter sequence to amplify single-stranded cDNA template to double-stranded cDNA. PCR products were sequenced on a Solexa sequencer (Illumina) at the Beijing Genomics Institute (BGI), Shenzhen, China.
Analysis of high-throughput sequencing data
The raw reads obtained from the Solexa sequencer were cleaned by removing contaminant reads including those reads with 5′-primer contaminants, reads without 3′-primer, reads with poly A, and reads with length less than 18 nt. Clean reads were then used to analyze length distribution. Thereafter, clean reads were mapped to Medicago truncatula genome Mt3.5.1 (http://www.medicagohapmap.org/downloads_genome/Mt3.5/Mt3.5.1_pseudomolecules.tar.gz) using SOAP (Li et al. 2008b), with no mismatches being allowed.
All clean reads were annotated using different databases. The known miRNAs were annotated by comparing to miRBase 16 (ftp://mirbase.org/pub/mirbase/CURRENT/miRNA.dat.gz). rRNAs, scRNAs, snoRNAs, snRNAs and tRNAs were annotated by BLASTn to NCBI Genbank database and Rfam database (e = 0.01). Small interfering RNA (siRNA) is a double-strand RNA with 22–24 nt in length, each strand is 2 nt longer than the other on the 3′ end. According to this structural feature, we aligned tags from clean reads to each other to find those sRNAs that meet this criterion. These tags may be potential siRNA candidates and were removed from new miRNA analysis. Reads that were not annotated were used to predict new miRNA.
Pre-miRNA candidates were predicted using MIREAP (http://sourceforge.net/projects/mireap/). Parameters were set as follows: minimal miRNA sequence length of 18 nt; maximal miRNA sequence length of 25 nt; minimal miRNA reference sequence length of 20 nt; and maximal miRNA reference sequence length of 23 nt. Maximal copy number of miRNAs on reference was set to be 20 nt, and maximal free energy allowed for a miRNA precursor was −18 kcal/mol. Maximal space between miRNA and miRNA* was set to be 300 nt. Minimal base pairs of miRNA and miRNA* were taken as 16; maximal bulge of miRNA and miRNA* was 4. Maximal asymmetry of miRNA/miRNA* duplex was 4 nt. Flank sequence length of miRNA precursors was 20 nt. New miRNAs’ pre-miRNA stem-loop structure was constructed by m-fold (Zuker 2003).
Target predicting was carried out following the rules proposed by Allen et al. (2005): (1) no more than four mismatches between sRNA and target (G–U bases count as 0.5 mismatches); (2) no more than two adjacent mismatches in the miRNA/target duplex; (3) no adjacent mismatches in positions 2–12 of the miRNA/target duplex (5′ of miRNA); (4) no mismatches in positions 10–11 of miRNA/target duplex; (5) no more than 2.5 mismatches in positions 1–12 of the miRNA/target duplex (5′ of miRNA); and (6) minimum free energy (MFE) of the miRNA/target duplex should be >75% of the MFE of the miRNA bound to its perfect complement.
In miRNA expression analysis, P value was calculated as:
Normalized read count was calculated as:
If one miRNA has no read in a library, the normalized read count of this miRNA in the library was arbitrarily set to be 0.001 for further calculation.
Quantitative real-time PCR
Total RNA was isolated from M. truncatula root tips exposed to different solutions identical to those described above using Trizol (Invitrogen) according to the manufacturer’s protocols. For determination of miRNA expression, RNAs were reverse-transcribed by One Step PrimeScript® miRNA cDNA Synthesis Kit (TaKaRa), which added a ploy (A) tail to the 3′-end of miRNA and with transcription leading by a known oligo-dT ligate. SYBR Premix Ex Tag II (TaKaRa) was used for qRT-PCR. Small nuclear RNA U6 was used as an internal reference. All primers used in this paper were given in Supplemental Table S1. qRT-PCR experiments were performed on Mx3000P™ PCR system (Agilent-Stratagene, USA). PCR program was set as: (1) 95°C 30 s; (2) 95°C, 5 s; thereafter 60°C, 30 s, 40 cycles.
Results
Deep-sequencing results of M. truncatula sRNA
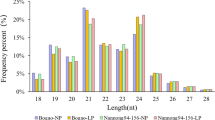
To identify miRNAs that were responsive to toxic Al3+, three sRNA libraries were constructed from M. truncatula root tips exposed to solution without Al3+ (control, CK) and solution containing 10 μM ACl3 for 4 h (Al4) and 24 h (Al24), respectively. A high-throughput sequencing technology, Solexa, was employed to sequence these libraries. This led to the generation of 17.1, 14.1 and 17.4 M raw reads from libraries of CK, Al4 and Al24, respectively (Table 1). After removal of contaminant reads, clean reads with length in the range between 18 and 30 nt were obtained from the raw reads. Most clean reads were those with a length of 19–26 nt (Fig. 1). Reads with length of 24 nt were the most abundant, followed by the reads with length of 21 nt (Fig. 1).

