
Abstract
As the limits of fetal viability have increased over the past 30 years, there has been a growing body of evidence supporting the idea that chronic disease should be taken into greater consideration in addition to survival after preterm birth. Accumulating evidence also suggests there is early onset of biologic aging after preterm birth. Similarly, chronic kidney disease (CKD) is also associated with a phenotype of advanced biologic age which exceeds chronologic age. Yet, significant knowledge gaps remain regarding the link between premature biologic age after preterm birth and kidney disease. This review summarizes the four broad pillars of aging, the evidence of premature aging following preterm birth, and in the setting of CKD. The aim is to provide additional plausible biologic mechanisms to explore the link between preterm birth and CKD. There is a need for more research to further elucidate the biologic mechanisms of the premature aging paradigm and kidney disease after preterm birth. Given the emerging research on therapies for premature aging, this paradigm could create pathways for prevention of advanced CKD.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Globally, one in every ten infants is born preterm and up to 15% of all babies born require special care in the neonatal intensive care unit (NICU) [1]. The rates of preterm birth are also rising in many countries making preterm birth a significant focus of the United Nations Sustainable Development Goals [2].
With the remarkable advances in technology and modern medicine, the number of former preterm infants living into adulthood is increasing. However, the pattern of major morbidity and the proportion of survivors affected remains unchanged [3]. Adult survivors of preterm birth develop chronic health conditions typically seen among individuals of advanced age, suggesting that the advanced aging process occurs at an earlier timepoint in life [4]. Many studies have replicated associations between lower birth weight and gestational age and higher risks of earlier life cardiovascular disease, obesity, insulin resistance, and hypertension, albeit among more homogenous populations [5]. Similarly, an increasing number of studies have demonstrated a relationship between both preterm birth or low birth weight and chronic kidney disease (CKD) [6,7,8]. Infants born preterm, as compared to infants born at term, are nearly twice as likely to develop CKD over the life course [6, 7].
Questions remain about the natural history of the development of CKD after preterm birth and the factors which might increase the risk for CKD during childhood. There is substantial research supporting the concept of the Developmental Origins of Health and Disease hypothesis, and an emergence of research on the role of nephron endowment, and impaired postnatal nephrogenesis in the risk of CKD after preterm birth [9]. In addition, studies are emerging identifying accelerated cellular senescence and a premature-aging phenotype independently associated with a wide range of kidney diseases [10]. In survivors of preterm birth, there is also increasing evidence demonstrating acceleration of cellular aging compared to term born controls [11]. It is not yet known whether these accelerated aging mechanisms after preterm birth directly contribute to an increased risk for CKD. In this review, we will summarize the literature regarding the general molecular mechanisms of cellular aging experienced among all with advancing chronologic age and examine the relationship between the premature aging phenotype known to occur after preterm birth as a potential paradigm for understanding poor kidney outcomes after preterm birth. Lastly, we will highlight how the mechanisms of premature aging may present important opportunities for the design of targeted therapeutic strategies to slow cellular aging and reduce kidney disease in this high-risk population.
Molecular mechanisms of aging
Aging begins in utero and continues across the lifespan. Children with Trisomy 21 experience a premature aging related to an aged oocyte with chromosomal folding changes that contribute to accelerated cellular aging in utero. The premature aging phenotype among younger persons with Trisomy 21 includes early-onset dementia, early loss of immune function, increased incidence of neoplasia, and mortality similar to individuals of advanced age [12]. It is well established that premature aging contributes to poor health outcomes in early life among many chronic diseases [13]. Thus, studying premature aging may lead to better understanding of the mechanisms and potential therapeutic targets of chronic diseases associated with premature aging, including pediatric CKD and cardiovascular diseases.
The four broad pillars of aging include inflammation, macromolecular dysfunction, stem cell progenitor alterations, and cellular senescence. These pillars categorize the aging process at a cellular and molecular level. To better understand premature aging as a potential pathway between preterm birth to CKD, we will briefly review the pillars of chronologic aging, a subject that is less well-studied in the context of pediatric age individuals.
Inflammation
Low-grade sterile inflammation is a prominent alteration in cellular communication associated with fibrosis, chronic disease, and premature aging. Inflammation may result from multiple pathways including enhanced activation of NF-κB transcription factor, defective autophagy, accumulation of pro-inflammatory tissue damage, ineffective immune system clearance of pathogens or dysfunctional host cells, or secretion of pro-inflammatory cytokines [14]. These dysfunctional responses can result in an enhanced pro-inflammatory cascade culminating in increased production of IL-1β, tumor necrosis factor, and interferons [14]. When constitutively active, these processes are involved in the pathogenesis of obesity, type 2 diabetes, and atherosclerosis which are associated with the aging phenotype [15]. Pro-inflammatory monocytes/macrophages (CD14 + /CD16 +), for example, have been shown to be specifically increased in younger individuals with inflammatory conditions such as atherosclerosis and inflammatory bowel disease [16]. A study analyzed the proportion of pro-inflammatory monocytes/macrophages in perirenal and perivascular fat of healthy living kidney donor women. Compared to healthy kidney donors ≤ 50 years, healthy kidney donors (age > 51 years) were found to have a significantly higher proportion of CD14 + /CD16 + monocyte/macrophages [17]. These data suggest there is a relationship between increasing monocyte/macrophage burden and chronologic aging among healthy individuals.
Macromolecular dysfunction
Macromolecular dysfunction, which can include proteins, DNA, or lipids, can also contribute to aging. Studies from the 1970s to 1980s demonstrate oxidative damage to proteins and DNA increased with age. Accumulation of damaged proteins, and DNA were thought to decrease cell turnover and ultimately decrease an organ’s ability to perform its normal functions [18]. Telomeres are critically important for protecting genomic integrity by preventing chromosomal end-to-end fusions and are well-researched in aging [19]. The length of telomeric DNA is inherited and varies among individuals and typically shortens with advancing chronologic age [20]. When telomeres become critically short, they lose their proliferative capacity and genomic integrity which can lead to damage to DNA via apoptosis and replicative senescence or an irreversible arrest of cell proliferation [19]. Mean telomere length shortening has been shown to be associated with age-related disease such as CKD, cardiovascular disease, dementia, cancer, and diabetes [21,22,23].
Progenitor cell alterations
Changes in the ability of stem or progenitor cells to replicate, differentiate, and acquire specialized function can also lead to more rapid shifts toward cellular arrest associated with breakdown in tissue function and the aging phenotype [24]. Progenitor cell aging can be tissue-specific or generalized to most adult progenitor cells. Age-associated alterations in progenitor cells can be driven by (1) elevated pro-inflammatory cytokines, (2) increased reactive oxygen species, and (3) dysregulation of autophagy. The end result is a reduction of function and proliferative potential in progenitor cells leading to cellular arrest associated with chronic disease [25]. An example of this can be found in studies of the numerical and functional capacity of circulating endothelial progenitor cells (EPCs). Reduced number and functional impairment of EPCs occurs with advancing age and various stressors [26]. Reduced numbers of EPCs have been shown to be associated with vascular aging and an increased risk for cardiovascular disease [26].
