
Abstract
Background
This study was performed to determine the clinical features and outcomes of childhood-onset anti-neutrophil cytoplasmic antibody (ANCA)–associated vasculitis (AAV), particularly microscopic polyangiitis (MPA).
Methods
A retrospective Japanese multicenter study was performed in patients diagnosed with AAV before 16 years of age.
Results
Of 49 patients with AAV, 36 were female. The diagnoses were as follows: MPA (n = 38, 78%), granulomatosis with polyangiitis (GPA; n = 9, 18%), eosinophilic granulomatosis with polyangiitis (EGPA; n = 1, 2%), and other (n = 1, 2%). The median age at onset was 10.7 years, and median time to diagnosis was 2.0 months. Twenty-seven (55%) patients were identified through a school urinary screening program. Initial symptoms included fever and fatigue (45%), and renal (71%), pulmonary (29%), ocular (20%), and mucocutaneous involvement (22%). Although 27 (55%) patients achieved remission and none had died at the last follow-up, at least one recurrence occurred in 13 (48%) patients after a median of 48 months and was more common in patients with GPA (P < 0.01). After a median follow-up of 43 months, seven (14%) patients (all with MPA) progressed to end-stage renal disease (ESRD).
Conclusions
Childhood-onset AAV has an estimated prevalence of 3.41–4.28 per million children and is characterized by female predominance and high frequency of detection in school urinary screening programs. More than 10% of patients with childhood-onset AAV still progress to ESRD without achieving remission. Histological chronicity is a factor associated with ESRD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Anti-neutrophil cytoplasmic antibody (ANCA)–associated vasculitis (AAV) is a group of disorders characterized by necrotizing inflammation of small- to medium-sized vessels. It has been classified into three entities: granulomatosis with polyangiitis (GPA, previously Wegener’s granulomatosis), microscopic polyangiitis (MPA), and eosinophilic granulomatosis with polyangiitis (EGPA, previously Churg–Strauss syndrome). AAV generally occurs in adults and rarely in pediatric populations, with peak age at onset commonly between the fifth and seventh decades of life. Therefore, AAV has generally been studied in adults. Reports of large pediatric series are limited, and those that have been published to date have focused mainly on GPA [1,2,3,4]. Our knowledge of AAV in pediatric patients is based mostly on small cohort studies and case series [5,6,7,8,9,10,11]. In addition, reliable epidemiological data on pediatric patients with MPA are scarce because these diseases rarely occur in children. Therefore, the epidemiology of AAV in pediatric patients, such as annual incidence, prevalence, and disease course, is poorly understood.
The present study was a nationwide survey conducted to investigate the principal clinical and demographic features of pediatric AAV at presentation, to determine the course of the disease, and to identify factors associated with progression to end-stage renal disease (ESRD).
Materials and methods
Study design and population
We conducted a cross-sectional, nationwide retrospective survey in patients diagnosed with AAV before 16 years of age. Two questionnaires were sent in 2014 and 2015 to 1701 institutions in Japan, including all institutions that are members of the Japanese Society for Pediatric Nephrology and the Pediatric Rheumatology Association of Japan, all university and children’s hospitals, and all general hospitals with > 200 beds. The inclusion criteria were all children < 16 years of age with AAV treated between 2012 and 2014.
The first questionnaire was designed to record the approximate number of children with AAV in each institution, whereas the second questionnaire was designed to collect data of each patient, including sex, age at initial symptoms, age at diagnosis, clinical and laboratory characteristics at disease onset, treatments received, duration of follow-up, relapse, and final outcome. Clinical characteristics included symptoms, signs, and organ involvement at presentation. The results of histological investigations were also collected.
Diagnosis and classification of AAV
AAV was diagnosed and classified based on the American College of Rheumatology or the 2012 Revised International Chapel Hill Consensus Conference on the Nomenclature of Systemic Vasculitis [12] and the Endorsed Consensus Criteria for the Classification of Childhood Vasculitides of the European League Against Rheumatism (EULAR)/Paediatric Rheumatology European Society (PRES) [13]. The EULAR/Paediatric Rheumatology International Trials Organisation (PRINTO)/PRES proposed validated classification criteria were also used to update the classification criteria for childhood GPA [14].