The length distribution of sRNAs in the libraries of control (CK), treatment with Al for 4 h (Al4) and 24 h (Al24)
About 11.8 M clean reads from CK (2,869,846 unique reads), 9.7 M clean reads from Al4 (1,598,588 unique reads) and 11.7 M clean reads (2,967,233 unique reads) from Al24 were mapped to M. truncatula genome sequence (Mt3.5.1, released in November 2010) with SOAP (Li et al. 2008b). miRNA, tRNA, siRNA, snRNA, snoRNA, rRNA, repeat regions, exon and intron RNA reads were annotated, respectively. Reads that were used for prediction of new miRNAs for CK, Al4 and Al24 were 6,179,361, 3,556,311 and 6,589,467, respectively (Table 1).
Identification of known miRNA in M. truncatula
To identify known miRNAs from the three libraries constructed in this work, clean reads were used to compare known M. truncatula miRNA precursors or mature miRNA sequence using miRBase 16.0 (Griffiths-Jones et al. 2008). There were 291, 262 and 282 miRNAs that were matched to the known M. truncatula miRNA from the libraries of CK, Al4 and Al24, respectively (Table 1; Supplemental Table S2). A total of 326 known miRNAs was identified in the three libraries. Some of the miRNAs exhibited extremely low expression levels in the three libraries (normalized count less than 10), i.e. mtr-miR171g and mtr-miR2588a. As the use of low expression miRNAs is prone to cause false results in express analysis, these miRNAs were not used for further analysis. After removing these miRNAs, a total of 67 known miRNAs belonging to 24 families was used for further analysis (Supplemental Table S3). The read counts differed among the 67 known miRNAs. For instance, mtr-miR166 was sequenced 391, 456 and 494K times in the libraries of CK, Al4 and Al24, respectively. This was the highest frequency among the miRNAs detected, and the frequency of mtr-miR156g was the second in terms of its sequenced frequency (Fig. 2).

Frequency distribution for those miRNAs that had the most reads from the three libraries (CK, Al4 and Al24)
Eight known miRNAs*, including mtr-miR1507*, mtr-miR1509*, mtr-miR1510a*, mtr-miR1510b*, mtr-miR2086*, mtr-miR2087*, mtr-miR2088a* and mtr-miR2089* were sequenced in the three libraries. Interestingly, the sequences of mtr-miR1510a* and mtr-miR2088a* occurred more frequently than those of their corresponding mature miRNAs, while the frequency of mtr-miR2089* sequence was comparable to its corresponding mtr-miR2089 (Table 2). A similar situation has been reported (Zhang et al. 2009). miRNAs are thought to be more stable than miRNA* in vivo due to their combination with RISC (Jones-Rhoades et al. 2006). There is a hypothesis that the amounts of miRNA are ten times more than those of miRNA* (Rajagopalan et al. 2006). Accordingly, our results indicate that mtr-miR1510a*, mtr-miR2088a* and mtr-miR2089* are likely to be true mature miRNAs.
Identification of novel miRNAs in M. truncatula
Based on the criteria for annotation of novel miRNA (Ambros et al. 2003; Meyers et al. 2008), a stem-loop precursor is a prerequisite for the annotation of new miRNA. The secondary structures of potential miRNA precursors were obtained using m-fold (Zuker 2003). There were 252 potential pre-miRNAs that met the requirements for new miRNAs (Supplemental Table S4). miRNA* has been used as strong evidence to identify miRNA (Meyers et al. 2008). We identified 21 candidates that had complementary miRNAs* (Table 3), suggesting that these candidates may be new miRNAs. The lengths of the 21 new miRNAs ranged from 21 to 23 nt with 85.7% being 21 nt in length, a classical length of miRNA. Among the 21 new miRNAs, 14 started with a 5′-uridine, which is also a hallmark of miRNA (Yao et al. 2007). The stem-loop structures of 21 new miRNA precursors are shown in Fig. 3 and Supplementary Figure S1. Several new miRNAs potentially expressed from multiple miRNA precursors, i.e. pmiR-007 and pmiR-017 (Table 3). Similarly to the known miRNAs, new miRNAs with normalized read-count less than ten in the three libraries were removed from the expression analysis, leading to nine miRNAs that were further analyzed.