Cellular senescence
The last and most well recognized pillar of aging is cellular senescence. Cellular senescence is defined as a quiescent state, signifying the loss of a cell’s ability to replicate and differentiate. Senescence can be beneficial for protection against replication of deleterious damaged cells but unhealthy if interfering with healthy cellular replication and regeneration. Traditionally, cellular senescence has been divided into two types including the Hayflick limit of replicative senescence caused by repeated cellular replication, and stress-induced premature senescence caused by cellular exposure to damage and reactive oxygen species [27]. Progression into senescence is mediated by cyclin-dependent kinase inhibitors such as p16ink4a, retinoblastoma protein, p53/p21CIP1, telomere shortening, and other factors which induce extensive changes in gene expression and chromatin organization leading to irreversible cell cycle arrest and inhibition of apoptosis [28]. Senescence can be acute or chronic. The chronic sustained pro-inflammatory state is known as the senescence-associated secretory phenotype (SASP). SASP is associated with upregulation and secretion of many pro-inflammatory proteins and cytokines that spread senescence signals to other cells contributing to injury and senescence in adjacent cells collectively impacting organ function [29]. Ample evidence implicates chronic cellular senescence throughout all cells in aging and chronic disease pathways in both preclinical and clinical studies [29]. In young adult survivors of childhood cancer (median age 37.9 years), peripheral blood T-lymphocyte p16ink4a expression was significantly higher as compared to age-matched healthy controls. The high p16ink4a expression was associated with earlier onset low walking speed, type 2 diabetes, and Alzheimer’s disease. The authors estimated that the young adults surviving childhood cancer had a 25-year age acceleration beyond their chronologic age [30].
According to the Unitary Theory of Fundamental Aging, inflammation, macromolecular damage, progenitor cell dysfunction, and cellular senescence are highly interrelated such that if one process occurs, all occur [27]. They are biologic mechanisms which can change the trajectory of normal physiologic tissue capacity, thereby increasing the risk for premature aging phenotype and chronic disease in earlier life. Fortunately, intervening against one process can affect the other aging processes offering us new opportunities to impact disease related to premature aging in early life [27].
In the following sections, we will discuss research on the association between premature aging and both preterm birth and kidney disease. Given the increasing evidence regarding premature cellular aging in persons born preterm and in persons with kidney disease, we propose that investigating premature aging may lead to additional research opportunities and interventions to improve kidney outcomes after preterm birth.
Premature aging in preterm born survivors
Research suggests that cellular aging begins in the oocyte and placenta. In utero, the placenta undergoes natural senescence, however, abnormally accelerated placental aging has been shown to play a critical role in pregnancy and fetal growth pathologies including preeclampsia, fetal growth restriction, fetal death, and preterm birth [31]. Maternal oxidative stress is considered a critical mechanism for offspring cellular aging, telomere shortening, embryo development, and offspring disease [32]. Accelerated aging in offspring has been demonstrated via DNA methylation of fetal cord blood in the offspring of mothers over 40 years of age at delivery and with preeclampsia [33]. To date, there are no studies evaluating the direct impact of maladaptive senescence on kidney embryogenesis; however, in preclinical models, exposure to senescent fibroblasts disrupts mouse alveolar epithelial cell functional differentiation and branching morphogenesis [34]. Perhaps maladaptive senescence during kidney development similarly impacts kidney epithelial cell branching morphogenesis.
After birth, compelling evidence indicates that individuals born preterm are prone to experience accelerated biological aging throughout their lifespan. The accelerated aging process after preterm birth manifests with organ dysfunction and chronic diseases at an earlier stage in life compared to geriatric individuals [4, 35].
In animal model studies, premature vascular aging has been shown to occur in animals affected by fetal growth restriction and preterm birth. One study examined ex vivo reactivity of the carotid and femoral arteries and an umbilical artery endothelial cell epigenetic marker of vascular aging (i.e., LINE-1 DNA methylation) in fetal guinea pigs compared to adult guinea pigs. The arteries from growth-restricted guinea pig fetuses showed increased contractility and decreased relaxation responses, typically associated with chronologic aging. Additionally, the epigenetic marker of vascular aging in the growth-restricted fetuses showed decreased methylation compared to the adult animals [36]. While infants born with fetal growth restriction may be at uniquely increased risk for vascular aging, fetal growth restriction is highly prevalent among individuals born preterm.
Among humans, there are many studies demonstrating significant changes in arterial structure and stiffness in children and young adults born preterm without growth restriction compared to term born controls, suggesting premature vascular aging may be a responsible pathway among the entire preterm born population [37, 38]. Together, these animal and human studies suggest a suboptimal perinatal period contributes to a premature aging phenotype. The sequelae of preterm birth are now also being considered contributors to impaired grip strength, exercise tolerance, and sleep disordered breathing in children and young adults [39, 40]. Young adult men aged 30–35 years with a history of extremely low birth weight (ELBW) had a significantly older epigenetic age, calculated from DNA methylation at 353 cytosine-phosphate-guanine sites, compared to normal birth weight men by 4.6 years (p = 0.01) [4]. The older epigenetic age among men born ELBW persisted even after adjustment for neurosensory impairment and the presence of other chronic health conditions [4].
To explore beyond the molecular aspect of aging, studies have focused on the combination of molecular and functional aging after preterm birth as well. A study comparing epigenetic age at two time points (at 23 and 32 years) among ELBW young adults and normal birth weight young adults found that the resting heart rate, blood pressure, basal cortisol levels, grip strength, and body mass index were biologically 2.2 years older for ELBW born young adults [41]. In another clinical and molecular study of premature aging, healthy young adults born preterm had a higher proportion of shortened telomeres compared to adults born term (29% vs. 40%, respectively). The study also found significantly higher blood pressure profiles among the preterm born adults compared to the term born adults (systolic blood pressures 133 mmHg vs. 123 mmHg, p < 0.01) [11]. These findings have also been replicated in children with low birth weight and have been associated with shorter telomere length and higher blood pressures during early childhood (mean age 4.6 ± 0.4 years at follow-up) [42, 43].
Exposures during the neonatal period shortly after preterm birth have been shown to impact premature aging as well. Hyperoxia (30–90% supplemental O2) is a common intervention for premature born infants and has been associated with postnatal alterations in kidney development and kidney injury [44]. Sprague–Dawley neonatal rats exposed to hyperoxia for 21 postnatal days had decreased expression of the anti-aging kidney protein Klotho, increased glomerular hypertrophy, and tubular injury on histopathology compared to the kidneys of the control neonatal rats exposed to normoxia [44]. Decreased kidney Klotho expression has also been associated with CKD and advanced biological age in many human and animal studies [45].
Rapid postnatal catch-up growth is another exposure after preterm birth potentially associated with premature aging. It is associated with the senescent phenotypes of obesity, cardiovascular disease, hypertension, diabetes, and kidney disease in human studies [46]. In an animal study evaluating mechanisms for heart and kidney disease, rats born low birth weight and fed a high calorie diet for rapid postnatal growth, had significantly increased expression of p16ink4a and p21CIP1 in kidney and heart tissue at 3 and 6 months, compared to rats fed a normal diet with a normal growth trajectory [47]. This research potentially indicates an important link between early catch-up growth and subsequent kidney disease in persons born preterm.
