
Abstract
A simple, rapid and eco-friendly deep eutectic solvent-modified magnetic nanoparticles extraction method has been reported for ligand-less separation/preconcentration of lead(II) and cadmium(II) for the first time. The metal ions interact with the deep eutectic solvent adhering to the magnetic nanoparticles. Trapped analytes can be easily desorbed with 1.0 M nitric acid and determined by flame atomic absorption spectrometry. The effects of pH value, type of deep eutectic solvent, sample volume, nature and concentration of desorbing solution, ionic strength and extraction time on the extraction were optimized. Under the optimized experimental conditions, the detection limits (defined as 3Sb/m) were 0.4 and 0.1 μg L−1 and the linear dynamic ranges were 2 to 250 μg L−1 and 0.5 to 30 μg L−1 for lead and cadmium, respectively. The relative standard deviations for six replicate measurements at 150 and 20 μg L−1 levels of lead and cadmium were 1.8 and 2.1 %, respectively. A sample volume of 60 mL resulted in a preconcentration factor of 100. The sorbent showed high capacity for lead (25.0 mg g−1) and cadmium (23.7 mg g−1). The method was successfully applied to the determination of lead and cadmium in soil, hair and several water samples.

Deep eutectic solvent (DES) and Fe3O4 nanoparticles were added to the sample solution, and the DES containing the target ions was trapped on the sorbent and separated by means of a strong magnet. The analytes were desorbed by nitric acid, and the extracted metal ions were then directly submitted to FAAS for quantification
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
It is well known that lead and cadmium are toxic, and children are more sensitive to these metals than adults. U.S. Environmental Protection Agency (EPA) sorted lead and cadmium as a Group B2 (probable) human carcinogen [1]. Lead and cadmium are not necessary elements in the body. In recent years, concern has increased over the concentration of lead and cadmium in soil, drinking and natural waters. The World Health Organization (WHO) recommends the maximum allowable concentration limits of 10 and 3 ng mL−1 in drinking water [2] and 300 and 3 μg g−1 in soil for lead and cadmium, respectively [3] which requires a much greater sensitivity in measurement than is obtainable by flame atomic absorption spectrometry, the most attractive analytical method for metal ions determination. Therefore, a preliminary preconcentration of lead and cadmium is usually a necessary step. Procedures reported in the literature for the separation and preconcentration of lead and cadmium are generally solid-phase extraction (SPE) [1, 2], liquid-liquid extraction [4], cloud point extraction (CPE) [5], dispersive liquid–liquid microextraction (DLLME) [6], coprecipitation [7] and solidified floating organic drop microextraction (SFODME) [8].
SPE is one of the most attractive methods used for the separation and preconcentration of metal ions from complex matrices, mainly because trace analyte preconcentration and sample matrix removal are achieved at the same time [9]. However, the simplicity, selectivity and flexibility of working conditions of SPE are dependent on the proper choice of a sorbent. Among different sorbents, used for SPE, magnetic sorbents such as Fe3O4 have recently attracted much attention [10–12]. This is due to such advantages as high surface area, rapid isolation of sorbents from a large sample volume using a strong magnet, short extraction process, ease of preparation of sorbents and low cost [10–12]. However, bare MNPs suffer from the basic disadvantage of having no affinity for the target analyte. To overcome this problem, modification of magnetic nanoparticles with different organic ligands [12], imprinted polymers [13], and surfactants [14, 15] has been carried out. Implementation of these modifications, however, leads to serious risks for operators as well as damages to the environment due to the toxic reagents consumption and generation of the secondary waste. For this reason, recently, the modification of magnetic nanoparticles has been done with ionic liquids [11, 16]. Ionic liquids (ILs) are classified as green solvents and have excellent properties. However, most ILs suffers from some limitations such as poor biodegradability and varying toxicity and stability. Also, synthetic processes are non-environment friendly and relatively expensive [17].