Stem-loop structures for six novel miRNA precursors. Mature miRNAs of the remaining stem-loop structures were given in Supplemental Fig. S1. Mature miRNAs were marked by red and miRNA stars were marked by blue. The numbers indicate the nucleotide sites form 5′ of miRNA precursors
Identification of Al3+-responsive miRNAs
Depending on their read counts, results obtained from the high-throughput sequencing can also be used to compare the difference in expression of miRNAs among different treatments. For comparison among the inter-libraries, the reads of the three libraries had to be normalized. In the libraries of Al4 and Al24, miRNAs were designated as ‘up-regulated’ if their normalized read counts were greater than those in the control library, the fold-changes were greater than 2 and P < 0.05. Similarly, miRNAs were designated as ‘down-regulated’ if their normalized read counts in Al4 and Al24 libraries were lower than those in the control library, the fold-changes were less than 0.5 and P < 0.05. We identified 20 miRNAs belonging to 15 families as down-regulated and 3 miRNAs belonging to 3 families as up-regulated in Al4 library by these criteria (Fig. 4; Supplemental Table S5). There were four down-regulated miRNAs belonging to four families, and one up-regulated in the Al24 library (Fig. 4; Supplemental Table S5). These results indicate that the number of Al3+-induced down-regulated miRNAs is greater than that of up-regulated miRNAs.

Comparison of miRNA expression among the three libraries (CK, Al4 and Al24). To identify Al3+-responsive miRNAs, we compared the normalized expression of miRNAs in the three libraries (CK, Al4 and Al24). The relative change in response to Al treatment was expressed as log2 (Al4/CK) and log2 (Al24/CK). A positive value indicates a higher miRNA expression in the Al4 and Al24 than in the CK library, while a negative value means a lower miRNA expression. Star marks mean that the |log2 (fold change in Al4) | or |log2 (fold change in Al24)| >1, and P < 0.05
Samples were collected after exposure of M. truncatula seedlings to solutions containing toxic Al3+ for 4 and 24 h, and referred to the two treatments as ‘short-term’ and ‘long-term’ Al3+ treatment, respectively. Among the identified Al3+-responsive miRNAs, we further classified them into three groups based on their expression patterns. The changes in miRNA expression in response to treatment with 4 or 24-h Al treatment exclusively were referred to as ‘rapid responsive’ and ‘late responsive’ miRNAs, respectively, while the changes in miRNA expression in response to both 4 and 24-h Al treatment were referred to as ‘sustained responsive’ miRNAs. Among the Al3+-responsive miRNAs, 18 miRNAs belonging to 13 families were classified as ‘rapid responsive’, 4 miRNAs belonging to 4 families were categorized as ‘sustained responsive’ (Table 4). In contrast to the ‘rapid’ and ‘sustained responsive’ miRNAs, only one miRNA was found to be ‘late responsive’ miRNA (Table 4). These results reveal that the majority of Al3+-responsive miRNAs is likely to be associated with rapid changes in physiological processes in response to Al3+. miR390 was the only late responsive miRNA (Table 5). Recent reports demonstrated that miR390 regulates lateral root development (Marin et al. 2010; Yoon et al. 2010).
Verification of high-throughput sequencing data by quantitative real-time PCR
To verify the data obtained from the high-throughput sequencing, we studied the responses of the Al3+-responsive mRNAs obtained by the high-throughput sequencing to Al3+ treatment by qRT-PCR (Table 6). Only one member of each Al3+-responsive miRNA family was selected for qRT-PCR analysis because miRNAs within the same family have similar mature miRNA sequences and qRT-PCR can hardly distinguish them. In general, the changes in Al3+-responsive miRNAs obtained by qRT-PCR were comparable to those obtained by the deep-sequencing. The time-course of many miRNAs examined by qRT-PCR in response to Al3+ showed a rapid decrease with short-term Al3+ treatment (1–12 h) and recovered after long-term (12–48 h) exposure to Al3+, i.e. mtr-miR159a, mtr-miR162, mtr-miR396a, mtr-miR1507, mtr-miR2088a, mtr-miR2089, mtr-miR2597, mtr-miR2668, pmiR-003 and pmiR-008. These results were correlated with the miRNAs that exhibited rapid response to Al3+ measured by the high-throughput sequencing. Furthermore, expression of sustained Al3+-responsive miRNAs such as mtr-miR1510a, mtr-miR2199, pmiR-012 pmiR-014 and pmiR-021 showed steady changes when treated with Al3+. In addition, the only late Al3+-responsive miRNA, mtr-miR390, displayed slight down-regulation by treatment with short-term Al3+ and significant up-regulation after long-term expose to Al3+.
Expression of several miRNAs in response to Al3+ obtained by the high-throughput sequencing differed from that obtained by the qRT-PCR. For example, deep-sequencing results showed that there was a significant change in expression of miR160 in response to Al3+ treatment, while their expression was little responsive to Al3+ treatment when determined by the qRT-PCR (Table 6). Note that no read-counts were found for pmiR-021 in the control library, leading to very high values for the fold changes in Al4 and Al24 libraries. However, the results from the qRT-PCR revealed that the Al3+-induced changes in expression were much lower than those from the deep-sequencing results. We speculate that these differences may result from the higher sensitivity and less specificity of qRT-PCR than those of the deep-sequencing technology. Despite of the differences in response of miRNAs to Al3+ treatment, the majority of miRNAs displayed comparable expression patterns in response to Al3+ by the qRT-PCR and the high-throughput sequencing.
