
Abstract
Published data on various versions of the homogeneous liquid–liquid microextraction (HLLME) of organic compounds are generalized and systematized, and a classification of these versions is given. The main HLLME procedures are discussed. Examples of the application of HLLME to the separation of organic compounds in the analysis of environmental samples, food products, and biological fluids are given.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
The determination of organic compounds in various materials is becoming a more and more important problem under the conditions of production and use of a huge number of chemicals [1, 2]. As a rule, the determination of organic compounds is preceded by sample preparation, which includes the isolation of test compounds from an analyzed matrix, their separation from interfering components, and their preconcentration [3–6]. For the miniaturization of sample preparation methods, various versions of liquid–liquid microextraction (LLME), which are consistent with the principles of green analytical chemistry and are an effective way to increase the sensitivity of analytical methods [6–8], are currently being actively developed. Extraction from an aqueous solution into a small amount of a water-immiscible solvent (up to 100 μL) is referred to as LLME. The use of LLME makes it possible to considerably simplify sample preparation, reduce or completely eliminate the use of toxic solvents, shorten the analysis time, and combine extraction with sample injection into an instrument within a single stage [9].
Several methods for the LLME of organic compounds are well known: dispersive liquid–liquid microextraction, single drop liquid-phase microextraction, hollow fiber microextraction [10, 11], and homogenous liquid–liquid microextraction (HLLME) [12, 13]. Unlike the first three methods, hydrophilic solvents that are miscible with water are used as extractants in HLLME. The HLLME method has found application for the separation of polar organic compounds from various objects and their subsequent determination directly in the extracts by HPLC and capillary electrophoresis. This review summarizes publications describing various HLLME versions, provides a classification of these versions, and describes the procedures and their combinations with other sample preparation methods and techniques for the subsequent determination of organic compounds in various samples.
PRINCIPLE, GENERAL CHARACTERISTIC, AND CLASSIFICATION OF HOMOGENEOUS LIQUID–LIQUID EXTRACTION
Homogeneous liquid–liquid extraction (HLLE) or extraction with hydrophilic solvents was proposed in 1973 on the wave of interest in the separation of polar organic compounds and biologically active substances that are poorly extracted with hydrophobic organic solvents [14]. In this method, polar solvents that are completely (acetonitrile, acetone, ethanol, propanol, and isopropanol) or partially (butanol, isobutanol, pentanol, isopentanol, and methyl ethyl ketone) miscible with water are used as extractants [15]. In the first works devoted to HLLE, the phase separation of two- or three-component solvent systems was carried out by introducing large amounts of neutral salts (salting-out agents) into an aqueous sample [16–19]. Salts decrease the solubility of hydrophilic solvents in water to result in the formation of a separate organic phase. In this case, a binary system with unlimited miscibility turns into a ternary system with limited solubility of an organic solvent in an aqueous solution of a salting-out agent [20]. The nitrates, sulfates, chlorides, and carbonates of potassium, sodium, or ammonium are used as salting-out agents. More recently, it was found that the phase separation of two- or three-component solvent systems can also be carried out by adding sugars [21], changing the pH [22] and temperature [23], or introducing a small amount of a hydrophobic solvent into a homogeneous solution [24].
Homogeneous extraction also includes extraction with polyethylene glycols and various water-soluble polymers. In this method, aqueous solutions of environmentally friendly polyethylene glycols, polyvinyl alcohol, polyvinylpyrrolidone, and other water-soluble polymers, which are completely miscible with water and easily decomposed by microorganisms, are used as extractants [25–27]. The system also separates into two immiscible aqueous phases due to the introduction of large amounts of inorganic salts as phase-forming agents. The upper layer is a saturated aqueous polymer solution (which plays the role of an organic phase), whereas the lower aqueous layer is saturated with a phase-forming salt. The advantages of using water-soluble polymers as extractants include an insignificant effect of hydration on the transfer of the extractable substance from one phase to another (both phases contain significant amounts of water) and a high complexing ability of polymers in relation to biologically active substances.
In recent years, the HLLE method has been increasingly implemented in the form of homogeneous liquid–liquid microextraction (HLLME, 2009) [28]. In HLLME, small volumes (microliters) of hydrophilic organic solvents are used as extractants. As in the classical HLLE, the phase separation of a homogeneous solution and the extraction of organic compounds into an extractant phase occur simultaneously in HLLME. In this method, equilibrium is established very quickly due to an extremely large interface between the aqueous and organic phases. HLLME is a simple and versatile preconcentration method, which decreases the consumption of reagents and solvents, the extraction time, and the cost of analysis. There is no need for back extraction because the resulting concentrate is miscible with water to greatly simplify the subsequent determination. Depending on the reason for phase separation, salting-out assisted liquid–liquid extraction (SALLE), sugaring-out assisted liquid–liquid extraction (SULLE), pH-assisted homogeneous liquid–liquid microextraction, subzero-temperature assisted liquid–liquid extraction (STLLE), and hydrophobic-solvent assisted liquid–liquid extraction (HSLLE) are distinguished. Table 1 summarizes the classification of HLLME versions depending on the factor causing the formation of an extractant phase.
HOMOGENEOUS SALTING-OUT ASSISTED LIQUID–LIQUID MICROEXTRACTION
Homogeneous salting-out assisted liquid–liquid microextraction is the most widespread version of HLLME. In this case, an individual organic phase is formed as a result of the addition of inorganic salts (salting-out agents), which decrease the solubility of polar solvents in water. In the English-language literature, this method is referred to as a number of terms such as salting-out assisted liquid–liquid extraction, miniaturized salting-out liquid–liquid extraction, salting-out homogenous extraction, and salting-out homogenous liquid–liquid extraction, which often make it difficult to find necessary information. The method has become widespread, primarily, in the analysis of biological samples, as evidenced by information summarized in reviews [29, 30].
Salting-out assisted HLLME is most often carried out in centrifuge tubes, and small amounts of an organic phase are sampled using microsyringes or miniature glass vessels. Here are some examples. Figure 1 shows a schematic diagram of HLLME used to separate lamivudine and zidovudine, which are the constituents of an antiviral drug, from blood plasma [31]. A 500-μL portion of blood plasma and 400 μL of a phosphate buffer solution were placed in a microtube (2 mL). Then, 200 μL of acetonitrile and 0.25 g of sodium sulfate were added as an extractant and a salting out agent, respectively. The mixture was shaken using a vortex mixer for 2 min. For the determination, 10 μL of an acetonitrile extract was used, which was directly injected into an HPLC system with an ultraviolet detector (HPLC-UV). The limits of detection were 6 and 3 ng/mL for lamivudine and zidovudine, respectively.

Schematic diagram of the implementation of homogeneous liquid–liquid microextraction with salting-out in a microtube [31].
An HLLME procedure proposed by Liu et al. [32] was used for the preconcentration of sulfanilamides from river water, urine, and honey diluted with water; an organic phase was taken with a microsyringe. A 0.5-mL portion of the analyzed solution was placed in a test tube (1 mL), and 0.5 g of sodium chloride and 100 μL of acetonitrile were added; the contents were stirred for 1 min and transferred into a 1-mL syringe. The syringe was turned upside down and left in this position for 10 min to form two separate phases. Thereafter, the organic phase was squeezed out with a syringe plunger, and a 10-μL sample was taken and diluted with a mobile phase in a ratio of 1 : 2 for the subsequent determination of sulfanilamides by HPLC-UV. The detection limits of sulfanilamides were 1.4–4.5 ng/mL.