Deep eutectic solvents (DESs), a new generation of green solvents, introduced by Abbot et al. (2003) have physico-chemical properties similar to those of ILs but overcome the limitations of ILs. These solvents are simply formed by mixing two or more safe, cheap, renewable, biocompatible and biodegradable organic components that are capable of associating with each other through hydrogen bonding and forming a compound that has a melting point far below that of either component. A number of DESs are prepared by simply mixing and heating organic halide salts such as choline chloride (i.e., a very cheap, biodegradable and non-toxic quaternary ammonium salt) as the hydrogen bond acceptor (HBA) with urea, organic acids, alcohols, amines or amides as the hydrogen bond donor (HBD). Thus, production of DES is much simpler and cheaper than that of ILs [18]. The physical properties of DESs such as freezing point, density and viscosity can simply be varied in a broad range by changing the relative amount of HBD and HBA. Among most DESs, choline chloride:urea (with approximate freezing point, density and viscosity of 12 °C, 1.25 g cm−3 and 750 cP, respectively) is the most popular one and have already been used successfully in various studies. The use of DES as a green solvent is growing in different research fields of chemistry including organic and polymer synthesis, electrochemistry, preparation, analysis and separation process [19–21]. However, its use in separation and analysis of metal ions is limited. In 2004, Abbott et al. [22] characterized the physical properties and phase behavior of a wide variety of DESs, and investigated the possibility of dissolution of metal oxides in them. In 2013, Tang et al. [23] used DESs as the extraction solvent in the headspace-microextraction technique and developed a procedure for the extraction of bioactive compounds. Habibi et al. designed a digestive method using DES for the determination of Cu, Fe and Zn in fish samples [24]. Soylak et al. (2014) [25] used DES for the extraction and determination of iron in liver samples. We have recently used DESs as the extraction solvent for separation and preconcentration of Pb and Cd from edible oil prior to their determination by electrothermal atomic absorption spectrometry [26]. Thus, DESs have a strong potential for routine trace metal analysis in biological samples, but their applicability as the extraction solvent from aqueous phase is limited due to their miscibility with water. According to our literature survey, there are only two reports on the use of DESs as the extraction solvent for the separation of proteins from aqueous media, but there is no report for the extraction of metal ions. In 2015, Wang et al. used DESs as the extraction solvent for the separation of proteins from aqueous solutions. In one study, they used the concept of salting out effect for the recovery of DES from aqueous solutions and added a relatively high amount of salt to the extraction vessel [27] while in another study, DES was sorbed on the surface of Fe3O4 modified with graphene oxide through an electrostatic interaction and then used for the extraction of protein from aqueous solutions [10].
In this study, another approach was considered for the possibility of using DESs in the extraction of metal ions from aqueous samples. In this attempt, the advantages of DESs and magnetic nanoparticles were combined, and a novel, rapid and simple deep eutectic solvent-mediated magnetic nanoparticles (DES-MNPs) extraction method was developed for the ligandless separation and preconcentration of lead and cadmium from aqueous samples. Thus, DES and Fe3O4 nanoparticles were simply added to the sample solution of analytes, and the DES containing the target ions was trapped on the sorbent and separated by the means of a strong magnet. The analytes were desorbed by nitric acid, and the extracted metal ions were then directly introduced to flame atomic absorption spectrometry (FAAS) for quantification. Compared with previous reports of using DES as the solvent for aqueous-phase extraction, the method is much simpler, improves the extraction time and cost of the process and combines the advantages of both DES and magnetic nanoparticles.
Experimental
Reagents and standard solution
All the chemicals used throughout this study were of the highest purity available and analytical reagent grades. Choline chloride (ChCl, > 98 %), iron (II) chloride tetrahydrate and iron (III) chloride hexahydrate were supplied by Sigma-Aldrich (http://www.sigmaaldrich.com/china-mainland/chemistry-product.html). High-purity ethylene glycol (EG), oxalic acid (Ox) and urea were purchased from Merck Company (Darmstadt, Germany, http://www.merck.de). Double distilled deionized water was used throughout the study. All the glassware was cleaned with 1 % (v/v) nitric acid and deionized water. Stock standard solutions (1000 mg L−1) of Pb(II) and Cd(II) were prepared from Pb(NO3)2 and Cd(NO3)2.4H2O respectively. Working solutions were prepared on a daily basis by serial dilutions of the stock solutions with double distilled water.