Recognition of premature biological aging among children after preterm birth could be critically important for reversal or prevention to improve quality of life, reduce chronic disease, and extend wellness throughout the lifespan in this increasingly prevalent population [1].
Premature aging and kidney disease
The aging process has a significant impact on the kidneys. This is partly due to the kidney’s high vascular blood flow rates, metabolic activity, and oxidative stress. Nephrosclerosis is a hallmark of the aging kidney and is characterized by nephron loss, hypertrophy of the remaining nephrons, global glomerulosclerosis, tubular atrophy, interstitial fibrosis, and arteriosclerosis [48]. Large-scale studies also demonstrate that kidney volume progressively declines with age. One particular study assessed kidney volume by computed tomography in over 1300 adult kidney donors and found that kidney volume was 22 cm3 less when subjects were stratified by age in decades [49]. The same macro- and microscopic structural features seen in the naturally advancing age kidney, such as decreased kidney volume and nephrosclerosis, are also seen in children and young adults with kidney disease. Thus, decreased kidney volumes and nephrosclerosis could also be considered markers of premature kidney aging as well. Table 1 summarizes some of the studies in adults and children highlighting associations between aging biomarkers and kidney disease. We will review some of the evidence from human and animal studies of premature aging and kidney disease below.
Human studies of premature aging and kidney disease
In cohorts of young adults and children with IgA nephropathy, kidney biopsies show a greater expression of p21CIP1, p16ink4a, and lower Klotho expression compared to healthy control kidneys. Moreover, greater expression of the age-related biomarkers in IgA nephropathy kidney biopsies was associated with greater IgA nephropathy-related kidney fibrosis [50]. This finding has been replicated in other glomerular diseases (i.e., focal segmental glomerulosclerosis), where senescence and other age-related biomarkers are increasingly expressed in the kidney tissue of young adults in association with a greater degree of kidney fibrosis [51].
Advanced glycation end (AGE) products are accumulated in advanced aging individuals, as well as in individuals with CKD due to increased generation and decreased clearance. The AGE receptor is known to activate the NK-ƙB pathway that promotes p16INK4a and p21CIP1 expression known to be associated with increased kidney senescence in human histopathology [52]. Studies have also shown that serum AGE levels are significantly increased in children with CKD compared to healthy age–matched controls [53].
Klotho is primarily expressed in the proximal and distal tubules of the kidney and downregulated in advanced age as well as in acute and chronic kidney injury. Circulating levels decline with age [45]. Low Klotho concentrations have been associated with premature aging and cellular senescence in kidneys. Mechanistically, Klotho affects intracellular signaling pathways for aging biomarkers p53/p21CIP1 [54]. In human studies of genetic Klotho deficiency, a phenotype of early bone disease, vascular calcification, hypertension, impaired angiogenesis, and left ventricular hypertrophy (features of early vascular aging) are seen [55]. There are many parallels between CKD progression and the phenotypes of Klotho deficiency or dysregulation. Low Klotho expression, phosphate retention, progressive hyperphosphatemia, and rising FGF23 levels are all observed in patients as CKD progresses and are also associated with endothelial dysfunction and advanced aged cardiovascular disease even among younger aged individuals [45]. As it relates to preterm birth and kidney health, young adults born preterm have been shown to have higher systolic blood pressure in association with lower urine α-Klotho/creatinine excretion suggesting suppression of kidney α-Klotho expression as a potential mechanism for induction of higher blood pressure and other correlates of advanced age in young persons after preterm birth [56].
In the context of CKD, vascular calcifications are well known to occur in association with complications of kidney disease. The vascular calcifications of CKD occur in younger age groups and are also a model example of the premature aging phenotype. A study of vascular aging in children, found in comparison to healthy age–matched controls, children with CKD stage 5 pre-dialysis and those on dialysis demonstrated significantly increased vessel oxidative damage, increased p16INK4a and p21CIP1 expression, and elevated circulating levels of SASP factors (characterized by secretion of pro-inflammatory cytokines) [57]. These changes correlated with increased vascular stiffness and coronary artery calcifications in the children with CKD stage 5 similar to individuals of advanced biological age, and all of the biomarkers of aging studied were more enhanced among children on dialysis compared to children with CKD stage 5 pre-dialysis.
Animal studies of premature aging and kidney disease
Similar to human studies, there are a number of animal studies demonstrating a relationship between premature aging and kidney disease. Related to the accelerated cardiovascular and kidney aging, among rats with and without hypertension, p16INK4a expression was significantly increased in kidney tubular, glomerular, and interstitial cells as well as in the vascular cells of the myocardium and cardiac arteries of hypertensive rats [58]. This study suggests a relationship between kidney as well as cardiovascular disease and cellular senescence and has been replicated in human studies.
Animal studies have also shown that cellular senescence occurs in kidney tubular epithelial cells even in early stages of acute kidney injury (AKI) [59]. If an AKI episode is prolonged or severe, it is associated with chronic cellular senescence and increased susceptibility to further injury and progression to kidney fibrosis [59]. For example, mice with ischemia–reperfusion induced AKI experience critically shortened telomeres and increased expression of kidney tissue p21CIP1 [60]. These cellular senescence findings occur in association with increased acute and chronic kidney injury histopathology [60]. A recently published review nicely discusses the relationship between cellular senescence and AKI [59]. Future research is needed to evaluate whether preterm born infants who experience neonatal AKI also demonstrate increased expression of senescence biomarkers.
In genetic kidney disease animal models, such as the Kif3a knockout mouse model of cystic kidney disease, Glis2/NPHP7 gene loss of function mediates cellular senescence pathways that are the central feature of the tubular cell fibrosis and progressive kidney atrophy in nephronophthisis at younger ages [61].
In animal models of kidney transplant, studies have examined the impact of premature aging on kidney transplant outcomes. Young mouse recipients of older aged donor mouse kidneys were found to have increased expression of the senescence marker p16INK4a in allografts from older mouse kidney donors compared to younger aged donor mouse kidneys, despite normal kidney allograft histopathology at time of transplant. At 7 days post-transplant, there was even greater expression of p16INK4a among the older aged donor allografts while expression remained absent in the younger aged donor kidney allografts. Following onset of severe acute rejection, p16INK4a expression increased in both young and older age donor kidneys but expression was greater among the older aged allografts [62]. Thus, the premature aging phenotype could be induced in the kidney and worsened in the setting of kidney injury even in young animal recipients of older allografts. In rat models of chronic allograft rejection, sustained increases of p16INK4a and substantial telomere shortening have also been observed supporting the association in mammals between p16INK4a expression, allograft injury, and the aging phenotype [63].