Sereshti et al. [33] proposed an interesting version of HLLME based on the use of a coupled-syringe system for the extraction of sulfanilamide from various complex matrices (river water, blood plasma, urine, and milk). In the first syringe, 0.5 mL of the analyzed sample, from which solid particles and proteins were previously removed, was taken; sodium chloride (250 mg/mL) was added, and the contents were stirred for 10 s until a homogeneous solution was formed. Then, 250 μL of acetonitrile was taken into the second syringe. The syringes were connected and their contents were alternately pumped from one to another five times. After the completion of the last cycle, the syringes were placed vertically and disconnected, and the phases were separated for 2 min. Then, the upper layer (an acetonitrile concentrate) was squeezed out with a syringe plunger to the narrow tip of the syringe, 20 μL of the organic phase was taken, and sulfanilamides were determined by HPLC-UV with a detection limit of 0.3 ng/mL.
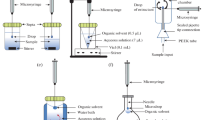
To facilitate extract collection after HLLME, a special vessel with a built-in glass capillary was used [34, 35]. Figure 2 illustrates the HLLME procedure used to separate pesticides from juices and shows an image of the corresponding device with specified dimensions.

(a) Schematic diagram of homogeneous liquid–liquid microextraction used for the separation of pesticides from juices and (b) a collector for the organic phase [34].
The use of 96-well plates with removable tubes or plates with deep 2.0–2.2 mL wells was proposed to increase the productivity of HLLME in combination with the subsequent determination of analytes by HPLC–tandem mass spectrometry (HPLC–MS/MS) [36, 37]. From 5 to 500 μL of test samples (plasma and urine), from 50 to 200 μL of a salt solution, and from 200 μL to 1 mL of an organic solvent were placed in test tubes. The ratio between the components was chosen so that the total volume of the mixture did not exceed 1.5 mL. Although two phases were formed within a few minutes after salt addition, centrifugation was performed to facilitate phase separation.
The main parameters varied in the selection of HLLME conditions are the nature of a solvent [28, 31–35, 38–50] and a salting out agent [28, 31, 34, 35, 39, 41–45, 48, 49, 51, 52]. Acetonitrile is mainly used as a polar solvent extractant in HLLME [28, 31–33, 36–42, 45, 48–51, 53–59]. A smaller volume of acetonitrile is required for phase separation, as compared with acetone [43, 47], isopropyl alcohol [34, 35, 60], tetrahydrofuran (THF) [44], and ethanol [61], which are less often used as extractants. Not only hydrophilic but also some hydrophobic organic compounds are well extracted in acetonitrile. Acetonitrile extracts contain less water and salts; because of this, the matrix components of a sample contaminate them to a lesser extent. Compared to other solvents, acetonitrile is better suitable for the subsequent determination of analytes by HPLC or capillary electrophoresis (CE) because it is often a mobile phase component. In addition, acetonitrile is often used to separate proteins at the first stage in the analysis of biological samples [29, 30, 40].
Salting-out agents are usually selected empirically for each specific system, but there are a number of general requirements that should be considered in this selection. Salting-out agents should be highly soluble in water and almost insoluble in a polar organic solvent. It is desirable that the ions of salting-out salts have high hydration energies. It is well known that the salting-out ability of cations and anions decreases in the lyotropic series Mg2+ > Ca2+ > Sr2+ > Ba2+ > Li+ > Na+ > K+ \( \gg \) Rb+ > Cs+ and > H3COO– > Cl– \( \gg \) Br–> I– > CNS– [29, 30, 62]. In addition, the nature of extracted organic compounds, the type of an analyzed sample, and the method of the subsequent determination are taken into account in the selection of salting-out agents. The salting-out effect is manifested at salt concentrations close to saturation, and the salt concentration ranges from 1–3 M depending on the salt used.
Among the described solvent/salting-out agent combinations, the following can be noted: acetonitrile/ammonium sulfate [28, 38, 39, 42, 52, 59], acetonitrile/sodium chloride [32, 33, 40, 56], acetonitrile/sodium sulfate [31, 53], acetonitrile/ammonium formate [37, 54], acetonitrile/ammonium acetate [48, 58], acetonitrile/sodium carbonate [57], acetonitrile/magnesium sulfate [45], acetonitrile/magnesium chloride [46], isopropanol/sodium sulfate [34, 60], isopropanol/ammonium sulfate [35], acetone/magnesium chloride [43], acetone/sodium sulfate [47], tetrahydrofuran/sodium sulfate [44], and ethanol/ammonium sulfate [61]. In some cases, combinations of two salts are used as salting-out agents: acetonitrile/sodium chloride/magnesium sulfate [41] and acetonitrile/sodium sulfate/sodium chloride [55] (and sodium hydroxide [49, 50]).
For biological materials to be analyzed by HPLC–MS, organic salts that do not interfere with mass-spectrometric detection, such as ammonium formate [37, 54] or ammonium acetate [58], are preferred as salting-out agents, and the use of sodium chloride is undesirable because the presence of sodium ions complicates the interpretation of mass spectra. On the contrary, sodium chloride is an ideal salting-out agent in a combination of HLLME with CE [40] because it is often used as a component of mobile phases in this method. Ammonium sulfate [28, 35, 38, 42, 52, 59] and sodium chloride [32, 33, 56] are most often used in combination with HPLC-UV.
Along with water-soluble organic solvents, ionic liquids (ILs) have begun to be used in HLLME [63–68]. This version is often referred to as extraction in ionic liquid–based aqueous biphasic systems or ionic liquid–based aqueous two-phase systems [68]. As an example, we consider a procedure used for the preconcentration of fluoroquinolones and sulfanilamides from milk [65].
The volumes of an analyzed sample and an extractant are important factors in the selection of sample preparation conditions for organic compounds with the use of HLLME because a ratio between these factors primarily affects the preconcentration factors. The analyzed sample and extractant volumes are varied depending on the analyzed material, the polar solvent chosen as the extractant, and the sensitivity of the subsequent determination method. Thus, in the analysis of blood plasma, the analyzed sample volume varied from 25 to 500 µL and the volume of an acetonitrile extractant varied from 50 to 200 µL, whereas these volumes were 100–1000 and 150–500 µL, respectively, in the analysis of urine. In the analysis of aqueous solutions, the analyzed sample volume can vary from 1 to 10 mL and the extractant volume, from 100 to 800 μL. When choosing the volume of an extractant, it is important to take into account a minimum volume that can result from a homogeneous solution after adding a salting-out agent. Liu et al. [32] found that, for the formation of a second phase, 500, 300, and 800 μL of isopropyl alcohol, acetonitrile, and acetone, respectively, should be added to 1 mL of an aqueous phase.
To increase the efficiency of preconcentration, HLLME is combined with dispersive LLME [69, 70] or a dispersive solid-phase extraction [71, 72]. In some cases, HLLME is combined with derivatization, which is carried out to convert analytes into colored or fluorescent derivatives [28, 46, 49, 52, 53]. In this case, very small volumes of derivatizing reagents and solvents are required to obtain derivatives.
Most often, salting-out assisted HLLME is used to separate medicinal compounds from various biological samples: plasma [29–31, 36–38, 46–48, 50, 51, 64] and human blood serum [38, 43, 44], urine [29, 30, 32, 33, 38, 40, 44, 47, 50, 56, 58], saliva [49], animal blood plasma [45, 54], and tissues [55]. The following compounds were separated from the above biological materials with the use of HLLME: lamivudine and zidovudine [31], sulfanilamides [32, 33, 64], lopinavir and ritonavir [36], simvastatin and simvastatinic acid [37], amoxapine and nortriptyline in the form of thiourea derivatives [38], atorvastatin [43], biomarkers of nerve agents [44], vitamin K and its homologues [46], warfarin enantiomers [40], entecavir [51], fluoroquinolone antibiotics [45, 47], febuxostat [48], diclofenac [49], metformin, buformin, and phenformin [50], trimetazidine [54], methoxetamine [55], amphetamines [56], and β-blockers and their metabolites [58]. The separated analytes were determined directly in concentrates by HPLC with ultraviolet [31–33, 38, 42, 50, 56, 58, 64], fluorescence [45–47], diode array [48], mass-spectrometric [51] and tandem mass-spectrometric [36, 37, 54, 55] detectors, gas chromatography with a flame-ionization detector [34], and CE [40, 73]. Salting-out assisted HLLME was also used to separate carbonyl compounds [28], sulfanilamides [32, 33], fluoroquinolones [42, 45], herbicides (sulfonylurea derivatives) [42], and dyes [39] from natural waters. Pesticides [34, 60] and herbicides [42, 66] from juices; biogenic amines from drinks [52]; sulfanilamides [32, 33, 59], fluoroquinolones [45] and neonicotinoid insecticides [58] from honey; and fluoroquinolones [45, 63] and sulfanilamides [64] from milk were separated using this technique. The automation of both the HLLME procedure itself and the entire analysis performed using HLLME is an important and still unresolved problem. Only a few publications on this subject matter are available [74–76].