Prediction of target for new miRNAs
Plant miRNAs are highly complementary to their targets (Jones-Rhoades and Bartel 2004), and this feature has been used to predict targets of miRNAs in plants (Rhoades et al. 2002). The targets for the new miRNAs identified in the present study were predicted using Mt3.5.1 according to the rules set by Allen et al. (2005). The predicted targets for the new miRNAs were involved in many plant physiological processes, including plant development, defense, ion transport (Supplemental Table S6). For example, the predicted target for pmiR-003 was a TIR-NBS-LRR resistance protein which is involved in plant defense (Noutoshi et al. 2005; Mestre and Baulcombe 2006; Yang et al. 2008).
Discussion
Al3+ phytotoxicity is a major factor limiting plant growth in acidic soils worldwide (Kochian et al. 2005). Although extensive studies on identification of genes responsible for Al phytotoxicity and tolerance have been conducted (see review Ryan et al. 2011), there has been little information on the roles of miRNAs in Al3+ phytotoxicity, especially at whole genome level. In the present study, we constructed three libraries: control, without Al3+ treatment (CK); treatment with Al3+ for 4 h (Al4); and 24 h (Al24) from M. truncatula root tips. Identification of miRNAs in M. truncatula by the high-throughput sequencing technology such as Solexa or Roche 454 has been reported in several recent studies (Szittya et al. 2008; Jagadeeswaran et al. 2009; Lelandais-Briere et al. 2009). 375 miRNAs in M. truncatula have been identified using the database of miRBase (Griffiths-Jones et al. 2008). In the present study, we sequenced M. truncatula root tips at the whole-genome level by Solexa technology. The three libraries with greater than 45 M reads in total were the most abundant for the identification of miRNAs in M. truncatula. The obtained reads allowed us to analyze the miRNAs with low abundance. In addition, the use of latest M. truncatula genomic database (Mt3.5.1) in the present study also facilitates identification of miRNAs as the database contains more sequence information for M. truncatula. In contrast to other studies of identification of miRNAs in M. truncatula, roots particularly root tips, were used to isolate miRNAs.
In the present study, a total of 326 known miRNA and 33 new miRNA was identified; among them, 23 miRNAs were identified to be responsive to Al3+. Zhou et al. (2008) identified a number of Al3+-responsive miRNAs by bioinformatic approach. However, our findings differed from those reported by Zhou et al. (2008). For instance, Zhou et al. (2008) reported that miR171, miR319, miR393, and miR519 are up-regulated, and that miR166 and miR398 are down-regulated in response to Al3+ treatment. In contrast, we found that expression of miR393 and miR398 were too low to be used for further expression analysis in the three libraries. Furthermore, we failed to sequence miR519, and found that miR519 was not recorded in the miRBase database. In the present study, we observed that miR166 and miR171 were not responsive to Al3+. Moreover, we also found that miR319 was a rapid responsive miRNA such that it was down-regulated after treatment with Al3+ for 4 h, while its expression was not responsive to the treatment with Al3+ for 24 h. Several possible explanations may account for the differences between our findings and those reported by Zhou et al. (2008). The miRNAs were predicted with a bioinformatic approach by Zhou et al. (2008). This approach has been suggested to be insufficient for the identification of miRNAs (Meyers et al. 2008). The application of high-throughput sequencing technology to identify miRNAs in the present study can overcome some problems associated with bioinformatic approach. In addition, since the root tip is the critical site for sensing toxic Al3+ (Ryan et al. 1993), we used root tips to construct the sRNA library, while leaves rather than root tips were used in studies on the effect of Al3+ on the expression of miRNAs by Zhou et al. (2008).
In addition to Al toxicity, plants grown in acidic soil also suffer from Mn toxicity (Taylor et al. 1998). There are reports demonstrating the cross-talk between Al3+ and Mn toxicity. For instance, Yang et al. (2009) found that excess Mn can increase the accumulation of Al3+ in soybean roots, and that high Al3+ concentration alleviates Mn toxicity. More recently, Valdes-Lopez et al. (2010) reported that the expression of miR319, and miR159 is up-regulated in roots and nodules upon exposure of Phaseolus vulgaris to toxic level of Mn. Our results showed that the Mn-responsive miRNAs were also responsive to Al3+ such that mtr-miR319 and mtr-miR159a were rapid responsive, down-regulated miRNAs. These findings indicate that the metal toxicity associated with Al3+ and Mn may affect plant by targeting the same miRNAs.