Apparatus
All the measurements were done with an Analytik Jena novAA 300 (model 330, Germany, http://www.analytik-jena.de) flame atomic absorption spectrometer. Hollow cathode lamps of lead and cadmium and air-acetylene flame were used in all the measurements. The operating conditions were as follows: wavelength of 283.3 nm for Pb and 228.8 nm for Cd, spectral slit of 1.2 nm and lamp current of 10 mA. The samples were introduced to FAAS using a single line flow injection system consisting a peristaltic pump (Ismatic, MS-REGLO/8-100, Switzerland) with silicone rubber tubing, and a rotary injection valve (Rheodyne, CA, USA) equipped with a loop of 100 μL capacity. A personal computer was used to record the absorbance signal profile. The pH measurements were carried out with a Metrohm pH meter (model 691, Switzerland, http://metrohm.com) using a combined glass-calomel electrode. In addition, for magnetic separations a strong neodymium-iron-boron (Nd2Fe12B) magnet (1.31 T) was used.
Synthesis of magnetite (Fe3O4) nanoparticles (MNPs)
Fe3O4 MNPs were prepared by the coprecipitation method [11]. Fifty mL of an aqueous solution containing FeCl3.6H2O (5.2 g) and FeCl2.4H2O (2.0 g) was heated at 80 °C for 15 min. Then, 10 mL of concentrated NH3 was added dropwise. N2 gas was used as a protective gas in the whole experiment. After completion of the reaction, the black precipitate was collected by an external magnetic field, washed with water and ethanol and dried in an oven at 80 °C. The average diameter of the obtained MNPs was approximately in the range of 53.0 ± 4.1 nm.
Preparation of deep eutectic solvents (DESs)
The DESs were synthesized by mixing mixture of ChCl with a different hydrogen bond donor including ethylene glycol, oxalic acid or urea, in a round-bottom flask [28]. The eutectic mixtures were prepared by stirring the two components at 100 °C until homogeneous colorless liquids ware formed.
Sample preparation
The human hair sample was belongs to a 28-year-old man (not colored, living in city of Yazd, Iran) and was prepared as described elsewhere [29] i.e., it was washed with chloroform, acetone and doubly distilled water, and dried at 60 °C. An exact amount of the dried sample (1.0 g) was weighed in a beaker and digested by 5 mL of concentrated HNO3 on a hot plate (initially at 100 °C for 45 min and then at 150 °C for 15 min). The mixture was cooled down to 70 °C, 20 mL of 30 % H2O2 was added to it and was heated to dryness to yield a whitish residue. Approximately, 10.0 mL of 0.1 mol L−1 HNO3 was added to the beaker and the contents were heated at 100 °C for several minutes. After cooling it to an ambient temperature, some water was added to it, the pH was adjusted to ~6.5, the mixture was diluted to 100 mL in a volumetric flask, and 60 mL of it was treated according to the given procedure.
A certain amount of the dried soil sample (0.2 g) was placed in the beaker and 10 mL of concentrated nitric acid was added to it. The content was heated on a hot plate to dryness. After cooling, a second 10-mL portion of the concentrated nitric acid was added, and the procedure of heating was repeated. Then, 10 mL of a concentrated hydrochloric acid was added to the beaker, and the solution was gently heated to complete dryness. After cooling, the residue was dissolved in 10 mL of 1 mol L−1 HCl, the mixture was filtered through a 0.45 μm Millipore filter, and the filtrate was diluted with distilled water [30]. The pH was adjusted to ~6.5 and treated according to the given procedure. For the recovery study of solid samples a precise amount of analytes was added to the samples prior to the digestion procedure.
Tap water, well water (Yazd, Iran), river water (Zayandeh Rood River, Esfahan, Iran), and Caspian Sea water were filtered through a 0.45 μm Millipore filter. The pH was adjusted and treated according to the given procedure.
General procedure
For the preconcentration of Pb(II) and Cd(II), the pH of 60 mL of aliquots of the standard or sample solution was adjusted to ~6.5 upon addition of diluted HCl or NaOH. Then, 200 μL of DES (ChCl-urea 1:2.5) and 20.0 mg of MNPs were added, and the mixture was stirred thoroughly for 10 min. At this stage, there occurs the interaction of DES with metal ions as well as its sorption to the MNPs. Subsequently, a strong magnet was placed at the bottom of the beaker, and the sorbent containing the analytes was collected. The bulk aqueous phase was easily decanted, and the analytes were desorbed upon addition of 600 μL of nitric acid (1.0 mol L−1). Finally, the sorbent was retained by means of a magnet, and the analytes in the supernatant solution were determined by flame atomic absorption spectrometry using a single line flow injection system.