The advantages of salting-out assisted HLLME include fast equilibration, ease of implementation, and low cost. The disadvantages of this HLLME version include the fact that phases are separated at a high concentration of salts, the presence of which in the extraction system can lead to undesirable chemical reactions, cause problems related to the contamination and corrosion of equipment, and complicate the subsequent determination, for example, to affect the ionization of compounds under conditions of mass-spectrometric detection. As an alternative to traditional inorganic salting-out salts, the use of saccharides, which are referred to as sugaring-out agents by analogy with salting-out agents, was proposed.
HOMOGENEOUS SUGARING-OUT ASSISTED LIQUID–LIQUID MICROEXTRACTION
Homogeneous sugaring-out assisted liquid–liquid microextraction is based on the extraction of substances into a polar organic solvent (usually, acetonitrile) and phase separation by adding large amounts of mono- or disaccharides (sugaring-out agents), which decrease the solubility of polar solvents in water. In 2008, it was found that acetonitrile, which is miscible with water in any proportion, forms biphasic systems in the presence of glucose, fructose, xylose, maltose, arabinose, and sucrose [21, 77–80]. The sugaring-out effect is explained by the ability of saccharides—polar molecules containing a large number of hydroxyl groups—to form hydrogen bonds with water, which are stronger than those with acetonitrile, and they are extractable organic compounds. Upon the addition of saccharides to a homogeneous water–acetonitrile–analyte solution, the abstraction of water molecules from hydrated acetonitrile molecules occurs. Dehydrated acetonitrile molecules aggregate to form a new phase containing extracted organic molecules.
It was noted above that acetonitrile is mainly used as an extractant in the sugaring-out assisted HLLME [81–90]. A few works published in recent years indicate that the sugaring-out effect was also observed in water–isopropanol [91], water–1-butanol [92], and water–ethyl acetate [93] mixtures; however, these solvents have not yet found application in HLLME.
The type of a sugaring-out agent [82–85], the volume of acetonitrile in an aqueous acetonitrile solution, and the concentration of a saccharide [82–86, 90] are the main parameters varied in the selection of extraction conditions. Glucose is mainly used as a sugaring-out agent in this HLLME version because of a lower viscosity of its solution [81–87]. As a rule, solutions containing equal volumes of acetonitrile and water are used because no phase separation occurs with a smaller amount of acetonitrile, whereas the concentration of target analytes in the acetonitrile extract decreases as the amount of acetonitrile increases to 70%, and this leads to a decrease in the determination sensitivity. In an aqueous acetonitrile solution with a volume ratio of 1 : 1 between the components, phase separation was observed at a glucose concentration of 15 mg/mL or higher. As a rule, the concentration of glucose is chosen empirically because its amount affects not only phase separation but also the degree of analyte extraction into an acetonitrile extract. In most cases, HLLME is performed at a glucose concentration of 100–200 mg/mL. In the determination of organic compounds in honey, additional sugars are not required because the chemical composition of honey is composed of almost 80% mono- and oligosaccharides [88–90]. In some cases, salts are introduced into the extraction system to increase the efficiency of HLLME [84, 86, 89, 90].
Homogeneous sugaring-out assisted liquid–liquid microextraction was used to separate lopinavir and ritonavir from human blood plasma [81]; procainamide from urine [83]; honokiol and magnolol [82] and 2,4-dichlorobenzyl alcohol, amylmetacresol, and dyes [85] from drugs; pesticides from juices [84]; 10-hydroxy-2-decenoic acid [86] and bisphenols [87] from royal jelly; and sulfanilamides [88], phenolic compounds [89], and polycyclic aromatic hydrocarbons (PAHs) [90] from honey. The analytes separated after HLLME were determined directly in the extracts by HPLC with mass-spectrometric [81], spectrophotometric [82, 86], fluorescence [87, 88, 90], or electrochemical [89] detection. To increase the productivity of analysis, fully automated methods have been developed based on an on-line combination of extraction with the subsequent determination of analytes by HPLC-UV [83, 85] and HPLC–MS/MS [84].
The sugaring-out assisted HLLME has a number of advantages over salting-out assisted HLLME. Biodegradable and nontoxic sugars do not react with the analytes to be determined, and they have less impact on the environment; their presence in the extraction system does not cause corrosion and contamination of equipment. Moreover, the acetonitrile/water mixture is one of the most commonly used mobile phases in HPLC, which provides an opportunity to directly inject an acetonitrile phase enriched in analyte into the HPLC system after the sugaring-out procedure. The disadvantage of using acetonitrile in the above HLLME versions is the need for extraction with a volume ratio of 1 : 1 between the aqueous and organic phases, which leads to a low preconcentration coefficient. Switchable-hydrophilicity solvents, which have a unique ability to change the hydrophilicity depending on the pH of solution, were proposed as extractants to eliminate this disadvantage.
HOMOGENEOUS LIQUID–LIQUID MICROEXTRACTION BASED ON pH CHANGES
Switchable-hydrophilicity solvents (SHSs), which were also described as “smart solvents” in 2010 [94], are used as extractants in HLLME based on changes in pH. They can occur in aqueous solutions in two forms, one of which is hydrophobic and immiscible with water, and the other is hydrophilic and completely soluble in water. Some organic bases—amidines, secondary or tertiary amines, and diamines—possess such properties [94–99]. The conversion of one form into another is initiated by passing carbon dioxide through the system or by adding dry ice. An acid–base reaction between the hydrophobic form of an amine and hydrated carbon dioxide leads to the formation of protonated amine bicarbonate, which is completely soluble in water. The reverse transition of the hydrophilic form of a solvent to the hydrophobic one occurs either upon the removal of CO2 from the solution by bubbling nitrogen/air or adding a mineral acid or upon amine deprotonation due to the addition of an alkali solution. In some cases, a salt or a more hydrophilic amine is introduced into the system in order to decrease the amine solubility [94–98].
Despite the fact that the number of SHSs constantly increases [97–99], only dimethylcyclohexylamine (DMCHA) [100–103], dipropylamine [104–107], diisobutylamine [108], triethylamine [109], and octylamine [110] have found application in this version of HLLME. In most studies, amines were mixed with an equal volume of water in order to obtain a soluble form; thereafter, the mixture was repeatedly treated with dry ice, and a solution of NaOH was used to separate the phases [100–104, 106, 109]. The disadvantage of this version of HLLME is associated with the need to use dry ice in order to obtain the hydrophilic form of a solvent. With the use of dipropylamine, a biodegradable commercially available and inexpensive secondary amine, as an example, it was found that a relatively long procedure for the production of a soluble form of an extractant using carbon dioxide can be replaced by a simpler one based on the addition of hydrochloric acid [105]. More recently, Shahvandi et al. [107] found that a homogeneous solution was formed in the water–dipropylamine system when the temperature was lowered to 5°C, and the subsequent heating to 25°C led to the appearance of an extractant phase. Acetonitrile was used for phase separation with the use of water-soluble octylamine as an extractant [110].