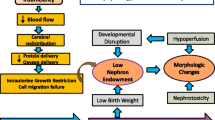
In summary, accelerated biological aging beyond chronological age is known to occur after preterm birth, as well as among individuals with kidney disease, through several potential pathways. Figure 1 depicts the pathways via the four broad pillars of premature aging after preterm birth that may contribute to premature kidney disease. Whereas cellular senescence has been shown to be important in general for embryogenesis, organ remodeling, wound healing, and tissue regeneration transiently, the chronic maladaptive responses of inflammation, macromolecular dysfunction, progenitor cell alterations, and cellular senescence are important pathways to understand in children born preterm [64]. These pathways are potential targets for future research, monitoring, and interventions to preserve kidney health in children at higher risk for kidney disease.

Created with BioRender.com
Pathways of premature aging after preterm birth associated with chronic kidney disease (CKD) and potential non-pharmacologic and pharmacologic interventions.
The potential future of biomarkers of aging for kidney disease
Beyond serum creatinine and cystatin C as the most readily available clinical biomarkers of kidney dysfunction and aging, a broad spectrum of biomarkers of aging and cellular senescence have been identified and are being explored in research to determine usefulness in clinical settings. Commonly reported biomarkers of aging such as p16INK4a and telomere length can be measured in peripheral whole blood to indicate the presence of accelerated aging.
Biomarker p21CIP1 has been studied in both plasma and urine as a potential marker of kidney aging and AKI [65]. Although, p21CIP1 was originally considered a less specific biomarker of aging because it is also expressed as a result of general tissue injury, more recently, direct expression of p21CIPI mRNA in kidney tissue has shown promise as a surrogate biomarker of aging [65]. Urinary and serum Klotho are also well-studied biomarkers known to be associated with both physiologic and pathologic kidney aging [45]. Future research is needed to identify the utility of these biomarkers at baseline after preterm birth to determine if they are useful predictive tools for CKD progression in childhood.
Potential future interventions for preterm birth and kidney disease
Considering the potential involvement of premature aging in kidney disease and the association between accelerated aging and preterm birth, it is conceivable that targeting premature aging pathways such as cellular senescence pathways, could serve as potential therapeutic targets for the identification, prevention, and treatment of kidney disease in general and specifically in the context of preterm birth. In the current era, there are multiple novel therapies which have been developed to halt aging-associated organ dysfunction which could perhaps change the trajectory of kidney health among individuals born preterm at high risk for kidney disease [66].
Potential non-pharmacologic interventions
Evidence from both animal and human models suggests that components of the premature aging phenotype and the biomarkers to identify the phenotype are responsive to caloric restriction and exercising. Caloric restriction refers to limiting caloric intake without causing damage to the body. This is an important clinical distinction as the risk of death has been shown to be increased after strict protein reduction in patients with advanced CKD [67]. Certainly, in pediatrics, where nutrition is critical for growth and development, avoidance of obesity among persons born preterm is likely a more appropriate focus of non-pharmacologic interventions to prevent premature aging. Future research should evaluate the impact of reduction in pediatric obesity on premature aging and kidney disease after preterm birth. However, current literature consistently demonstrates that caloric restriction and exercise reduce oxidative stress, systemic inflammation, and age-related telomere shortening, increase Klotho concentrations, and influence DNA methylation patterns. Each of these processes has been shown to delay cellular senescence changes within the kidney such as glomerulosclerosis, tubular atrophy, and interstitial fibrosis [68,69,70]. Further studies are needed to characterize safe, appropriate approaches to caloric restriction, perhaps in young adults born preterm.
Finally, allostatic load from chronic stress and adversity in childhood (such as violence, lower socioeconomic status, maternal depression, family disruption, and institutionalization) have been repeatedly shown to impact cellular resiliency, telomere length, and senescence [33, 71]. Large-scale studies are needed to monitor childhood stress reduction as a non-pharmacologic intervention to address premature cellular aging and chronic health outcomes.
Potential pharmacologic interventions
Hypertension has been shown to induce cellular senescence in kidney and heart in animal models. Likewise, antihypertensive therapies, such as angiotensin receptor blockers, have been shown to prevent increases in p16INK4a expression and ameliorate telomere shortening [58, 72]. This change in aging biomarker expression with antihypertensive therapy (i.e., angiotensin receptor blockers) is also associated with milder histopathological features such as reduced tubular atrophy and interstitial fibrosis [72]. In newborn animal models, evidence of alterations of aging can occur with antihypertensive medication exposures. For example, renin–angiotensin system (RAS) blockade with enalapril (30 mg/kg/day) for the first 7 days after birth in newborn rat pups significantly altered cellular senescence in neonatal rat kidneys. Compared to control rat pup kidneys, intrarenal expression of cell cycle regulators p21CIP1 and p16INK4a by immunohistochemistry were significantly decreased throughout the renal cortex and medulla at postnatal day eight after the 7 days of RAS blockade [73]. Thus, interruption of the RAS during postnatal nephrogenesis can disrupt physiologic cellular senescence in the developing rat kidney. Other animal studies have shown that blockade of angiotensin II type 1 receptor during the first 12 days of postnatal life in rats resulted in reduced number of glomeruli, reduced kidney function, and increased arterial pressure [74]. More work is necessary to determine the short- and long-term clinical implications of RAS manipulation for the purposes of aging.
Klotho may also have future therapeutic potential given that it has been shown to play a role in regulating cellular senescence in the kidney. Exogenous Klotho administration to Spraque-Dawley neonatal rats exposed to hyperoxia was associated with increased kidney perfusion and decreased vascular stiffness as defined by decreased renal artery resistance and pulsatility by Doppler ultrasound [44]. It has also been studied as a novel therapeutic agent in the prevention of AKI and CKD progression in animal models [75]. More specifically, direct exogenous Klotho has been proven effective in increasing endogenous Klotho levels and protective against kidney injury in animal models [76]. Many other drugs, such as angiotensin II receptor antagonists, PPAR-γ agonists, and paricalcitol, have also been shown to increase endogenous Klotho expression [77, 78]. Human studies research is needed to determine whether exogenous Klotho or upregulation of endogenous kidney Klotho are potential future therapeutic options for premature aging and kidney disease.
Clinical studies are emerging regarding the benefits of anti-diabetic medications, such as metformin and sodium-glucose cotransporter-2 (SGLT2) inhibitors, on vascular and kidney aging prevention. An in vitro study using human proximal tubular kidney cells demonstrated that an SGLT2 inhibitor, canagliflozin, reduced the levels of senescence-associated-inflammatory proteins TNF receptor 1, interleukin-6, matrix metalloproteinase 7, and fibronectin 1 [79]. In vitro research also suggests that canagliflozin impedes molecular aging of the kidney tubular cells by downregulating pathways involved in synthesis of reactive oxygen species and blocking inappropriate proliferation of vascular smooth muscles cells typically leading to atherosclerosis [79]. In patients with diabetic kidney disease, SGLT2 inhibitors improve cardiovascular biomarkers of aging, such as arterial stiffness and pulse wave velocity [80]. Trials are currently ongoing exploring use of SGLT2 inhibitors in pediatric CKD patients and may become a promising intervention to be studied for children born preterm with microalbuminuria.