Results and discussion
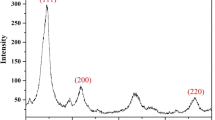
It was observed that, when nanoparticles of bare Fe3O4, alumina or TiO2 are added to the aqueous solution containing DES (ChCl-urea 1:2.5), the eutectic solvent acts as a hydroxy functionalized IL. It loses its liquid state and is sorbed on the surface of nanoparticles probably through strong hydrogen bonding and electrostatic interaction with the functional groups on the surface of particles, and separates from the aqueous solution. This was confirmed by comparing the FTIR spectra of pure DES with Fe3O4, alumina and TiO2 separated from the aqueous phase containing DES. Thus, when the extraction process was completed, the sorbents were collected and dried in an oven, and then their FT-IR spectra were recorded by the KBr pellet method (Fig. 1). As it emerges from the FTIR spectra, the appearance of N-H, C=O and C-N at 3319, 1661 and 1605 cm−1 of DES on the Fe3O4, alumina and TiO2 nanoparticles with some shift indicates the sorption of DES on the surface of the sorbents. However, as the magnetic sorbent can reduce the processing time through the elimination of the centrifugation or filtration step, the Fe3O4 nanoparticles were selected as the sorbent for further studies. The modified and unmodified Fe3O4 sorbent was also characterized by thermal gravimetric analysis (TGA). It was observed (as in Fig. 2) that, when the temperature was raised from room temperature to 400 °C the weight loss of DES sorbed on Fe3O4 was about 14 % due to the loss of water and decomposition of DES whereas the weight loss of unmodified Fe3O4 was about 2 %. However, the sorbent withstood further increase at a temperature up to 700. Thus, based on the TGA curves, the amount of DES on Fe3O4 was found to be about 12 %. Furthermore, the sorbent were characterized by XRD, SEM and VSM. The XRD pattern of Fe3O4 magnetite nanoparticles (Fig. S1) shows six characteristic peaks for Fe3O4 (2θ = 30.1, 35.5, 43.1, 53.4, 57.0, and 62.6°), marked by their indices ((220), (311), (400), (422), (511), and (440)) which are consistent with those reported in the literature. This revealed that the resultant nanoparticles are pure Fe3O4 with a spinel structure. The surface morphology of the magnetic nanoparticles before and after adsorption of DES was characterized by scanning electron microscopy (SEM) (Fig. S2). As it is demonstrated the DES is uniformly distributed on the magnetic nano particles, the size of magnetic nanoparticles after adsorption of DES is not significantly changed and is still in nano dimension. The magnetic properties of magnetic nanoparticles before and after adsorption of DES were studied by vibrating sample magnetometer (VSM). It was found that (Fig. S3) the curve of magnetic hysteresis loop of Fe3O4 and DES-modified magnetic nanoparticles are both S shape and the saturation magnetization value of the DES-modified magnetic nanoparticles is 58.0 emu g−1 which is slightly lower than that of pure Fe3O4 (65.7 emu g−1). Thus, the saturation magnetization of DES-modified magnetic nanoparticles is adequate to ensure its convenient separation from solutions by external magnetic field.

FT-IR spectra of MNPs (a), DES (b), DES mediated Fe3O4(C), DES mediated alumina (d) and DES mediated TiO2 (e)

TGA curves of Fe3O4 (a) and DES modified Fe3O4 (b)
Finally, a simple, fast deep eutectic solvent-mediated magnetic nanoparticles (DES-MNPs) extraction method was developed for the ligandless separation and preconcentration of lead and cadmium, as the model analytes, from aqueous samples. The factors affecting the process were optimized by a univariable approach.
Optimization of experimental variables in deep eutectic solvent-mediated magnetic nanoparticle (DES-MNP) based extraction
The pH of a sample solution usually plays an important role in the separation/preconcentration of metal ions. A proper pH has a unique role in the interaction of DES with metal ions and its chemical stability. It can enhance sorption efficiency and reduce matrix interference. The effect of the sample pH on the extraction of Pb(II) and Cd(II) ions was investigated by the designed method in the pH range of 3.0 to 9.0. The results (Fig. S4) demonstrated that the extraction recovery of Pb(II) and Cd(II) ions increased by an increase in pH from 3.0 to 6.0 and became maximum and constant at a pH above the isoelectric pH of Fe3O4, i.e., in the range of 6.0 to 7.5. A pH of approximately 6.5 was chosen for the subsequent works, as it was more convenient.