Basic solvents with switchable hydrophilicity were used in HLLME to separate PAHs [100], triazine herbicides [101], ibuprofen, ketoprofen, diclofenac, mefenamic acid, naproxen, 17-β-estradiol [103], nitrotoluene, 2,4-dinitrotoluene, 2,6-dinitrotoluene [104], and phthalates [107] from natural waters; bisphenols from milk and juices [102]; methamphetamine [105], methadone, and tramadol from urine [106]; pesticides from juices [108]; paraquat from river water, juice, urine, and blood plasma [109]; and meropenem from urine and blood plasma [110]. The separated analytes were determined by gas chromatography with mass-spectrometric [101, 105, 107] or flame ionization [104, 106, 108] detectors; HPLC with spectrophotometric [102, 109, 110] or diode array [103] detectors; and fluorescence analysis [100].
Octanoic [111, 112], heptanoic [113, 114], nonanoic [115], pivalic [116], and di-(2-ethylhexyl)phosphoric [117] acids have found applications as acidic SHSs in the HLLME method; these acids are completely soluble in aqueous alkaline solutions, but they form a two-phase system upon the addition of concentrated mineral acids. Homogeneous solutions of octanoic acid were obtained by dissolving it in a phosphate buffer solution [111, 112], and the solutions of heptanoic [113], pivalic [116], and di-(2-ethylhexyl)phosphoric [117] acids were obtained in ammonia. The organic phase formed after the addition of mineral acids was separated by centrifugation.
To eliminate the stage of centrifugation, a new version of the method was used [114, 115], which combined HLLME and microextraction by dispersing an extractant: homogeneous liquid–liquid microextraction with phase separation by carbon dioxide. In the English-language literature, this version of microextraction is referred to as effervescence-assisted liquid phase microextraction [118]. Sodium carbonate is used to transfer a higher carboxylic acid into a homogeneous phase. An excess of sodium carbonate makes it possible to perform phase separation due to carbon dioxide, which is released as a result of the interaction of carbonate ions with a mineral acid. In this case, there is no need to carry out an additional step of centrifugation. Figure 3 schematically shows HLLME with phase separation by carbon dioxide. The advantage of this approach is the possibility of automating the procedures of preconcentration and subsequent determination [114].

Procedure proposed for the homogeneous liquid–liquid microextraction with phase separation using carbon dioxide [114].
In the selection of conditions for HLLME based on pH changes, a ratio between the analyzed solution and SHS volumes and the volumes and concentrations of strong acids or bases required for phase separation are most often varied.
Acid SHSs were used in HLLME for the separation of chlorophenols [111, 117], chlorobenzenes [112], phenols [113], and steroid hormones [115] from natural waters; ofloxacin from urine [114]; and pyrethroid insecticides from juices [116]. Compounds were determined by HPLC with spectrophotometric [111–113, 115, 117] and fluorescence [114] detectors and by gas chromatography with mass-spectrometric detection [116].
OTHER VERSIONS OF HOMOGENEOUS LIQUID–LIQUID MICROEXTRACTION
In addition to salts, sugars, and changes in pH, phase separation in HLLME can be initiated by changing the temperature or adding another hydrophobic solvent. Among other versions, HLLME with the use of homogeneous three-component systems and HLLME with deep eutectic solvents can be distinguished.
Homogeneous liquid–liquid microextraction based on cooling. This version of HLLME is based on a phase separation phenomenon in a homogeneous aqueous solution of acetonitrile at low temperatures (–20°C or lower) [23, 119–122]. The upper layer formed on phase separation is a phase rich in acetonitrile and analytes, and the lower phase is a frozen phase rich in water. The HLLME based on cooling does not require the use of additional reagents, and the extracts are much cleaner than those obtained upon salting-out. However, the stage of cooling lasts from 30 to 60 min to significantly increase the analysis time. Moreover, the recoveries of organic compounds are lower than those in other described HLLME versions. In the Russian-language literature, this method was referred to as extractive freezing-out [123–131]. To increase the efficiency of separating an extract from a frozen water portion, it was proposed to perform preconcentration simultaneously with sample centrifugation [129, 130].
The homogeneous liquid–liquid microextraction based on cooling was used to separate benzodiazepines [23], thiamylal barbiturate [119], and caffeine [125] from blood plasma and serum; anthraquinone derivatives from liquid dosage forms [120]; polyphenols from propolis [121]; nitrophenols [122] and phenols [124, 126] from waters; and 1,4-benzodiazepines [125] and pyrovalerone from urine [131].
Homogeneous liquid–liquid microextraction based on the introduction of a small amount of a hydrophobic solvent into a homogeneous solution. Another method of phase separation in a homogeneous aqueous acetonitrile solution consists in the addition of small amounts of nonpolar solvents, such as methyl tert-butyl ether [132] and chloroform [133, 134]. Chloroform is soluble in acetonitrile but insoluble in water; because of this, the solubility of acetonitrile in water decreases in its presence to induce its release in the form of its own phase. In addition, the recovery of hydrophobic compounds increases in the presence of a hydrophobic solvent. In addition to chloroform, small toluene additives cause phase separation in an aqueous acetonitrile solution [135].
As an example, we can consider a procedure for the determination of pharmaceutical preparations (andrographolide, sildenafil, and finasteride) in blood plasma by gas chromatography–mass spectrometry. The procedure included the following operations: 1 mL of plasma was mixed with 700 μL of acetonitrile; then, 70 μL of chloroform was added to the solution. After centrifugation, 10 μL was taken from the upper organic phase with a volume of 370 µL and directly injected into an HPLC–MS system [134]. The limits of detection of andrographolide, sildenafil, and finasteride were 40, 2, and 0.5 ng/mL, respectively.
Three-phase homogeneous liquid–liquid microextraction. In addition to two-phase homogeneous systems containing a polar organic solvent and water, homogeneous three-component systems of the polar organic solvent/nonpolar organic solvent/water type are also used in HLLME [136–144]. In these systems, a polar solvent ensures the solubility of a nonpolar one and a homogeneous state of the entire system. In this version of HLLME, phase separation is caused by the addition of salts [136–139] or water [140–142], which initiate the release of a nonpolar organic compound into a separate phase.
The following systems were proposed to separate organic compounds from various samples: acetone/CCl4/water/NaCl [136], methanol/chloroform/water/NaCl [137], methanol/toluene/ water/NaCl [138], and methanol/hexane/water/ Na2-SO4 [139]. Here are some examples. For the separation of malathion, lambda-cyhalothrin, and cypermethrin pesticides from soils [136], at the first stage, the pesticides were extracted from 4-g soil samples using 10 mL of acetone with stirring for 30 min on a mechanical shaker. To 1 mL of the acetone extract transferred into a 10-mL glass centrifuge tube, 40 μL of CCl4 was added and the mixture was stirred until a homogeneous solution was formed. Then, 0.3 g of NaCl was added to separate the extractant phase. After centrifugation for 4 min, 1 μL was taken from the 22 μL of the bottom organic phase for the determination of pesticides by gas chromatography with an electron capture detector. The detection limits of pesticides were 0.01–0.04 ng/g.
To determine organochlorine pesticides in milk [139], 10 mL of methanol was added to 5 g of milk, and the contents were stirred for 60 s and centrifuged at 3500 rpm for 5 min for the sedimentation of proteins. Then, 1.0 mL of n-hexane was added to 5.0 mL of the methanol extract, and the mixture was vigorously shaken for 30 s. A saturated solution of sodium sulfate (4 mL) was added to the extract solution; then, the n-hexane phase (an upper layer) was separated. For the determination of pesticides by gas chromatography with an electron capture detector, a 2.0-μL portion of the extract was used, which was directly injected into a chromatographic system. The limits of detection were 0.03–0.7 ng/mL.