Senolytics are a class of drugs that selectively remove or transiently disable senescent cell anti-apoptotic pathways leading to apoptosis of senescent cells while selectively sparing nonsenescent cells. In animal models of kidney disease, senolytic therapies have shown promise in reducing kidney injury. For example, senolytic treatment in the Glis2 knockout mouse model with nephronophthisis type 7 has been shown to reduce kidney tubular damage, fibrosis, cystic area, and inflammation [81]. Dasatinib, a tyrosine kinase inhibitor, decreases suppression of apoptosis, and quercetin, a natural flavonoid, suppresses senescent cell viability via restraint of the PI3K intracellular signaling pathway directly related to cellular senescence [52]. These two agents are often administered in combination and referred to as “D + Q.” In animal models, “D + Q” administration has been shown to reduce expression of senescence marker proteins such as p16INK4a and p21CIP1 in mouse renal tubular epithelial cells suggesting the drug combination could inhibit progression of renal fibrosis by delaying renal tubular epithelial cell senescence [82]. In an open label phase 1 pilot study in humans with diabetic kidney disease, 3 days of oral “D + Q” therapy known as a “hit-and-run” senolytic therapeutic approach was associated with reduced adipose tissue senescent cell burden within 11 days marked by decreases in p16INK4A- and p21CIP1-expressing cells, and cells with senescence-associated β-galactosidase activity, and decreased adipose tissue fibrosis [83].
There are over 30 trials across the Translational Geoscience Network currently underway to target the fundamental aging processes and chronic disease in humans. These therapies are particularly promising because senescent cell drug resistance is unlikely, given that senescent cells do not divide and take weeks or months to re-accumulate if the underlying drivers of senescence remain active. Senolytic drugs present a promising and compelling option for the future, as they can effectively eliminate senescent cells with brief exposure, without the need for continuous circulation. Thus, intermittent administration (i.e., once monthly) of senolytic therapies may improve adherence for high-risk individuals compared to, or even along with daily dietary restrictions and exercise.
More research is emerging demonstrating individuals born preterm are also likely to have a premature burden of frailty and advanced biological age-related mechanisms of disease, such as CKD, in early life. These non-therapeutics and therapeutic anti-aging discoveries thus have the potential to unlock an additional therapeutic route for the early prevention and treatment of advanced CKD in children born preterm (Fig. 1).
Conclusion
Premature aging is associated with inflammation, macromolecular dysfunction, stem cell progenitor alterations, and cellular senescence that leads to fibrosis and kidney disease and other age-related disorders. Preterm birth and CKD share common characteristics and mechanisms of premature aging and senescence. Kidney senescence is a promising target for therapeutic intervention of kidney disease in general and perhaps specifically in a high-risk population with a burden of advanced molecular aging such as individuals born preterm. Preclinical and early pilot data have shown the efficacy of senolytics. In the future, judicious research directly linking preterm birth and kidney disease molecular mechanisms of aging will provide opportunities for implementation of senotherapies for prevention of CKD progression (Fig. 2). Moreover, starting to address the molecular mechanisms of aging early in life is likely to have a greater impact on reducing advanced kidney disease burden.

Extrapolated figure hypothesizing the cellular integrity (i.e., telomere length, cellular replication potential) and glomerular filtration rate (GFR) for persons born at term and preterm birth which is worsened by adverse early life exposures (A); and the hypothesized potential to improve molecular integrity and GFR in persons born preterm with early therapeutic interventions to reduce premature aging (B). RAASi, renin, angiotensin, aldosterone inhibitors; SGLT2, sodium glucose cotransporter-2
Abbreviations
- CKD:
-
Chronic kidney disease
- AKI:
-
Acute kidney injury
References
Division of Reproductive Health Division of Reproductive Health, National Center for Chronic Disease Prevention and Health Promotion (2022) Preterm birth. Centers for Disease Control and Prevention. https://www.cdc.gov/reproductivehealth/maternalinfanthealth/pretermbirth.htm. Accessed 30 Oct 2023
Walani SR (2020) Global burden of preterm birth. Int J Gynaecol Obstet 150:31–33. https://doi.org/10.1002/ijgo.131953
Costeloe KL, Hennessy EM, Haider S, Stacey F, Marlow N, Draper ES (2012) Short term outcomes after extreme preterm birth in England: comparison of two birth cohorts in 1995 and 2006 (the EPICure studies). BMJ 345:e7976. https://doi.org/10.1136/bmj.e7976
Van Lieshout RJ, McGowan PO, de Vega WC, Savoy CD, Morrison KM, Saigal S, Mathewson KJ, Schmidt LA (2021) Extremely low birth weight and accelerated biological aging. Pediatrics 147:e2020001230. https://doi.org/10.1542/peds.2020-001230
Barker DJ, Winter PD, Osmond C, Margetts B, Simmonds SJ (1989) Weight in infancy and death from ischaemic heart disease. Lancet 2:577–580. https://doi.org/10.1016/s0140-6736(89)90710-1
Crump C, Sundquist J, Winkleby MA, Sundquist K (2019) Preterm birth and risk of chronic kidney disease from childhood into mid-adulthood: national cohort study. BMJ 365:l1346. https://doi.org/10.1136/bmj.l1346
White SL, Perkovic V, Cass A, Chang CL, Poulter NR, Spector T, Haysom L, Craig JC, Salmi IA, Chadban SJ, Huxley RR (2009) Is low birth weight an antecedent of CKD in later life? A systematic review of observational studies. Am J Kidney Dis 54:248–261. https://doi.org/10.1053/j.ajkd.2008.12.042
Gjerde A, Reisæter AV, Skrunes R, Marti HP, Vikse BE (2020) Intrauterine growth restriction and risk of diverse forms of kidney disease during the first 50 years of life. Clin J Am Soc Nephrol 15:1413–1423. https://doi.org/10.2215/cjn.04080320