The selection of type and composition of DES may have a significant role in the extraction efficiency and selectivity of metal ions. Two hundred μL of three types of DES including ChCl-Ox, ChCl-Urea and ChCl- EG at the mole ratio of 1:2 was tested to find the best type of DES for DES-MNP extraction. The highest extraction recovery was obtained when urea was used as a hydrogen bond donor (Fig. S5). Therefore, ChCl-urea was selected as the best type of DES for the extraction of target ions. The relative amount of HBD and HBA of DES was found to have a significant effect on the recovery of analytes. To find the best composition of ChCl-urea for extraction, several experiments were performed with 200 μL of ChCl-urea solvents with different mole ratios of ChCl and urea. It was observed that the extraction recovery of Pb(II) and Cd(II) was maximized at the molar ratio of 1 to 2.5 of ChCl and urea and then remained constant at higher molar ratios. The increase in extraction efficiency at higher molar ratios of urea can be attributed to the increase in the interaction of nitrogen donor moiety of urea in the ChCl-urea solvent with analytes. Therefore, a 1:2.5 ChCl-urea DES was chosen as the extracting solvent for the subsequent work.
In order to select an optimum volume of DES, several experiments were performed in which 60 mL of each sample was subjected to the recommended extraction procedure with varying amounts of DES but in other experimental constant conditions. It is observed that the extraction recovery of lead and cadmium were low (approximately 25–30 %) in the absence of DES but remarkably increased when the amount of DES was increased up to 200 μL and then reached a plateau. This observation can be explained based on the fact that an increase in the amount of DES to a certain volume causes further metal solvents interaction as well as the sorption of metals onto the MNPs.
The effect of the mass of MNPs on the recovery of Pb(II) and Cd(II) was also studied. It was found that the recovery of both analytes would increase by an increase in the MNPS mass up to 15 mg. However, for higher amounts of MNPs, the extraction efficiency was approximately constant. So, a mass of 20 mg of MNP was selected as the optimum amount for further studies. Another significant factor affecting the extraction efficiency is the contact time between MNPs, DES and metal ions. Experiments revealed that the recovery of the analytes increased with an increase in the extraction time up to 10 min and leveled off at a higher extraction time. Therefore, an extraction time of 10 min was selected for the subsequent experiments.
The nature, concentration and volume of the desorbing solution can have an important effect on the recovery and measurement process. The desorbing solutions must be capable of complete desorption of analytes in a minimum volume in a short time, and it must not interfere in the measurement process. In order to find the best desorbing solution, the possibility of desorption of analytes by 600 μL of various desorbing solutions including nitric acid, thiourea and acetic acid was tested. The recoveries were found to be higher with nitric acid. Then, the effect of the concentration of the desorbing solution on the recovery of analytes was studied by varying its concentration in the range of 0.2–2.0 mol L−1. It was found that the recovery of analytes increased with an increase in the nitric acid concentration up to 1.0 mol L−1 and became constant at a higher concentration. Therefore, nitric acid with a concentration of 1.0 mol L−1 was selected for further experiments. Moreover, the effect of the volume of eluent was investigated in the range of 400.0–1000.0 μL. The results revealed that 600.0 μL of nitric acid was sufficient for the quantitative recoveries of analytes. Thus, to achieve a high preconcentration factor, 600.0 μL of nitric acid (1 mol L−1) was chosen as the optimum desorbing solution in the subsequent experiments.
The effect of desorption time was also studied by varying it between 0 and 60.0 s. It was found that desorption of analytes from the sorbent was very fast, and the recovery of analytes was maximum and almost independent of the desorption time. Therefore, the desorption time of 5 s was selected for the complete desorption of analytes from the sorbent.
The influence of ionic strength on DES-MNP performance was studied by varying the NaCl concentration from 0 to 40 % (w/v) under other constant experimental conditions. The results confirmed that the recovery of analytes was not significantly influenced by increasing the NaCl concentration. This indicates the capability of the method for the extraction of analytes from saline samples such as sea water.
Sample volume is one of the most important key parameters that influences the achievable preconcentration factor and demonstrates the possibility of extraction of low amounts of analytes from a large sample volume. Thus, the effects of sample volume on the extraction efficiency of 9 of Pb and 1.2 μg of Cd from different sample volumes (20–80 mL) were investigated. It was found that the recoveries of analytes were quantitative up to 60 mL, and then they decreased with further increase in the sample volume. By considering the sample volume of 60 mL and the volume of the desorbing solution (600 μL), a preconcentration factor of 100 was determined for both analytes.