Recent studies revealed that Al3+-induced inhibition of root elongation may result from disruption of auxin distribution in roots by targeting auxin polar transporters PIN2 and AUX1 (Kollmeier et al. 2000; Doncheva et al. 2005; Sun et al. 2010). The Al3+-responsive miR160 and miR390 have been shown to modulate root growth by regulating auxin response factor (ARF) (Wang et al. 2005; Marin et al. 2010; Yoon et al. 2010). We found that mtr-miR160 family was rapidly, down-regulated by Al3+. It has been reported that targets of miR160 are ARF10 and ARF16 that regulate the development of root cap (Wang et al. 2005). Therefore, the rapid down-regulation of miR160 in response to Al3+ treatment may be involved in inhibition of root elongation by up-regulating ARF.
miR390 was the only late responsive miRNA such that its expression was down-regulated in the Al24 library. miR390 has been reported to control the generation of tasiRNA (TAS3-derived trans-acting short-interfering RNA), which regulates lateral root emergence by targeting transcription factors such as ARF2, ARF3 and ARF4 (Marin et al. 2010; Yoon et al. 2010). As root elongation in M. truncatula can be rapidly suppressed by Al3+ (Sun et al. 2007), the down-regulation of miR390 after 24 h of exposure to Al3+ may be an indirect effect of Al3+.
We identified five new Al3+-responsive miRNAs in M. truncatula. Two of them, pmiR-003 and pmiR-008 were predicted to target mRNA of the TIR-NBS-LRR resistance protein. NBS-LRR proteins are considered to be involved in resistance to many types of pathogens (Hammond-Kosack and Parker 2003). Furthermore, the latest research revealed that this family plays an important role in the symbiotic process in legume species (Colebatch et al. 2004). We found that pmiR-003 and pmiR-008 were rapidly responsive miRNAs, and that both were down-regulated in Al4. These results may suggest that pathogens resistance protein may also be involved in Al3+ toxicity.
In summary, we identified a group of new miRNAs in the legume model plant of M. truncatula by a deep-sequencing method, and discovered a number of miRNAs that were responsive to Al3+. These findings provide valuable information for functional characterization of miRNAs in response of legume plants to Al3+ phytotoxicity.
Abbreviations
- miRNA:
-
MicroRNA
- pre-miRNA:
-
MicroRNA precursor
- pri-miRNA:
-
MicroRNA primary transcript
- miRNA*:
-
MicroRNA star
- qRT-PCR:
-
Quantitative real-time PCR
References
Achard P, Herr A, Baulcombe DC, Harberd NP (2004) Modulation of floral development by a gibberellin-regulated microRNA. Development 131:3357–3365
Allen E, Xie Z, Gustafson AM, Carrington JC (2005) MicroRNA-directed phasing during trans-acting siRNA biogenesis in plants. Cell 121:207–221
Alonso-Peral MM, Li JY, Li YJ, Allen RS, Schnippenkoetter W, Ohms S, White RG, Millar AA (2010) The microRNA159-regulated GAMYB-like genes inhibit growth and promote programmed cell death in Arabidopsis. Plant Physiol 154:757–771
Ambros V, Bartel B, Bartel DP, Burge CB, Carrington JC, Chen X, Dreyfuss G, Eddy SR, Griffiths-Jones S, Marshall M, Matzke M, Ruvkun G, Tuschl T (2003) A uniform system for microRNA annotation. RNA 9:277–279
Bari R, Pant BD, Stitt M, Scheible WR (2006) PHO2, microRNA399, and PHR1 define a phosphate-signaling pathway in plants. Plant Physiol 141:988–999
Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116:281–297
Chen X (2004) A microRNA as a translational repressor of APETALA2 in Arabidopsis flower development. Science 303:2022–2025
Colebatch G, Desbrosses G, Ott T, Krusell L, Montanari O, Kloska S, Kopka J, Udvardi MK (2004) Global changes in transcription orchestrate metabolic differentiation during symbiotic nitrogen fixation in Lotus japonicus. Plant J 39:487–512
Ding D, Zhang L, Wang H, Liu Z, Zhang Z, Zheng Y (2009) Differential expression of miRNAs in response to salt stress in maize roots. Ann Bot 103:29–38
Doncheva S, Amenos M, Poschenrieder C, Barcelo J (2005) Root cell patterning: a primary target for aluminum toxicity in maize. J Exp Bot 56:1213–1220
Fang Y, Spector DL (2007) Identification of nuclear dicing bodies containing proteins for microRNA biogenesis in living Arabidopsis plants. Curr Biol 17:818–823
Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ (2008) miRBase: tools for microRNA genomics. Nucleic Acids Res 36:D154–D158
Guo HS, Xie Q, Fei JF, Chua NH (2005) MicroRNA directs mRNA cleavage of the transcription factor NAC1 to downregulate auxin signals for Arabidopsis lateral root development. Plant Cell 17:1376–1386
Gutierrez L, Bussell JD, Pacurar DI, Schwambach J, Pacurar M, Bellini C (2009) Phenotypic plasticity of adventitious rooting in Arabidopsis is controlled by complex regulation of AUXIN RESPONSE FACTOR transcripts and microRNA abundance. Plant Cell 21:3119–3132
Hafner M, Landgraf P, Ludwig J, Rice A, Ojo T, Lin C, Holoch D, Lim C, Tuschl T (2008) Identification of microRNAs and other small regulatory RNAs using cDNA library sequencing. Methods 44:3–12
Hammond-Kosack KE, Parker JE (2003) Deciphering plant-pathogen communication: fresh perspectives for molecular resistance breeding. Curr Opin Biotech 14:177–193
Jagadeeswaran G, Zheng Y, Li YF, Shukla LI, Matts J, Hoyt P, Macmil SL, Wiley GB, Roe BA, Zhang WX, Sunkar R (2009) Cloning and characterization of small RNAs from Medicago truncatula reveals four novel legume-specific microRNA families. New Phytol 184:85–98
Jian X, Zhang L, Li G, Wang X, Cao X, Fang X, Chen F (2010) Identification of novel stress-regulated microRNAs from Oryza sativa L. Genomics 95:47–55
Jones-Rhoades MW, Bartel DP (2004) Computational identification of plant microRNAs and their targets, including a stress-induced miRNA. Mol Cell 14:787–799
Jones-Rhoades MW, Bartel DP, Bartel B (2006) MicroRNAS and their regulatory roles in plants. Annu Rev Plant Biol 57:19–53
Kochian LV, Pineros MA, Hoekenga OA (2005) The physiology, genetics and molecular biology of plant aluminum resistance and toxicity. Plant Soil 274:175–195
Kollmeier M, Felle HH, Horst WJ (2000) Genotypical differences in aluminum resistance of maize are expressed in the distal part of the transition zone. Is reduced basipetal auxin flow involved in inhibition of root elongation by aluminum? Plant Physiol 122:945–956
Kurihara Y, Watanabe Y (2004) Arabidopsis micro-RNA biogenesis through Dicer-like 1 protein functions. Proc Natl Acad Sci USA 101:12753–12758
Lee RC, Ambros V (2001) An extensive class of small RNAs in Caenorhabditis elegans. Science 294:862–864
Lee HC, Li L, Gu W, Xue Z, Crosthwaite SK, Pertsemlidis A, Lewis ZA, Freitag M, Selker EU, Mello CC, Liu Y (2010) Diverse pathways generate microRNA-like RNAs and Dicer-independent small interfering RNAs in fungi. Mol Cell 38:803–814
Lelandais-Briere C, Naya L, Sallet E, Calenge F, Frugier F, Hartmann C, Gouzy J, Crespi M (2009) Genome-wide Medicago truncatula small RNA analysis revealed novel microRNAs and isoforms differentially regulated in roots and nodules. Plant Cell 21:2780–2796
Li WX, Oono Y, Zhu J, He XJ, Wu JM, Iida K, Lu XY, Cui X, Jin H, Zhu JK (2008a) The Arabidopsis NFYA5 transcription factor is regulated transcriptionally and posttranscriptionally to promote drought resistance. Plant Cell 20:2238–2251
Li R, Li Y, Kristiansen K, Wang J (2008b) SOAP: short oligonucleotide alignment program. Bioinformatics 24:713–714
Liang G, Yang FX, Yu DQ (2010) MicroRNA395 mediates regulation of sulfate accumulation and allocation in Arabidopsis thaliana. Plant J 62:1046–1057
Liu PP, Montgomery TA, Fahlgren N, Kasschau KD, Nonogaki H, Carrington JC (2007) Repression of AUXIN RESPONSE FACTOR10 by microRNA160 is critical for seed germination and post-germination stages. Plant J 52:133–146
Liu HH, Tian X, Li YJ, Wu CA, Zheng CC (2008) Microarray-based analysis of stress-regulated microRNAs in Arabidopsis thaliana. RNA 14:836–843
Liu DM, Song Y, Chen ZX, Yu DQ (2009) Ectopic expression of miR396 suppresses GRF target gene expression and alters leaf growth in Arabidopsis. Physiol Plant 136:223–236
Liu XD, Huang J, Wang Y, Khanna K, Xie ZX, Owen HA, Zhao DZ (2010) The role of floral organs in carpels, an Arabidopsis loss-of-function mutation in microRNA160a, in organogenesis and the mechanism regulating its expression. Plant J 62:416–428