Along with salts, the formation of a separate phase in three-phase solvent systems can be caused by the addition of water. A polar solvent is infinitely miscible with both water and a nonpolar solvent, and the solubility of the nonpolar solvent in the system decreases upon the addition of water, which is miscible only with the polar solvent; this leads to the release of the nonpolar solvent as a separate phase. In this version, the methanol/hexane/water [140, 141], acetonitrile/chloroform/water [142], acetonitrile/butyl acetate/water [143], and ethanol/dichloromethane/water [144] solvent systems were used to separate PAHs [140] and pesticides [141] from soils, pesticides from fish [142] and milk [143], and caffeine from tea and coffee [144].
Homogeneous liquid–liquid microextraction based on the use of deep eutectic solvents. In recent years, deep eutectic solvents (DESs) have been used as alternative solvents in liquid microextraction [145, 146]. They are obtained by mixing two compounds, one of which acts as an acceptor of hydrogen bonds, and the other is a donor of hydrogen bonds. As a result of specific interactions between these compounds (mainly due to the formation of hydrogen bonds), a eutectic mixture with a much lower melting point than that of either of the two components is formed.
Deep eutectic solvents and ionic liquids have similar physical and chemical properties. They have increased dissolving power and low vapor pressure and high electrical conductivity, viscosity, and surface tension. They are incombustible and readily regenerable and, as a rule, do not pose a threat to the environment. This new class of solvents has advantages over ionic liquids, such as ease of preparation, easy availability of relatively inexpensive and environmentally friendly components, and biodegradability [147].
In most cases, deep eutectic solvents based on choline chloride (2-hydroxyethyltrimethylammonium chloride) are used in various LLME methods. Choline chloride is widely used due to its nontoxicity, low cost, availability, and biodegradability. Choline chloride is conventionally referred to as a complex of B vitamins, and it is used as an additive in the production of premixes and feed components. In addition to choline chloride, other halides (methyltriphenylphosphonium bromide, benzyltriphenylphosphonium chloride, acetylcholine chloride, and tetramethylammonium chloride) are also used [146, 147].
Water-soluble solvents based on choline chloride and compounds that are donors of hydrogen bonds (phenol [148–151], ethylene glycol [152, 153], and oxalic acid [154]), tetrabutylammonium chloride and decanoic acid [155], and tetrabutylammonium bromide and heptanol [156] are used as extractants in HLLME. A ratio between components in these eutectic mixtures was 1 : 2, 1 : 3, or 1 : 4. Phase separation was carried out by adding small amounts of aprotic solvents: tetrahydrofuran [148–151, 155], hexane [152], ethyl acetate [153], and cyclohexane [154]. Usually, the formation of a turbid solution was observed upon the addition of an aprotic organic solvent; therefore, the ultrasonic treatment and centrifugation of analyzed samples was carried out to accelerate phase separation. Figure 4 schematically illustrates an HLLME procedure based on the use of a mixture of choline chloride with phenol (1 : 4) as an extractant, which was applied to the determination of malachite green in waters [149].

Schematic diagram of homogeneous liquid–liquid microextraction based on the use of a mixture of choline chloride with phenol (1 : 4) as an extractant for the determination of malachite green in waters [149].
Shishov et al. [156] proposed a new method for phase separation in this version of HLLME. They found that a deep eutectic solvent formed from tetrabutylammonium bromide and heptanol decomposed upon its addition to an analyzed aqueous phase, and this led to the in situ formation of a dispersed organic phase into which organic compounds were extracted.
Shishov with coauthors [157, 158] proposed to in situ synthesize a deep eutectic solvent by adding menthol to the analyzed solution for the separation of non-steroidal anti-inflammatory drugs (NSAIDs) from urine and milk. The procedure involved the separation of NSAIDs from the aqueous phase of a sample by the in situ formation of deep eutectic mixtures due to the formation of hydrogen bonds between the hydroxyl groups of menthol and the oxygen atoms of the carboxyl groups of NSAIDs [157, 158].
Deep eutectic solvents were used in HLLME for the separation of benzene, toluene, PAHs [148], malachite green [149], organochlorine pesticides [153], and rhodamine B [155] from natural waters; caffeine from beverages [150, 151]; ferulic, caffeic, and cinnamic acids from olive, almond, and sesame oils [152]; PAHs from marine fish and algae [154]; rhodamine B from cosmetic products [155]; steroidal estrogens from drugs [156]; and NSAIDs from urine [157] and milk [158]. Compounds were determined directly in extracts by HPLC with spectrophotometric [148, 150–152, 156, 157] and fluorescence [154] detectors, gas chromatography with mass-spectrometric detection [153], and spectrophotometric analysis [149, 155].
* * *
Homogeneous liquid–liquid microextraction is a miniature version of homogeneous liquid–liquid extraction. Unlike other LLME methods, this method uses water-miscible hydrophilic solvents (mainly acetonitrile) as extractants. In different versions of HLLME, various ways to achieve extractant phase formation are used: salting-out, sugaring-out, changes in pH or temperature, and addition of a hydrophobic solvent in a small amount. The HLLME has found applications primarily in bioanalysis for the separation of polar organic compounds and their subsequent direct determination in extracts (without additional purification) by currently available analytical techniques: HPLC–MS, HPLC–MS/MS, HPLC-UV, and CE. A simple procedure of sample preparation with the use of HLLME and the possibility of its adaptation to automated analysis systems, a rapid equilibration, a low consumption of extractants, and a decrease in the duration of extraction and the cost of analysis are responsible for increasing interest in this method. The current stage in the development of this method is associated with the appearance of “green” extractants—switchable-hydrophilicity solvents and deep eutectic solvents—and their use in analysis. This sample preparation method is increasingly used in the analysis of biological samples, environmental materials, and food products for the preconcentration of organic compounds in a wide range of polarities.
REFERENCES
Xu, W., Wang, X., and Cai, Z., Anal. Chim. Acta, 2013, vol. 790, p. 1.
Lorenzo, M., Campo, J., and Picó, Y., TrAC, Trends Anal. Chem., 2018, vol. 103, p. 137.
Drugov, Yu.S., Rodin, A.A., and Kashmet, V.V., Probopodgotovka v ekologicheskom analize (Sample Preparation in Environmental Analysis), Moscow: Lab-Press, 2005.
Drugov, Yu.S. and Rodin, A.A., Analiz zagryaznenii pochvy i opasnykh otkhodov (Analysis of Soil Contamination and Hazardous Waste), Moscow: Binom, 2007.
Drugov, Yu.S. and Rodin, A.A., Kontrol’ bezopasnosti i kachestva produktov pitaniya i tovarov detskogo assortimenta (Control of the Safety and Quality of Food and Baby Products), Moscow: Binom, 2012.
Tobiszewski, M., Mechlińska, A., Zygmunt, B., and Namieśnik, J., TrAC, Trends Anal. Chem., 2009, vol. 28, p. 943.
Carasek, E., Merib, J., Mafra, G., and Spudeit, D., TrAC, Trends Anal. Chem., 2018, vol. 108, p. 203.
Armenta, S., Garrigues, S., Esteve-Turrillas, F.A., and de la Guardia, M., TrAC, Trends Anal. Chem., 2019, vol. 116, p. 248.
Yamini, Y., Rezazadeh, M., and Seidi, S., TrAC, Trends Anal. Chem., 2019, vol. 112, p. 264.
Hashemi, B., Zohrabi, P., Kim, K.-H., Shamsipur, M., Deep, A., and Hong, J., TrAC, Trends Anal. Chem., 2017, vol. 97, p. 83.
Rutkowska, M., Płotka-Wasylka, J., Sajid, M., and Andruch, V., Microchem. J., 2019, vol. 149, 103 989. https://doi.org/10.1016/j.microc.2019.103989
Anthemidis, A.N. and Ioannou, K.I.G., Talanta, 2009, vol. 80, p. 413.