Raju TNK, Pemberton VL, Saigal S, Blaisdell CJ, Moxey-Mims M, Buist S (2017) Long-term healthcare outcomes of preterm birth: an executive summary of a conference sponsored by the National Institutes of Health. J Pediatr 181:309-318.e1. https://doi.org/10.1016/j.jpeds.2016.10.015
Ferenbach DA, Bonventre JV (2015) Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat Rev Nephrol 11:264–276
Parkinson JRC, Emsley R, Adkins JLT, Longford N, Ozanne SE, Holmes E, Modi N (2020) Clinical and molecular evidence of accelerated ageing following very preterm birth. Pediatr Res 87:1005–1010
Franceschi C, Garagnani P, Gensous N, Bacalini MG, Conte M, Salvioli S (2019) Accelerated bio-cognitive aging in Down syndrome: state of the art and possible deceleration strategies. Aging Cell 18:e12903
Ness KK, Kirkland JL, Gramatges MM, Wang Z, Kundu M, McCastlain K, Li-Harms X, Zhang J, Tchkonia T, Pluijm SMF (2018) Premature physiologic aging as a paradigm for understanding increased risk of adverse health across the lifespan of survivors of childhood cancer. J Clin Oncol 36:2206–2215. https://doi.org/10.1200/JCO.2017.76.7467
Salminen A, Kaarniranta K, Kauppinen A (2012) Inflammaging: disturbed interplay between autophagy and inflammasomes. Aging 4:166–175. https://doi.org/10.18632/aging.100444
Barzilai N, Huffman DM, Muzumdar RH, Bartke A (2012) The critical role of metabolic pathways in aging. Diabetes 61:1315–1322. https://doi.org/10.2337/db11-1300
Pamukcu B, Lip GY, Devitt A, Griffiths H, Shantsila E (2010) The role of monocytes in atherosclerotic coronary artery disease. Ann Med 42:394–403. https://doi.org/10.3109/07853890.2010.497767
Králová Lesná I, Poledne R, Fronek J, Králová A, Sekerková A, Thieme F, Pitha J (2015) Macrophage subsets in the adipose tissue could be modified by sex and the reproductive age of women. Atherosclerosis 241:255–258. https://doi.org/10.1016/j.atherosclerosis.2015.03.018
Kirkwood TB (2005) Understanding the odd science of aging. Cell 120:437–447. https://doi.org/10.1016/j.cell.2005.01.027
Engin AB, Engin A (2021) The connection between cell fate and telomere. Adv Exp Med Biol 1275:71–100. https://doi.org/10.1007/978-3-030-49844-3_3
Farrukh S, Baig S, Hussain R, Imad R, Khalid M (2022) Parental genetics communicate with intrauterine environment to reprogram newborn telomeres and immunity. Cells 11:3777. https://doi.org/10.3390/cells11233777
Zhao J, Miao K, Wang H, Ding H, Wang DW (2013) Association between telomere length and type 2 diabetes mellitus: a meta-analysis. PLoS One 8:e79993. https://doi.org/10.1371/journal.pone.0079993
Ma H, Zhou Z, Wei S, Liu Z, Pooley KA, Dunning AM, Svenson U, Roos G, Hosgood HD III, Shen M (2011) Shortened telomere length is associated with increased risk of cancer: a meta-analysis. PLoS One 6:e20466. https://doi.org/10.1371/journal.pone.0020466
Haycock PC, Heydon EE, Kaptoge S, Butterworth AS, Thompson A, Willeit P (2014) Leucocyte telomere length and risk of cardiovascular disease: systematic review and meta-analysis. BMJ 349:g4227. https://doi.org/10.1136/bmj.g4227
Pilzecker B, Buoninfante OA, van den Berk P, Lancini C, Song J-Y, Citterio E, Jacobs H (2017) DNA damage tolerance in hematopoietic stem and progenitor cells in mice. Proc Natl Acad Sci 114:E6875–E6883. https://doi.org/10.1073/pnas.1706508114
Spehar K, Pan A, Beerman I (2020) Restoring aged stem cell functionality: current progress and future directions. Stem Cells 38:1060–1077. https://doi.org/10.1002/stem.3234
Hill JM, Zalos G, Halcox JP, Schenke WH, Waclawiw MA, Quyyumi AA, Finkel T (2003) Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N Engl J Med 348:593–600. https://doi.org/10.1056/NEJMoa022287
Kirkland JL, Tchkonia T (2017) Cellular senescence: a translational perspective. EBioMedicine 21:21–28. https://doi.org/10.1016/j.ebiom.2017.04.013
Ogrodnik M (2021) Cellular aging beyond cellular senescence: markers of senescence prior to cell cycle arrest in vitro and in vivo. Aging Cell 20:e13338. https://doi.org/10.1111/acel.13338
Di Micco R, Krizhanovsky V, Baker D, d’Adda di Fagagna F (2021) Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nat Rev Mol Cell Biol 22:75–95. https://doi.org/10.1038/s41580-020-00314-w
Smitherman AB, Wood WA, Mitin N, Ayer Miller VL, Deal AM, Davis IJ, Blatt J, Gold SH, Muss HB (2020) Accelerated aging among childhood, adolescent, and young adult cancer survivors is evidenced by increased expression of p16(INK4a) and frailty. Cancer 126:4975–4983. https://doi.org/10.1002/cncr.33112
Kajdy A, Modzelewski J, Cymbaluk-Płoska A, Kwiatkowska E, Bednarek-Jędrzejek M, Borowski D, Stefańska K, Rabijewski M, Torbé A, Kwiatkowski S (2021) Molecular pathways of cellular senescence and placental aging in late fetal growth restriction and stillbirth. Int J Mol Sci 22:4186. https://doi.org/10.3390/ijms22084186
Richter T, von Zglinicki T (2007) A continuous correlation between oxidative stress and telomere shortening in fibroblasts. Exp Gerontol 42:1039–1042. https://doi.org/10.1016/j.exger.2007.08.005
Girchenko P, Lahti J, Czamara D, Knight AK, Jones MJ, Suarez A, Hämäläinen E, Kajantie E, Laivuori H, Villa PM, Reynolds RM, Kobor MS, Smith AK, Binder EB, Räikkönen K (2017) Associations between maternal risk factors of adverse pregnancy and birth outcomes and the offspring epigenetic clock of gestational age at birth. Clin Epigenetics 9:49. https://doi.org/10.1186/s13148-017-0349-z
Parrinello S, Coppe J-P, Krtolica A, Campisi J (2005) Stromal-epithelial interactions in aging and cancer: senescent fibroblasts alter epithelial cell differentiation. J Cell Sci 118:485–496. https://doi.org/10.1242/jcs.01635
Duke JW, Lovering AT, Goss KN (2020) Premature aging and increased risk of adult cardiorespiratory disease after extreme preterm birth. Getting to the heart (and lungs) of the matter. Am J Respir Crit Care Med 202:319–320. https://doi.org/10.1164/rccm.202004-1437ED
Paz AA, Arenas GA, Castillo-Galán S, Peñaloza E, Cáceres-Rojas G, Suazo J, Herrera EA, Krause BJ (2019) Premature vascular aging in guinea pigs affected by fetal growth restriction. Int J Mol Sci 20:3474. https://doi.org/10.3390/ijms20143474