Definitions
The estimated glomerular filtration rate (eGFR) was determined using the modified Schwartz formula [15]. Patients with urine protein to creatinine ratio of ≥ 0.15 g/gCr and of ≥ 2.0 g/gCr were classified as positive for proteinuria and nephrotic-range proteinuria, respectively. Hematuria was defined as ≥ 5 red blood cells/high-power field in centrifuged specimens. The Birmingham Vasculitis Activity Score (BVAS) was used to quantify disease activity [16]. For some data, such as serum creatine level and blood pressure, the Pediatric VAS was used in place of BVAS [17]. Remission was defined as a score of zero using the BVAS. Treatment failure was characterized as no improvement or deterioration of clinical symptoms and renal function. Relapse was diagnosed in patients showing reappearance or deterioration of clinical symptoms after achieving initial remission.
Statistical analysis
Basic demographic characteristics, clinical features, and laboratory findings were primarily extracted for patients who were uniquely classified as having MPA, GPA, or others. Depending on the type of data, patient characteristics at diagnosis were expressed as median with interquartile range (IQR) or percentage. Kaplan–Meier plots were used to assess time to ESRD, and time to first relapse. The log-rank test was used to compare relapse-free survival between different groups. Multiple logistic regression analysis was used to identify factors associated with ESRD.
The number of patients with AAV in Japan was estimated by dividing the number of cases reported in the first questionnaire by the response rate. However, as it is presumed that AAV is managed concentrating on the facilities of pediatric nephrologists and pediatric rheumatologists in each area, the patients were stratified based on the type of institution (i.e., university hospital, children’s hospital, and general hospital), the number of beds (< 500 vs. ≥ 500), and the presence of pediatric nephrologists on staff (yes vs. no), assuming that the response rate was independent of the number of patients in each stratified category [18, 19]. Next, we weighted the number of patients in each category by the reciprocal of the predicted response rate and summed to estimate the total number of patients in Japan. To account for the inverse probability weights, robust standard errors were estimated for confidence intervals (CIs). Prevalence rate was calculated by dividing the total estimated number of patients by the size of the population at risk in Japan at the end of 2016, based on the report of the Statistics Bureau of the Ministry of Internal Affairs and Communications of Japan [20]. All statistical analyses were performed using STATA 12.0 (StataCorp, College Station, TX). In all analyses, P < 0.05 was taken to indicate statistical significance.
This study was conducted in accordance with the ethical principles of the Declaration of Helsinki and with the ethical guidelines for epidemiological studies issued by the Ministry of Health, Labour and Welfare, Japan. The study was approved by the Ethics Review Committee of Graduate School of Medicine, Yokohama City University (institution of the principal investigator, SI, ID: B151201009) before commencement. Informed consent was not deemed necessary because the data were obtained retrospectively from the patient charts.
Results
Cohort description
Responses to the first questionnaire were obtained from 1077 of 1701 institutions (response rate, 63.3%), and 63 children with AAV were reported. Consent for the secondary survey was obtained for 49 children (13 males and 36 females), and they were included in the present study. Therefore, 49 children were included for detailed patient background, treatment, and prognostic surveillance studies, while prevalence rate was calculated from 49 (consent for the secondary survey) to 63 (all patients with responses to the first questionnaire) children. Therefore, the prevalence of reported cases was then estimated to be 3.41–4.28 (95% CI, 2.33–6.19) per million children. Tables 1 and 2 summarize the patient characteristics. Median age at onset was 10.7 years (interquartile range (IQR), 8.3–12.4); median time to diagnosis was 2.0 months (IQR, 1.0–4.0). The 49 patients included in the study consisted of 38 (78%) with MPA, nine (18%) with GPA, one (2%) with EGPA, and one with drug-induced AAV. Children with MPA were significantly younger than those with GPA (10.0 vs. 12.8 years, respectively, P < 0.01). MPA was detected by a school urinary screening program in 27 of the 38 patients with MPA (71%). On serological analysis, 37 patients (76%) were positive for MPO-ANCA, eight (16%) were positive for PR3-ANCA, and two (4%) were negative for ANCA. Data for the remaining two (4%) patients were not available.
Clinical features
Table 1 shows the differences between MPA and GPA presentation. GPA is frequently heralded by the presence of systemic symptoms, such as fever (78%) and weight loss (33%). Otolaryngological (78%), ocular (56%), and pulmonary (44%) involvements were also common in patients with GPA. In contrast, renal involvement was the most common manifestation in patients with MPA (82%) and these patients showed a more severe clinical course than those with GPA.