Moradi, M., Yamini, Y., and Ebrahimpour, B., J. Iran. Chem. Soc., 2014, vol. 11, p. 1087.
Matkovich, C.E., Anal. Chem., 1973, vol. 45, no. 11, p. 1915.
Tabata, M., Kumamoto, M., and Nishimoto, J., Anal. Sci., 1994, vol. 10, p. 383.
Leggett, D.C., Jenkins, T.F., and Miyares, P.H., Anal. Chem., 1990, vol. 62, p. 1355.
Korenman, Ya.I., Ermolaeva, T.N., and Kuchmenko, T.A., Zh. Prikl. Khim., 1991, vol. 64, p. 573.
Korenman, Ya.I., Kuchmenko, T.A., and Ermolaeva, T.N., Zh. Anal. Khim., 1991, vol. 46, p. 1530.
Parkin, J.E., J. Chromatogr. A, 1993, vol. 635, no. 1, p. 75.
Grover, P.K. and Ryall, R.L., Chem. Rev., 2005, vol. 105, no. 1, p. 1.
Wang, B., Ezejias, T., Feng, H., and Blaschek, H., Chem. Eng. Sci., 2008, vol. 63, p. 2595.
Igarashi, S. and Yotsuyanagi, T., Microchim. Acta, 1992, vol. 44, p. 37.
Yoshida, M. and Akane, A., Anal. Chem., 1999, vol. 71, p. 1918.
Ebrahimzadeh, H., Yamini, Y., Kamarei, F., and Shariati, S., Anal. Chim. Acta, 2007, vol. 594, p. 93.
Zvarova, T., Shkinev, V., Spivakov, B., and Zolotov, I., Dokl. Akad. Nauk SSSR, 1983, vol. 273, no. 1, p. 107.
Videira, M. and Aires-Ban-O, M.R., J. Chromatogr. A, 1994, vol. 668, p. 237.
Shkinev, B.M., Mokshina, N.Ya., and Khokhlov, V.Yu., Dokl. Ross. Akad. Nauk, 2013, vol. 448, p. 427.
Gupta, M., Jain, A., and Verma, K.K., Talanta, 2009, vol. 80, p. 526.
Tang, Y.Q. and Weng, N., Bioanalysis, 2013, vol. 5, p. 1583.
Valente, I.M. and Rodrigues, J.A., Bioanalysis, 2015, vol. 7, p. 2187.
Heydari, R., Lotfi, Z., and Ramezani, M., J. Anal. Chem., 2018, vol. 73, no. 11, p. 1105.
Liu, J., Jiang, M., Li, G., Xu, L.C., Xie, M., and Liu, J., Anal. Chim. Acta, 2010, vol. 679, p. 74.
Sereshti, H., Khosraviani, M., and Amini-Fazl, M.S., Talanta, 2014, vol. 121, p. 199.
Farajzadeh, M.A., Mohebbi, A., Mogaddam, M.R.A., Davaran, M., and Norouzi, M., Food Anal. Methods, 2018, vol. 11, p. 2497.
Chen, M.J., Liu, Y.T., Lin, C.W., Ponnusamy, V.K., and Jen, J.F., Anal. Chim. Acta, 2013, vol. 767, p. 81.
Myasein, F., Kim, E., Zhang, J., Wu, H., and El-Shourbagy, T.A., Anal. Chim. Acta, 2009, vol. 651, p. 112.
Zhang, J., Rodila, R., Gage, E., Hautman, M., Fan, L., King, L.L., Wu, H., and El-Shourbagy, T.A., Anal. Chim. Acta, 2010, vol. 661, p. 167.
Gupta, M., Jain, A., and Verma, K.K., J. Sep. Sci., 2010, vol. 33, p. 3774.
Razmara, R.S., Daneshfar, A., and Sahrai, R., J. Ind. Eng. Chem., 2011, vol. 17, no. 3, p. 533.
Wang, M., Cai, Z., and Xu, L., J. Chromatogr. A, 2011, vol. 1218, no. 26, p. 4045.
Valente, I.M., Moreira Gonçalves, L.M., and Rodrigues, J.A., J. Chromatogr. A, 2013, vol. 1308, p. 58.
Gure, A., Lara, F.J., Moreno-González, D., Megersa, N., Olmo-Iruela, M., and García-Campaña, A.M., Talanta, 2014, vol. 127, p. 51.
Hassan, J. and Bahrani, S.H., Arabian J. Chem., 2014, vol. 7, no. 1, p. 87.
Røen, B.T., Sellevåg, S.R., and Lundanes, E., Anal. Chem., 2014, vol. 86, p. 11 833.
Du, D., Dong, G., Wu, Y., Wang, J., Gao, M., Wang, X., and Li, Y., Anal. Methods, 2014, vol. 6, p. 6973.
Ahmed, S. and Mahmoud, A.M., Talanta, 2015, vol. 144, p. 480.
Wang, H., Gao, M., Wang, M., Zhang, R., Wang, W., Dahlgren, R.A., and Wang, X., J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2015, vol. 985, p. 62.
Tandel, D., Shah, P., Patel, K., Thakkar, V., Patel, K., and Gandhi, T., J. Chromatogr. Sci., 2016, vol. 54, p. 1827.
Pochivalov, A., Vakh, C., Andruch, V., Moskvin, L., and Bulatov, A., Talanta, 2017, vol. 169, p. 156.
Alshishani, A., Salhimi, S.M., and Saad, B., J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2018, vol. 1073, p. 51.
Zhao, F.-J., Tang, H., Zhang, Q.-H., Yang, J., Davey, A.K., and Wang, J., J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2012, vols. 881–882, p. 119.
Jain, A., Gupta, M., and Verma, K.K., J. Chromatogr. A, 2015, vol. 1422, p. 60.
Diuzheva, A., Balogh, J., Studenyak, Y., Cziáky Z., and Jekőb, J., Talanta, 2019, vol. 194, p. 446.
Xiong, X. and Yang, L., Biomed. Chromatogr., 2015, vol. 29, p. 268.
Hajkova, K., Jurasek, B., Sykora, D., Palenicek, T., Miksatkova, P., and Kuchar, M., Anal. Bioanal. Chem., 2016, vol. 408, p. 1171.
Akramipour, R., Fattahi, N., Pirsaheb, M., and Gheini, S., J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2016, vol. 1012, p. 162.
Heydari, R. and Mousavi, M., J. Chil. Chem. Soc., 2016, vol. 61, p. 3090.
Magiera, S., Kolanowska, A., and Baranowski, J., J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2016, vol. 1022, p. 93.
Giroud, B., Bruckner, S., Straub, L., Neumann, P., Williams, G.R., and Vulliet, E., Microchem. J., 2019, vol. 151, 104 249. https://doi.org/10.1016/j.microc.2019.104249
Farajzadeh, M.A., Mohebbi, A., Mogaddam, M.R.A., Davaran, M., and Norouzi, M., Food Anal. Methods, 2018, vol. 11, p. 2497.
Fu, H., Sun, Y., Teng, H., Zhang, D., and Xiu, Z., Sep. Purif. Technol., 2015, vol. 139, p. 36.
Hyde, A.M., Zultanski, S.L., Waldman, J.H., Zhong, Y.-L., Shevlin, M., and Peng, F., Org. Process Res. Dev, 2017, vol. 21, no. 9, p. 1355.
Gao, S.Q., Jin, H.Y., Ding, Y., Zhang, N., Wang, Y., Ren, R.B., Zhang, R., and Zhang, H., J. Chromatogr. A, 2011, vol. 1218, p. 7254.
Liu, Z., Yu, W., Zhang, H., Gu, F., and Jin, X., Talanta, 2016, vol. 161, p. 748.