Sorbent capacity
In order to determine the maximum amount of analytes that can be extracted by the sorbent, the pH of 100 mL of a sample solution containing 30 and 20 mg L−1 of lead and cadmium respectively was adjusted to ~6.5, and 1 mL of DES along with 90 mg of MNPs was added to it. Then, the mixture was stirred for sufficient time to guarantee the achievement of equilibrium in the extraction system. Then, the amount of analytes in the supernatant solution was measured by FAAS. The amount of analytes extracted by the sorbent was determined from the differences of the concentration of lead and cadmium in the initial and final solutions. The capacity of the sorbent was found to be 25.0 and 23.7 mg g−1 for lead and cadmium respectively.
Effect of interfering ions
The influence of common ions, often present in real samples, on the recovery of lead and cadmium was investigated. The tolerance limit of each interfering ion was taken as the largest amount of the added ion causing a ±5 % error on the recovery of 7.2 and 0.9 μg of Pb and Cd respectively. The results (Table S1) indicate, the presence of K+, Na+, Ca2+, NO3 −,Cl−, SO4 2−,Sb3+, Al3+, Mg2+ and Ba2+ at the mole ratio of 1000, Cr3+, Mn2+ and Ni2+ at the mole ratio of 800 and Cu2+ and Zn2+ at the mole ratio of 200 had no significant influence on the extraction and determination of analytes (Table S1). Thus, the method offers high selectivity for the determination of lead and cadmium. It should be noted that the selectivity of the method can be controlled through proper tailoring of DES.
Figures of merit
The analytical characteristics of this method including dynamic range, correlation coefficient (R2), limit of detection, relative standard deviation (RSD) and enhancement factor under optimized conditions were investigated for lead and cadmium. The method showed linearity in the wide range of 2 to 250 μg L−1 and 0.5 to 30 μg L−1 with correlation coefficients of 0.9993 and 0.9991 for Pb and Cd respectively. The limits of detection defined as 3Sb/m (where Sb is the standard deviation of the blank and m is the slope of the calibration graph) were 0.4 and 0.1 μg L−1 for lead and cadmium respectively. The enhancement factors, defined as the ratio of the slope of the calibration curves with and without preconcentration, were found to be 99 and 97 for Pb and Cd respectively. The closeness of enhancement factors to the preconcentration factor of 100 indicates that the extraction process is a quantitative one. There were only small deviations observed amongst consecutive determinations of lead and cadmium (i.e., R.S.D = 1.8 and 2.1 %, for six replicate measurements at the concentration level 150 and 20 μg L−1 for Pb and Cd respectively).
Comparison with other methods
The method was compared to various preconcentration techniques reported recently in the literature for the separation, preconcentration and determination of lead and cadmium by FAAS and the results are summarized in Table 1. As it can be seen the method provides lower LODs and higher preconcentration factors for Pb and Cd than most other methods. The sorbent capacity is also higher. Moreover, the DES-MNPs extraction method is fast and is capable of effective separation and preconcentration of lead and cadmium from complicated matrices.
Application
The applicability of the method was examined by analyzing a synthetic sea water sample (having the composition (mol L−1) NaCl 0.485, MgCl2 0.056 and CaSO4.2H2O 0.01), tap water, well water, river water, Caspian Sea water as well as soil and hair samples. The results are summarized in Table 2. The accuracy of the method was verified by spiking experiments and comparing the results with those obtained by independent analyses using electrothermal atomic absorption spectrometry (ETAAS). The recoveries of the spiked samples (Table 2) fluctuate in the range of 95.0 to 105.0 %, suggesting that the method is accurate, reliable and independent of the presence of salts. Furthermore, Table 2 indicates that, at the 95 % confidence level, there is satisfactory agreement between the results and the data obtained by the independent analysis. Thus, the method is reliable for the analysis of lead and cadmium in the sample types examined.
Conclusion
In this article, a new approach was examined for the use of DESs as the green solvents extract metal ions from aqueous samples. The advantages of DESs and magnetic nanoparticles were combined, and for the first time a rapid, simple, and eco-friendly deep eutectic solvent-mediated magnetic nanoparticles (DES-MNPs) extraction method was developed for the ligandless separation and preconcentration of lead and cadmium from aqueous samples. The selectivity of the method can be controlled through proper tailoring of DESs. In comparison with other reported methods, the method has a higher preconcentration factor and a lower detection limit. Also, due to the use of DES, the method is compatible with green chemistry.