Flahault A, Oliveira Fernandes R, De Meulemeester J, Ravizzoni Dartora D, Cloutier A, Gyger G, El-Jalbout R, Bigras J-L, Luu TM, Nuyt AM (2020) Arterial structure and stiffness are altered in young adults born preterm. Arterioscler Thromb Vasc Biol 40:2548–2556. https://doi.org/10.1161/ATVBAHA.120.315099
Chainoglou A, Sarafidis K, Chrysaidou K, Farmaki E, Kollios K, Economou M, Kotsis V, Stabouli S (2022) Arterial stiffness and nocturnal hypertension in preterm children and adolescents. J Hypertens 40:1751–1757. https://doi.org/10.1097/hjh.0000000000003209
Morrison KM, Gunn E, Guay S, Obeid J, Schmidt LA, Saigal S (2021) Grip strength is lower in adults born with extremely low birth weight compared to term-born controls. Pediatr Res 89:996–1003. https://doi.org/10.1038/s41390-020-1012-5
Lovering AT, Elliott JE, Laurie SS, Beasley KM, Gust CE, Mangum TS, Gladstone IM, Duke JW (2014) Ventilatory and sensory responses in adult survivors of preterm birth and bronchopulmonary dysplasia with reduced exercise capacity. Ann Am Thorac Soc 11:1528–1537. https://doi.org/10.1513/AnnalsATS.201312-466OC
Mathewson KJ, McGowan PO, de Vega WC, Morrison KM, Saigal S, Van Lieshout RJ, Schmidt LA (2021) Cumulative risks predict epigenetic age in adult survivors of extremely low birth weight. Dev Psychobiol 63:e22222. https://doi.org/10.1002/dev.22222
Martens DS, Sleurs H, Dockx Y, Rasking L, Plusquin M, Nawrot TS (2022) Association of newborn telomere length with blood pressure in childhood. JAMA Netw Open 5:e2225521. https://doi.org/10.1001/jamanetworkopen.2022.25521
Raqib R, Alam DS, Sarker P, Ahmad SM, Ara G, Yunus M, Moore SE, Fuchs G (2007) Low birth weight is associated with altered immune function in rural Bangladeshi children: a birth cohort study. Am J Clin Nutr 85:845–852. https://doi.org/10.1093/ajcn/85.3.845
Ali MF, Venkatarayappa SKB, Benny M, Rojas C, Yousefi K, Shehadeh LA, Kulandavelu S, Sharma M, Da Silva N, Freundlich M, Abitbol CL, DeFreitas MJ, Young KC (2020) Effects of Klotho supplementation on hyperoxia-induced renal injury in a rodent model of postnatal nephrogenesis. Pediatr Res 88:565–570. https://doi.org/10.1038/s41390-020-0803-z
Buchanan S, Combet E, Stenvinkel P, Shiels PG (2020) Klotho, aging, and the failing kidney. Front Endocrinol 11:560. https://doi.org/10.3389/fendo.2020.00560
Hales CN, Ozanne SE (2003) The dangerous road of catch-up growth. J Physiol 547:5–10. https://doi.org/10.1113/jphysiol.2002.024406
Luyckx VA, Compston CA, Simmen T, Mueller TF (2009) Accelerated senescence in kidneys of low-birth-weight rats after catch-up growth. Am J Physiol Renal Physiol 297:F1697-1705. https://doi.org/10.1152/ajprenal.00462.2009
Denic A, Lieske JC, Chakkera HA, Poggio ED, Alexander MP, Singh P, Kremers WK, Lerman LO, Rule AD (2017) The substantial loss of nephrons in healthy human kidneys with aging. J Am Soc Nephrol 28:313–320. https://doi.org/10.1681/ASN.2016020154
Wang X, Vrtiska TJ, Avula RT, Walters LR, Chakkera HA, Kremers WK, Lerman LO, Rule AD (2014) Age, kidney function, and risk factors associate differently with cortical and medullary volumes of the kidney. Kidney Int 85:677–685. https://doi.org/10.1038/ki.2013.359
Yamada K, Doi S, Nakashima A, Kawaoka K, Ueno T, Doi T, Yokoyama Y, Arihiro K, Kohno N, Masaki T (2015) Expression of age-related factors during the development of renal damage in patients with IgA nephropathy. Clin Exp Nephrol 19:830–837. https://doi.org/10.1007/s10157-014-1070-2
Sis B, Tasanarong A, Khoshjou F, Dadras F, Solez K, Halloran PF (2007) Accelerated expression of senescence associated cell cycle inhibitor p16INK4A in kidneys with glomerular disease. Kidney Int 71:218–226. https://doi.org/10.1038/sj.ki.5002039
Zhao JL, Qiao XH, Mao JH, Liu F, Fu HD (2022) The interaction between cellular senescence and chronic kidney disease as a therapeutic opportunity. Front Pharmacol 13:974361. https://doi.org/10.3389/fphar.2022.974361
Misselwitz J, Franke S, Kauf E, John U, Stein G (2002) Advanced glycation end products in children with chronic renal failure and type 1 diabetes. Pediatr Nephrol 17:316–321. https://doi.org/10.1007/s00467-001-0815-9
Richter B, Faul C (2018) FGF23 actions on target tissues-with and without klotho. Front Endocrinol 9:189. https://doi.org/10.3389/fendo.2018.00189
Iurciuc S, Cimpean AM, Mitu F, Heredea R, Iurciuc M (2017) Vascular aging and subclinical atherosclerosis: why such a “never ending” and challenging story in cardiology? Clin Interv Aging 12:1339–1345. https://doi.org/10.2147/CIA.S141265
South AM, Shaltout HA, Gwathmey TM, Jensen ET, Nixon PA, Diz DI, Chappell MC, Washburn LK (2020) Lower urinary α-Klotho is associated with lower angiotensin-(1–7) and higher blood pressure in young adults born preterm with very low birthweight. J Clin Hypertens (Greenwich) 22:1033–1040. https://doi.org/10.1111/jch.13897
Sanchis P, Ho CY, Liu Y, Beltran LE, Ahmad S, Jacob AP, Furmanik M, Laycock J, Long DA, Shroff R, Shanahan CM (2019) Arterial “inflammaging” drives vascular calcification in children on dialysis. Kidney Int 95:958–972. https://doi.org/10.1016/j.kint.2018.12.014
Westhoff JH, Hilgers KF, Steinbach MP, Hartner A, Klanke B, Amann K, Melk A (2008) Hypertension induces somatic cellular senescence in rats and humans by induction of cell cycle inhibitor p16 INK4a. Hypertension 52:123–129. https://doi.org/10.1161/HYPERTENSIONHA.107.099432
Lin X, Jin H, Chai Y, Shou S (2022) Cellular senescence and acute kidney injury. Pediatr Nephrol 37:3009–3018. https://doi.org/10.1007/s00467-022-05532-2
Westhoff JH, Schildhorn C, Jacobi C, Hömme M, Hartner A, Braun H, Kryzer C, Wang C, von Zglinicki T, Kränzlin B, Gretz N, Melk A (2010) Telomere shortening reduces regenerative capacity after acute kidney injury. J Am Soc Nephrol 21:327–336. https://doi.org/10.1681/asn.2009010072
Lu D, Rauhauser A, Li B, Ren C, McEnery K, Zhu J, Chaki M, Vadnagara K, Elhadi S, Jetten AM (2016) Loss of Glis2/NPHP7 causes kidney epithelial cell senescence and suppresses cyst growth in the Kif3a mouse model of cystic kidney disease. Kidney Int 89:1307–1323. https://doi.org/10.1016/j.kint.2016.03.006