Wang, H., Gao, M., Gao, J., Yu, N., Huang, H., Yu, Q., and Wang, X., Anal. Bioanal. Chem., vol. 408, p. 6105.
Zhang, L., Yu, R., Yu, Y., Wang, C., and Zhang, D., Microchem. J., 2019, vol. 146, p. 115.
Li, Y., Dai, J.-Y., and Xiu, Z.-L., Sep. Purif. Technol., 2020, vol. 240, 116 584. https://doi.org/10.1016/j.seppur.2020.116584
Pletnev, I.V., Smirnova, S.V., and Shvedene, N.V., J. Anal. Chem., 2019, vol. 74, p. 625.
Farajzadeh, M.A., Feriduni, B., and Mogaddam, M.R.A., Talanta, 2016, vol. 146, p. 772.
Shi, Z., Huai, Q., Li, X., Ma, H., Zhou, C., Chu, X., and Zhang, H., J. Chromatogr. Sci., 2020, vol. 58, p. 171.
Rezaei, F. and Hosseini, M.-R.M., Anal. Chim. Acta, 2011, vol. 702, p. 274.
Jia, C., Zhu, X., Wang, J., Zhao, E., He, M., Chen, L., and Yu, P., J. Sep. Sci., 2014, vol. 37, p. 1862.
Ahmed, O.S., Ladner, Y., Montels, J., Philibert, L., and Perrin, C., J. Chromatogr. A, 2018, vol. 1579, p. 121.
Vakh, Ch., Falkova, M., Timofeeva, I., Moskvin, A., Moskvin, L., and Bulatov, A., Crit. Rev. Anal. Chem., 2016, vol. 46, no. 5, p. 374.
Lezov, A., Vakh, C., Pochivalov, A., Bulatov, A., Lebedinets, S., Moskvin, L., and Cherkashina, K., Talanta, 2018, vol. 184, p. 122.
Ahmed, O.S., Ladner, Y., Xia, J., Montels, J., Philibert, L., and Perrin, C., Talanta, 2020, vol. 208, 120 391. https://doi.org/10.1016/j.talanta.2019.120391
Wang, B., Feng, H., Ezeji, T., and Blaschek, H., Chem. Eng. Technol., 2008, vol. 31, p. 1869.
Dhamole, P.B., Mahajan, P., and Feng, H., J. Chem. Eng. Data, 2010, vol. 55, no. 9, p. 3803.
Dhamole, P.B., Mahajan, P., and Feng, H., Process Biochem., 2010, vol. 45, no. 10, p. 1672.
de Brito Cardoso, G., Mourão, T., Menezes Pereira, F., Freire, M.G., Fricks, A.T., Faria Soares, C.M., and Silva Lima, Á., Sep. Purif. Technol., 2013, vol. 104, p. 106.
Zhang, J., Myasein, F., Wu, H.Q., and El-Shourbagy, T.A., Microchem. J., 2013, vol. 108, p. 198.
Shi, Z., Li, Z., Qiu, L., Sun, M., Zhang, D., and Zhang, H., J. Liq. Chromatogr. Relat. Technol., 2015, vol. 38, p. 722.
Nugbienyo, L., Malinina, Y., Garmonov, S., and Bulatov, A., Talanta, 2017, vol. 167, p. 709.
Timofeeva, I., Shishov, A., Kanashina, D., Dzema, D., and Bulatov, A., Talanta, 2017, vol. 167, p. 761.
Shishov, A., Nechaeva, D., Moskvin, L., Andruch, V., and Bulatov, A., J. Mol. Liq., 2017, vol. 233, p. 149.
Tu, X.J., Sun, F.Y., Wu, S.Y., Liu, W.Y., Gao, Z.S., Huang, S.K., and Chen, W.B., J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2018, vol. 1073, p. 90.
Tu, X.J., Wu, S.Y., Liu, W.Y., Gao, Z.S., Huang, S.K., and Chen, W.B., Food Anal. Methods, 2019, vol. 12, p. 705.
Tsai, W.H., Chuang, H.Y., Chen, H.H., Wu, Y.W., Cheng, S.H., and Huang, T.C., J. Chromatogr. A, 2010, vol. 1217, p. 7812.
Zhu, Z., Zhang, Y., Wang, J., Li, X., Wang, W., and Huang, Z., J. Chromatogr. A, 2019, vol. 1601, p. 104.
Koltsakidou, A., Zacharis, C.K., and Fytianosa, K., J. Chromatogr. A, 2015, vol. 1377, p. 46.
Yan, L., Sun, Y., and Xiu, Z., Sep. Purif. Technol., 2016, vol. 161, p. 152.
Ebrahimi, N. and Sadeghi, R., J. Chromatogr. A, 2018, vol. 1581–1582, p. 156.
Dai, J.-Y., Ma, L.-H., Wang, Z.-F., Guan, W.-T., and Xiu, Z.-L., Bioprocess Biosyst. Eng, 2017, vol. 40, no. 3, p. 423.
Jessop, P.G., Phan, L., Carrier, A., Robinson, S., Durr, C.J., and Harjani, J.R., Green Chem., 2010, vol. 12, p. 809.
Vanderveen, J.R., Durelle, J., and Jessop, P.G., Green Chem., 2014, vol. 16, p. 1187.
Durelle, J., Vanderveen, J.R., and Jessop, P.G., Phys. Chem. Chem. Phys., 2014, vol. 16, p. 5270.
Durelle, J., Vanderveen, J.R., Quan, Y., Chalifoux, C.B., Kostin, J.E., and Jessop, P.G., Phys. Chem. Chem. Phys., 2015, vol. 17, no. 7, p. 5308.
Jessop, P.G., Kozycz, L., Rahami, Z.G., Schoenmakers, D., Boyd, A.R., Wechsler, D., and Holland, A.M., Green Chem., 2011, vol. 13, p. 619.
Vanderveen, J.R., Zhang, S., Geng, J., and Jessop, P.G., RSC Adv., 2018, vol. 8, no. 48, p. 27 318.
Lasarte-Aragonés, G., Lucena, R., Cárdenas, S., and Valcárcel, M., Talanta, 2015, vol. 131, p. 645.
Lasarte-Aragonés, G., Lucena, R., Cárdenas, S., and Valcárcel, M., J. Sep. Sci., 2015, vol. 38, p. 990.
Wang, X., Gao, M., Zhang, Z., Gu, H., Liu, T., Yu, N., Wang, X., and Wang, H., Food Anal. Methods, 2018, vol. 11, p. 2093.
Lasarte-Aragonés, G., Álvarez-Lueje, A., Salazar, R., and Toledo-Neira, C., Molecules, 2020, vol. 25, no. 1, p. 86.
Rameshgar, J., Hasheminas, K.S., Adlnas, L., and Ahmar, H., J. Sep. Sci., 2017, vol. 40, p. 3114.
Shahvandi, S.K., Banitabab, M.H., and Ahmara, H., Talanta, 2018, vol. 184, p. 103.
Ahmar, H., Nejati-Yazdinejad, M., Najafi, M., and Hasheminasab, K.S., Chromatographia, 2018, vol. 81, p. 1063.
Shahvandi, S.K., Banitaba, M.H., Ahmar, H., and Karimi, P., Microchem. J., 2020, vol. 152, 104 300. https://doi.org/10.1016/j.microc.2019.104300
Farajzadeh, M.A., Mohebbi, A., and Feriduni, B., Anal. Methods, 2016, vol. 8, p. 5676.
Kakavandi, N.R., Ezoddin, M., Abdi, K., Ghazikhansari, M., Amini, M., and Shahtaheri, S.J., J. Sep. Sci., 2017, vol. 40, p. 3703.
Cherkashina, K., Lebedinets, S., Pochivalov, A., Lezov, A., Vakh, C., and Bulatov, A., Anal. Chim. Acta, 2019, vol. 1074, p. 117.