Llave C, Kasschau KD, Rector MA, Carrington JC (2002) Endogenous and silencing-associated small RNAs in plants. Plant Cell 14:1605–1619
Lobbes D, Rallapalli G, Schmidt DD, Martin C, Clarke J (2006) SERRATE: a new player on the plant microRNA scene. EMBO Report 7:1052–1058
Lu C, Fedoroff N (2000) A mutation in the Arabidopsis HYL1 gene encoding a dsRNA binding protein affects responses to abscisic acid, auxin, and cytokinin. Plant Cell 12:2351–2366
Marin E, Jouannet V, Herz A, Lokerse AS, Weijers D, Vaucheret H, Nussaume L, Crespi MD, Maizel A (2010) miR390, Arabidopsis TAS3 tasiRNAs, and their AUXIN RESPONSE FACTOR targets define an autoregulatory network quantitatively regulating lateral root growth. Plant Cell 22:1104–1117
Meng Y, Huang F, Shi Q, Cao J, Chen D, Zhang J, Ni J, Wu P, Chen M (2009) Genome-wide survey of rice microRNAs and microRNA-target pairs in the root of a novel auxin-resistant mutant. Planta 230:883–898
Mestre P, Baulcombe DC (2006) Elicitor-mediated oligomerization of the tobacco N disease resistance protein. Plant Cell 18:491–501
Meyers BC, Axtell MJ, Bartel B, Bartel DP, Baulcombe D, Bowman JL, Cao X, Carrington JC, Chen X, Green PJ, Griffiths-Jones S, Jacobsen SE, Mallory AC, Martienssen RA, Poethig RS, Qi Y, Vaucheret H, Voinnet O, Watanabe Y, Weigel D, Zhu JK (2008) Criteria for annotation of plant microRNAs. Plant Cell 20:3186–3190
Noutoshi Y, Ito T, Seki M, Nakashita H, Yoshida S, Marco Y, Shirasu K, Shinozaki K (2005) A single amino acid insertion in the WRKY domain of the Arabidopsis TIR-NBS-LRR-WRKY-type disease resistance protein SLH1 (sensitive to low humidity 1) causes activation of defense responses and hypersensitive cell death. Plant J 43:873–888
Pant BD, Buhtz A, Kehr J, Scheible WR (2008) MicroRNA399 is a long-distance signal for the regulation of plant phosphate homeostasis. Plant J 53:731–738
Park W, Li J, Song R, Messing J, Chen X (2002) CARPEL FACTORY, a Dicer homolog, and HEN1, a novel protein, act in microRNA metabolism in Arabidopsis thaliana. Curr Biol 12:1484–1495
Rajagopalan R, Vaucheret H, Trejo J, Bartel DP (2006) A diverse and evolutionarily fluid set of microRNAs in Arabidopsis thaliana. Genes Dev 20:3407–3425
Rengel Z, Zhang WH (2003) Role of dynamics of intracellular calcium in aluminum-toxicity syndrome. New Phytol 159:295–314
Reyes JL, Chua NH (2007) ABA induction of miR159 controls transcript levels of two MYB factors during Arabidopsis seed germination. Plant J 49:592–606
Rhoades MW, Reinhart BJ, Lim LP, Burge CB, Bartel B, Bartel DP (2002) Prediction of plant microRNA targets. Cell 110:513–520
Rodriguez RE, Mecchia MA, Debernardi JM, Schommer C, Weigel D, Palatnik JF (2010) Control of cell proliferation in Arabidopsis thaliana by microRNA miR396. Development 137:103–112
Ryan PR, Ditomaso JM, Kochian LV (1993) Aluminum toxicity in roots—an investigation of spatial sensitivity and the role of the root cap. J Exp Bot 44:437–446
Ryan PR, Tyerman SD, Sasaki T, Furuichi T, Yamamoto Y, Zhang WH, Delhaize E (2011) The identification of aluminum-resistance genes provides opportunities for enhancing crop production on acid soils. J Exp Bot 62:9–20
Schommer C, Palatnik JF, Aggarwal P, Chetelat A, Cubas P, Farmer EE, Nath U, Weigel D (2008) Control of jasmonate biosynthesis and senescence by miR319 targets. Plos Biol 6:1991–2001
Sivaguru M, Horst WJ (1998) The distal part of the transition zone is the most aluminum-sensitive apical root zone of maize. Plant Physiol 116:155–163
Sun P, Tian QY, Zhao MG, Dai XY, Huang JH, Li LH, Zhang WH (2007) Aluminum-induced ethylene production is associated with inhibition of root elongation in Lotus japonicus L. Plant Cell Physiol 48:1229–1235
Sun P, Tian QY, Chen J, Zhang WH (2010) Aluminum-induced inhibition of root elongation in Arabidopsis is mediated by ethylene and auxin. J Exp Bot 61:347–356
Sunkar R, Zhu JK (2004) Novel and stress-regulated microRNAs and other small RNAs from Arabidopsis. Plant Cell 16:2001–2019
Sunkar R, Kapoor A, Zhu JK (2006) Posttranscriptional induction of two Cu/Zn superoxide dismutase genes in Arabidopsis is mediated by down-regulation of miR398 and important for oxidative stress tolerance. Plant Cell 18:2051–2065