References
Wang Y, Gao S, Zang X, Li J, Ma J (2012) Graphene-based solid-phase extraction combined with flame atomic absorption spectrometry for a sensitive determination of trace amounts of lead in environmental water and vegetable samples. Anal Chim Acta 716:112–118
Baghban N, Haji Shabani AM, Dadfarnia S (2013) Solid phase extraction and flame atomic absorption spectrometric determination of trace amounts of cadmium and lead in water and biological samples using modified TiO2 nanoparticles. Int J Environ Anal Chem 93:1367–1380
Adelekan BA, Abegunde KD (2011) Heavy metals contamination of soil and groundwater at automobile mechanic villages in Ibadan, Nigeria. Int J Phys Sci 6:1045–1058
Amorim F, Ferreira S (2005) Determination of cadmium and lead in table salt by sequential multi-element flame atomic absorption spectrometry. Talanta 65:960–964
Dos Santos WNL, Cavalcante DD, Ferreira HS, Das Virgens CF, Borges AR, Silva MM, Vale MGR (2011) Cloud point extraction for the determination of cadmium and lead employing sequential multi-element flame atomic absorption spectrometry. Int J Environ Anal Chem 91:1447–1452
Kocúrová L, Balogh IS, Andruch V (2013) Dispersive liquid-phase microextraction procedure for spectrometric determination of cadmium. Microchem J 107:3–9
Hu X (2011) Rapid coprecipitation-separation and flame atomic absorption spectrometric determination of lead and cadmium in water with cobalt (II) and ammonium pyrrolidine dithiocarbamate. Int J Environ Anal Chem 91:263–271
Dadfarnia S, Haji Shabani AM, Kamranzadeh E (2009) Separation/preconcentration and determination of cadmium ions by solidification of floating organic drop microextraction and FI-AAS. Talanta 79:1061–1065
Ramandi NF, Shemirani F, Farahani MD (2014) Dispersive solid phase extraction of lead (II) using a silica nanoparticle-based ionic liquid ferrofluid. Microchim Acta 181:1833–1841
Huang Y, Wang Y, Pan Q, Wang Y, Ding X, Xu K, Li N, Wen Q (2015) Magnetic graphene oxide modified with choline chloride-based deep eutectic solvent for the solid-phase extraction of protein. Anal Chim Acta 877:90–99
Khan S, Kazi TG, Soylak M (2014) Rapid ionic liquid-based ultrasound assisted dual magnetic microextraction to preconcentrate and separate cadmium-4-(2-thiazolylazo)-resorcinol complex from environmental and biological samples. Spectrochim Acta A 123:194–199
Jiang HM, Yang T, Wang YH, Lian HZ, Hu X (2013) Magnetic solid-phase extraction combined with graphite furnace atomic absorption spectrometry for speciation of Cr(III) and Cr(VI) in environmental waters. Talanta 116:361–367
Kazemi E, Haji Shabani AM, Dadfarnia S (2015) Synthesis and characterization of a nanomagnetic ion imprinted polymer for selective extraction of silver ions from aqueous samples. Microchim Acta 182:1025–1033
Jalbani N, Soylak M (2014) Ligandless surfactant mediated solid phase extraction combined with Fe3O4 nano-particle for the preconcentration and determination of cadmium and lead in water and soil samples followed by flame atomic absorption spectrometry: multivariate strategy. Ecotoxicol Environ Saf 102:174–178
Baghban N, Haji Shabani AM, Dadfarnia S (2012) Solid phase extraction of trace amounts of cadmium with cetyltrimethylammonium bromide-coated magnetic nanoparticles prior to its determination by flame atomic absorption spectrometry. J Chin Chem Soc 59:782–787
Davudabadi Farahani M, Shemirani F (2012) Supported hydrophobic ionic liquid on magnetic nanoparticles as a new sorbent for separation and preconcentration of lead and cadmium in milk and water samples. Microchim Acta 179:219–226
Zeng C, Hu Y, Luo J (2012) Ionic liquid-based hollow fiber supported liquid membrane extraction combined with thermospray flame furnace AAS for the determination of cadmium. Microchim Acta 177:53–58
Tang B, Row KH (2013) Recent developments in deep eutectic solvents in chemical sciences. Monatsh Chem 144:1427–1454