Ebrahimpour, B., Yamini, Y., and Esrafili, A., J. Sep. Sci., 2013, vol. 36, no. 8, p. 1493.
Ebrahimpour, B. and Yamini, Y., J. Sep. Sci., 2014, vol. 37, no. 15, p. 2002.
Shih, H.K., Shu, T.Y., Ponnusamy, V.K., and Jen, J.F., Anal. Chim. Acta, 2015, vol. 854, p. 70.
Vakh, C., Pochivalov, A., Andruch, V., Moskvin, L., and Bulatov, A., Anal. Chim. Acta, 2016, vol. 907, p. 54.
Shishov, A., Sviridov, I., Timofeeva, I., Chibisova, N., Moskvin, L., and Bulatov, A., J. Mol. Liq., 2017, vol. 247, p. 246.
Torbati, M., Farajzadeh, M.A., Torbati, M., Nabil, A.A.A., Mohebbi, A., and Mogaddam, M.R.A., Talanta, 2018, vol. 176, p. 565.
Cabuk, H., Kokturk, M., and Ata, S., J. Sep. Sci., 2014, vol. 37, p. 1343.
Liu, X., Shen, Z., Wang, P., Liu, C., Zhou, Z., and Liu, D., J. Chromatogr. A, 2014, vol. 1371, p. 58.
Yoshida, M., Akane, A., Nishikawa, M., Watabiki, T., and Tsuchihashi, H., Anal. Chem., 2004, vol. 76, p. 4672.
Zhang, H.Y., Li, S.S., Liu, X.Z., Yuan, F.F., Liang, Y.H., and Shi, Z.H., J. Liq. Chromatogr. Relat. Technol., 2015, vol. 38, p. 584.
Shi, Z., Li, Z., Zhang, S., Fu, H., and Zhang, H., J. Chromatogr. Sci., 2015, vol. 53, no. 8, p. 1407.
Zhang, H., Wu, Z., Zhao, J., and Wang, Z., IERT Procedia, 2013, vol. 5, p. 277.
Bekhterev, V.N., Mendeleev Commun., 2007, vol. 17, p. 241.
Bekhterev, V.N., J. Anal. Chem., 2008, vol. 63, no. 10, p. 950.
Bekhterev, V.N., Gavrilova, S.N., and Koshkareva, E.V., Pharm. Chem. J., 2008, vol. 42, no. 2, p. 95.
Podolina, E.A., Rudakov, O.B., Fan Vin Thin, and Rudakova, L.V., J. Anal. Chem., 2010, vol. 65, no. 2, p. 117.
Bekhterev, V.N., Gavrilova, S.N., Kozina, E.P., and Maslakov, I.V., Sud.-Med. Ekspert., 2010, vol. 53, no. 5, p. 22.
Bekhterev, V.N., J. Anal. Chem., 2011, vol. 66, no. 6, 591.
Bekhterev, V.N., Sorbtsionnye Khromatogr. Protsessy, 2015, vol. 15, no. 5, p. 683.
Bekhterev, V.N., Russ. J. Phys. Chem. A, 2016, vol. 90, no. 10, p. 2055.
Bekhterev, V.N., Gavrilova, S.N., Koshkareva, E.V., and Shipanov, I.N., Sud.-Med. Ekspert., 2017, vol. 60, no. 3, p. 27.
Hu, L., Tao, Y., Luo, D., Feng, J., Wang, L., Yu, M., Li, Y., Covaci, A., and Mei, S., Chemosphere, 2019, vol. 233, p. 724.
Zhang, M.S., Li, S.J., Yao, S.Z., Liu, G.Z., and Chen, B., Chem. J. Chinese Univ., 2010, vol. 31, no. 8, p. 1517.
Liu, G.Z., Zhou, N.Y., Zhang, M.S., Li, S.J., Tian, Q.Q., Chen, J.T., Chen, B., Wu, Y.N., and Yao, S.Z., J. Chromatogr. A, 2010, vol. 1217, p. 243.
Cai, B.D., Ye, E.C., Yuan, B.F., and Feng, Y.Q., J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2015, vol. 1004, p. 23.
Wang, X., Zhao, X., Liu, X., Li, Y., Fu, L., Hu, J., and Huang, C., Anal. Chim. Acta, 2008, vol. 620, p. 162.
Yazdanfar, N., Yamini, Y., and Ghambarian, M., Chromatographia, 2014, vol. 77, p. 329.
Hosseini, M.H., Rezaee, M., Akbarian, S., Mizani, F., Pourjavid, M.R., and Arabieh, M., Anal. Chim. Acta, 2013, vol. 762, p. 54.
Amoli, J.S., Hassan, J., and Taleshi, M.S., Austin Chromatogr., 2014, vol. 1, no. 2, p. 2379.
Shamsipur, M. and Hassan, J., J. Chromatogr. A, 2010, vol. 1217, p. 4877.
Hassan, J., Farahani, A., Shamsipur, M., and Damerchili, F., J. Hazard. Mater., 2010, vol. 184, p. 869.
Rezaei, F. and Hosseini, M.M., Anal. Chim. Acta, 2011, vol. 702, no. 2, p. 274.
Hassan, J., J. Anal. Chem., 2014, vol. 69, p. 851.
Amini, T. and Hashemi, P., J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2018, vol. 1092, p. 252.
Shishov, A., Bulatov, A., Locatelli, M., Carradori, S., and Andruch, V., Microchem. J., 2017, vol. 135, p. 33.
Cunha, S.C. and Fernandes, J.O., TrAC, Trends Anal. Chem., 2018, vol. 105, p. 225.
Makoś, P., Słupek, E., and Gębicki, J., Microchem. J., 2020, vol. 152, 104 384. https://doi.org/10.1016/j.microc.2019.104384
Khezeli, T., Daneshfar, A., and Sahraei, R., J. Chromatogr. A, 2015, vol. 1425, p. 25.
Aydin, F., Yilmaz, E., and Soylak, M., Microchem. J., 2017, vol. 132, p. 280.
Shishov, A., Volodina, N., Nechaeva, D., Gagarinova, S., and Bulatov, A., Microchem. J., 2018, vol. 144, p. 469.
Sivrikaya, S., Food Addit. Contam., Part A, 2020, vol. 37, p. 488.
Khezeli, T., Daneshfar, A., and Sahraei, R., Talanta, 2016, vol. 150, p. 577.
Matong, J., Mpupa, A., and Nomngongo, P.N., Eurasian J. Anal. Chem., 2018, vol. 13, no. 5, em59. https://doi.org/10.29333/ejac/97219
Helalat-Nezhad, Z., Ghanemi, K., and Fallah-Mehrjardi, M., J. Chromatogr. A, 2015, vol. 1394, p. 46.
Yilmaz, E. and Soylak, M., Spectrochim. Acta, Part A, 2018, vol. 202, p. 81.
Shishov, A., Chromá, R., Vakh, C., Kuchár, J., Simon, A., Andruch, V., and Bulatov, A., Anal. Chim. Acta, 2019, vol. 1065, p. 49.
Shishov, A.Y., Chislov, M.V., Nechaeva, D.V., Moskvin, L.N., and Bulatov, A.V., J. Mol. Liq., 2018, vol. 272, p. 738.
Shishov, A., Nechaeva, D., and Bulatov, A., Microchem. J., 2019, vol. 150, 104 080. https://doi.org/10.1016/j.microc.2019.104080
Funding
This work was carried out within the framework of state contract no. AAAA-A16-116111750033-9.
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated by V. Makhlyarchuk
Rights and permissions
About this article

Cite this article
Dmitrienko, S.G., Apyari, V.V., Gorbunova, M.V. et al. Homogeneous Liquid–Liquid Microextraction of Organic Compounds. J Anal Chem 75, 1371–1383 (2020). https://doi.org/10.1134/S1061934820110052
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1061934820110052