Sunkar R, Chinnusamy V, Zhu J, Zhu JK (2007) Small RNAs as big players in plant abiotic stress responses and nutrient deprivation. Trends Plant Sci 12:301–309
Szittya G, Moxon S, Santos DM, Jing R, Fevereiro MPS, Moulton V, Dalmay T (2008) High-throughput sequencing of Medicago truncatula short RNAs identifies eight new miRNA families. BMC Genomics 9:593
Taylor GJ, Blamey FPC, Edwards DG (1998) Antagonistic and synergistic interactions between aluminum and manganese on growth of Vigna unguiculata at low ionic strength. Physiol Plant 104:183–194
Trindade I, Capitao C, Dalmay T, Fevereiro MP, Santos DM (2009) miR398 and miR408 are up-regulated in response to water deficit in Medicago truncatula. Planta 231:705–716
Trinh TH, Ratet P, Kondorosi E, Durand P, Kamate K, Bauer P, Kondorosi A (1998) Rapid and efficient transformation of diploid Medicago truncatula and Medicago sativa ssp. falcata lines improved in somatic embryogenesis. Plant Cell Rep 17:345–355
Valdes-Lopez O, Yang SS, Aparicio-Fabre R, Graham PH, Reyes JL, Vance CP, Hernandez G (2010) MicroRNA expression profile in common bean (Phaseolus vulgaris) under nutrient deficiency stresses and manganese toxicity. New Phytol 187:805–818
Wang JW, Wang LJ, Mao YB, Cai WJ, Xue HW, Chen XY (2005) Control of root cap formation by microRNA-targeted auxin response factors in Arabidopsis. Plant Cell 17:2204–2216
Wang TZ, Chen L, Zhao MG, Tian QY, Zhang WH (2011) Identification of drought-responsive microRNAs and their targets in Medicago truncatula by genome-wide high-throughput sequencing and degradome analysis. BMC Genomics 12:367
Wu F, Yu L, Cao W, Mao Y, Liu Z, He Y (2007) The N-terminal double-stranded RNA binding domains of Arabidopsis HYPONASTIC LEAVES1 are sufficient for pre-microRNA processing. Plant Cell 19:914–925
Wu L, Zhou H, Zhang Q, Zhang J, Ni F, Liu C, Qi Y (2010) DNA methylation mediated by a microRNA pathway. Mol Cell 38:465–475
Yang S, Gao M, Xu C, Gao J, Deshpande S, Lin S, Roe BA, Zhu H (2008) Alfalfa benefits from Medicago truncatula: the RCT1 gene from M. truncatula confers broad-spectrum resistance to anthracnose in alfalfa. Proc Natl Acad Sci USA 105:12164–12169
Yang ZB, You JF, Xu MY, Yang ZM (2009) Interaction between aluminum toxicity and manganese toxicity in soybean (Glycine max). Plant Soil 319:277–289
Yao YY, Guo GG, Ni ZF, Sunkar R, Du JK, Zhu JK, Sun QX (2007) Cloning and characterization of microRNAs from wheat (Triticum aestivum L.). Genome Biol 8:120
Yoon EK, Yang JH, Lim J, Kim SH, Kim SK, Lee WS (2010) Auxin regulation of the microRNA390-dependent transacting small interfering RNA pathway in Arabidopsis lateral root development. Nucleic Acids Res 38:1382–1391
Yu B, Bi L, Zheng B, Ji L, Chevalier D, Agarwal M, Ramachandran V, Li W, Lagrange T, Walker JC, Chen X (2008) The FHA domain proteins DAWDLE in Arabidopsis and SNIP1 in humans act in small RNA biogenesis. Proc Natl Acad Sci USA 105:10073–10078
Zhang J, Xu Y, Huan Q, Chong K (2009) Deep sequencing of Brachypodium small RNAs at the global genome level identifies microRNAs involved in cold stress response. BMC Genomics 10:449
Zhou ZS, Huang SQ, Yang ZM (2008) Bioinformatic identification and expression analysis of new microRNAs from Medicago truncatula. Biochem Biophys Res Comm 374:538–542
Zhou LG, Liu YH, Liu ZC, Kong DY, Duan M, Luo LJ (2010) Genome-wide identification and analysis of drought-responsive microRNAs in Oryza sativa. J Exp Bot 61:4157–4168
Zuker M (2003) M-fold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31:3406–3415
Acknowledgments
This work was supported by Natural Science Foundation of China (90817011, 30788003 and 30800706) and the State Key Basic Research Development Program of China (2007CB106800) and State Key Laboratory of Vegetation and Environmental Chang.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Chen, L., Wang, T., Zhao, M. et al. Identification of aluminum-responsive microRNAs in Medicago truncatula by genome-wide high-throughput sequencing. Planta 235, 375–386 (2012). https://doi.org/10.1007/s00425-011-1514-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00425-011-1514-9