Chen Z, Zhou B, Cai H, Zhu W, Zou X (2009) Simple and efficient methods for selective preparation of α-mono or α, α-dichloro ketones and β-ketoesters by using DCDMH. Green Chem 11:275–278
Abbott AP, Ttaib KE, Frisch G, McKenzie KJ, Ryder KS (2009) Electrodeposition of copper composites from deep eutectic solvents based on choline chloride. Phys Chem Chem Phys 11:4269–4277
Liao HG, Jiang YX, Zhou ZY, Chen SP, Sun SG (2008) Shape-controlled synthesis of gold nanoparticles in deep eutectic solvents for studies of structure-functionality relationships in electrocatalysis. Angew Chem Int Ed 47:9100–9103
Abbott AP, Boothby D, Capper G, Davies DL, Rasheed RK (2004) Deep eutectic solvents formed between choline chloride and carboxylic acids: versatile alternatives to ionic liquids. J Am Chem Soc 126:9142–9147
Tang B, Bi W, Zhang H, Row KH (2013) Deep eutectic solvent-based HS-SME coupled with GC for the analysis of bioactive terpenoids in chamaecyparis obtusa leaves. Chromatographia 77:373–377
Habibi E, Ghanemi K, Fallah-Mehrjardi M, Dadolahi-Sohrab A (2013) A novel digestion method based on a choline chloride-oxalic acid deep eutectic solvent for determining Cu, Fe, and Zn in fish samples. Anal Chim Acta 762:61–67
Yilmaz E, Soylak M (2015) Ultrasound assisted-deep eutectic solvent extraction of iron from sheep, bovine and chicken liver samples. Talanta 136:170–173
Karimi M, Dadfarnia S, Haji Shabani AM, Tamaddon F, Azadi D (2015) Deep eutectic liquid organic salt as a new solvent for liquid-phase microextraction and its application in ligandless extraction and preconcentraion of lead and cadmium in edible oils. Talanta 144:648–654
Xu K, Wang Y, Huang Y, Li N, Wen Q (2015) A green deep eutectic solvent-based aqueous two-phase system for protein extracting. Anal Chim Acta 864:9–20
Zhang Q, De Oliveira VK, Royer S, Jérôme F (2012) Deep eutectic solvents: syntheses, properties and applications. Chem Soc Rev 41:7108
Pournaghi-Azar M, Dastangoo H (2000) Differential pulse anodic stripping voltammetry of copper in dichloromethane: application to the analysis of human hair. Anal Chim Acta 405:135–144
Shamsipur M, Raoufi F, Sharghi H (2000) Solid phase extraction and determination of lead in soil and water samples using octadecyl silica membrane disks modified by bis [1-hydroxy-9, 10-anthraquinone-2-methyl] sulfide and flame atomic absorption spectrometry. Talanta 52:637–643
Gama E, Dasilvalima A, Lemos V (2006) Preconcentration system for cadmium and lead determination in environmental samples using polyurethane foam/Me-BTANC. J Hazard Mater 136:757–762
Ghanemi K, Nikpour Y, Omidvar O, Maryamabadi A (2011) Sulfur-nanoparticle-based method for separation and preconcentration of some heavy metals in marine samples prior to flame atomic absorption spectrometry determination. Talanta 85:763–769
Tuzen M, Saygi KO, Soylak M (2008) Solid phase extraction of heavy metal ions in environmental samples on multiwalled carbon nanotubes. J Hazard Mater 152:632–639
Narin I, Surme Y, Bercin E, Soylak M (2007) SP70-alpha-benzoin oxime chelating resin for preconcentration-separation of Pb(II), Cd(II), Co(II) and Cr(III) in environmental samples. J Hazard Mater 145:113–119
Soylak M, Divrikli U, Saracoglu S, Elci L (2007) Membrane filtration–atomic absorption spectrometry combination for copper, cobalt, cadmium, lead and chromium in environmental samples. Environ Monit Assess 127:169–176
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 1631 kb)
Rights and permissions
About this article

Cite this article
Karimi, M., Shabani, A.M.H. & Dadfarnia, S. Deep eutectic solvent-mediated extraction for ligand-less preconcentration of lead and cadmium from environmental samples using magnetic nanoparticles. Microchim Acta 183, 563–571 (2016). https://doi.org/10.1007/s00604-015-1671-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00604-015-1671-9