Melk A, Schmidt BMW, Braunc H, Vongwiwatana A, Urmson J, Zhu LF, Rayner D, Halloran PF (2009) Effects of donor age and cell senescence on kidney allograft survival. Am J Transplant 9:114–123. https://doi.org/10.1111/j.1600-6143.2008.02500.x
Joosten SA, van Ham V, Nolan CE, Borrias MC, Jardine AG, Shiels PG, van Kooten C, Paul LC (2003) Telomere shortening and cellular senescence in a model of chronic renal allograft rejection. Am J Pathol 162:1305–1312. https://doi.org/10.1016/S0002-9440(10)63926-0
Chevalier RL (2023) Bioenergetics: the evolutionary basis of progressive kidney disease. Physiol Rev 103:2451–2506. https://doi.org/10.1152/physrev.00029.2022
Johnson AC, Zager RA (2018) Plasma and urinary p21: potential biomarkers of AKI and renal aging. Am J Physiol Renal Physiol 315:F1329–F1335. https://doi.org/10.1152/ajprenal.00328.2018
Tan H, Xu J, Liu Y (2022) Ageing, cellular senescence and chronic kidney disease: experimental evidence. Curr Opin Nephrol Hypertens 31:235–243. https://doi.org/10.1097/mnh.0000000000000782
Selamet U, Tighiouart H, Sarnak MJ, Beck G, Levey AS, Block G, Ix JH (2016) Relationship of dietary phosphate intake with risk of end-stage renal disease and mortality in chronic kidney disease stages 3–5: the Modification of Diet in Renal Disease Study. Kidney Int 89:176–184. https://doi.org/10.1038/ki.2015.284
Ning Y-C, Cai G-Y, Zhuo L, Gao J-J, Dong D, Cui S, Feng Z, Shi S-Z, Bai X-Y, Sun X-F (2013) Short-term calorie restriction protects against renal senescence of aged rats by increasing autophagic activity and reducing oxidative damage. Mech Ageing Dev 134:570–579. https://doi.org/10.1016/j.mad.2013.11.006
Middelbeek RJW, Motiani P, Brandt N, Nigro P, Zheng J, Virtanen KA, Kalliokoski KK, Hannukainen JC, Goodyear LJ (2021) Exercise intensity regulates cytokine and klotho responses in men. Nutr Diabetes 11:5. https://doi.org/10.1038/s41387-020-00144-x
Qiu Z, Zheng K, Zhang H, Feng J, Wang L, Zhou H (2017) Physical exercise and patients with chronic renal failure: a meta-analysis. Biomed Res Int 2017:7191826. https://doi.org/10.1155/2017/7191826
Kim K, Yaffe K, Rehkopf DH, Zheng Y, Nannini DR, Perak AM, Nagata JM, Miller GE, Zhang K, Lloyd-Jones DM, Joyce BT, Hou L (2023) Association of adverse childhood experiences with accelerated epigenetic aging in midlife. JAMA Netw Open 6:e2317987. https://doi.org/10.1001/jamanetworkopen.2023.17987
Baumann M, Bartholome R, Peutz-Kootstra CJ, Smits JF, Struijker-Boudier HA (2008) Sustained tubulo-interstitial protection in SHRs by transient losartan treatment: an effect of decelerated aging? Am J Hypertens 21:177–182. https://doi.org/10.1038/ajh.2007.30
Yoo KH, Yim HE, Bae ES (2020) Angiotensin inhibition and cellular senescence in the developing rat kidney. Exp Mol Pathol 117:104551. https://doi.org/10.1016/j.yexmp.2020.104551
Woods LL, Ingelfinger JR, Nyengaard JR, Rasch R (2001) Maternal protein restriction suppresses the newborn renin-angiotensin system and programs adult hypertension in rats. Pediatr Res 49:460–467. https://doi.org/10.1203/00006450-200104000-00005
Christov M, Neyra JA, Gupta S, Leaf DE (2019) Fibroblast growth factor 23 and klotho in AKI. Semin Nephrol 39:57–75. https://doi.org/10.1016/j.semnephrol.2018.10.005
Hu M-C, Shi M, Zhang J, Quiñones H, Kuro-o M, Moe OW (2010) Klotho deficiency is an early biomarker of renal ischemia–reperfusion injury and its replacement is protective. Kidney Int 78:1240–1251. https://doi.org/10.1038/ki.2010.328
Neyra JA, Hu MC, Moe OW (2021) Klotho in clinical nephrology: diagnostic and therapeutic implications. Clin J Am Soc Nephrol 16:162–176. https://doi.org/10.2215/CJN.02840320
Sanchez-Niño MD, Fernandez-Fernandez B, Ortiz A (2020) Klotho, the elusive kidney-derived anti-ageing factor. Clin Kidney J 13:125–127. https://doi.org/10.1093/ckj/sfz125
Heerspink HJL, Perco P, Mulder S, Leierer J, Hansen MK, Heinzel A, Mayer G (2019) Canagliflozin reduces inflammation and fibrosis biomarkers: a potential mechanism of action for beneficial effects of SGLT2 inhibitors in diabetic kidney disease. Diabetologia 62:1154–1166. https://doi.org/10.1007/s00125-019-4859-4
Lunder M, Janić M, Japelj M, Juretič A, Janež A, Šabovič M (2018) Empagliflozin on top of metformin treatment improves arterial function in patients with type 1 diabetes mellitus. Cardiovasc Diabetol 17:153. https://doi.org/10.1186/s12933-018-0797-6
Jin H, Zhang Y, Liu D, Wang SS, Ding Q, Rastogi P, Purvis M, Wang A, Elhadi S, Ren C (2020) Innate immune signaling contributes to tubular cell senescence in the Glis2 knockout mouse model of nephronophthisis. Am J Pathol 190:176–189. https://doi.org/10.1016/ajpath.2019.09.013
Li C, Shen Y, Huang L, Liu C, Wang J (2021) Senolytic therapy ameliorates renal fibrosis postacute kidney injury by alleviating renal senescence. FASEB J 35:e21229. https://doi.org/10.1096/fj.202001855RR
Hickson LJ, Langhi Prata LGP, Bobart SA, Evans TK, Giorgadze N, Hashmi SK, Herrmann SM, Jensen MD, Jia Q, Jordan KL, Kellogg TA, Khosla S, Koerber DM, Lagnado AB, Lawson DK, LeBrasseur NK, Lerman LO, McDonald KM, McKenzie TJ, Passos JF, Pignolo RJ, Pirtskhalava T, Saadiq IM, Schaefer KK, Textor SC, Victorelli SG, Volkman TL, Xue A, Wentworth MA, Wissler Gerdes EO, Zhu Y, Tchkonia T, Kirkland JL (2019) Senolytics decrease senescent cells in humans: preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine 47:446–456. https://doi.org/10.1016/j.ebiom.2019.08.069
Funding
The primary author’s research efforts in this publication are supported by the NIDDK of the National Institutes of Health under award number K23DK131289 and L40DK130155. JRC is supported by R21DK134104-01, P50DK096373-11, R41DK129138, and R56DK110622. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Sanderson, K.R., Wekon-Kemeni, C. & Charlton, J.R. From premature birth to premature kidney disease: does accelerated aging play a role?. Pediatr Nephrol 39, 2001–2013 (2024). https://doi.org/10.1007/s00467-023-06208-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-023-06208-1