Histopathological findings
The results of renal biopsies were available for 40 children with AAV. The detailed histopathological findings are presented in Table 3. Among these 40 patients, 77%, 58%, 40%, 75%, 70%, and 53% showed cellular crescent, fibrocellular crescent, fibrous crescent, global sclerosis, interstitial inflammation, and interstitial fibrosis, respectively. In addition, a kidney biopsy showing chronic lesions (global sclerosis + fibrocellular crescent + fibrous crescent) involving > 50% of glomeruli was associated with a significantly higher rate of progression to ESRD (P < 0.01). With regard to tubulointerstitial lesions, 70% and 53% had interstitial inflammation and interstitial fibrosis, respectively.
Treatment
Almost all children received corticosteroid induction therapy, with 39/49 (80%) receiving intravenous methylprednisolone pulse therapy. Thirty-one (63%) patients received a combination of corticosteroid and cyclophosphamide (intravenously (iv) in 14 patients with median total dose of 3000 mg (IQR, 2500–4500 mg)). Other induction therapies included azathioprine (n = 18, 37%), mizoribine (n = 16, 33%), plasma exchange (n = 9, 19%), rituximab (n = 2, 6%), mycophenolate mofetil (n = 2, 6%), tacrolimus (n = 2, 6%), cyclosporin A (n = 2, 6%), and methotrexate (n = 2, 6%). Maintenance therapy consisted of oral corticosteroids alone or in combination with other immunosuppressive agents in seven (14%) and 42 (86%) patients, respectively. Three patients (6%) were treated with rituximab.
Outcome after induction therapy
Data regarding remission were available for 47 patients. Of these, 27 (55%) achieved remission after induction therapy. Thirteen patients (28%) had at least one relapse after a median follow-up period of 3.6 years. Median time to relapse was 48 months (IQR, 23–79). Although the relapse-free survival rate for MPA was higher than that for GPA (P < 0.01), renal prognosis was very poor in MPA (Figs. 1 and 2). When the patients relapsed, corticosteroid therapy alone or in combination with another immunosuppressant (mycophenolate mofetil (n = 3), azathioprine (n = 2), mizoribine (n = 2), methotrexate (n = 2), and tacrolimus (n = 1)) was prescribed. Eight of 13 patients (62%) received corticosteroid therapy for relapse, with 5/13 (38%) receiving intravenous methylprednisolone pulse therapy. Four patients received cyclophosphamide (iv in all four patients). Other therapies for relapse included rituximab (n = 7), azathioprine (n = 4), mycophenolate mofetil (n = 3), and mizoribine (n = 1). During follow-up, 11 (22%) patients had chronic kidney disease (CKD) stages 3–5. Although no patients died during the study period, seven of 38 (18%) patients with MPA progressed to ESRD. In contrast, none of the patients with GPA progressed to ESRD. Figure 3 shows the Kaplan–Meier curve for patients with ESRD. In univariate analysis, sex, age at onset, and diagnosis delay were not associated with risk of progression to ESRD. However, type of AAV, nephrotic-range proteinuria, and histological chronicity indices were factors associated with renal outcome. Histological chronicity was significantly correlated with nephrotic-range proteinuria (ρ = 0.50, P < 0.01) and was an independent factor associated with ESRD on logistic regression analysis (odds ratio, 24.20; 95% CI, 1.12–520.56; P = 0.042) (Table 4).

Disease course of childhood-onset AAV. AAV, anti-neutrophil cytoplasmic antibody–associated vasculitis; GPA, granulomatosis with polyangiitis; MPA, microscopic polyangiitis; CPM, cyclophosphamide; MPT, methylprednisolone pulse therapy; AZP, azathioprine; MMF, mycophenolate mofetil; MTX, methotrexate; RTX, rituximab; ESRD, end-stage renal disease

Kaplan–Meier curves for relapse-free survival in patients with MPA and GPA. Kaplan–Meier plots were used to assess time to first relapse. The log-rank test was used to compare relapse-free survival between different groups. GPA, granulomatosis with polyangiitis; MPA, microscopic polyangiitis

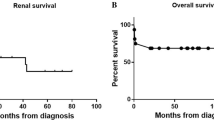
Kaplan–Meier curves for patients with ESRD (n = 49). Kaplan–Meier plots were used to assess time to ESRD. AAV, anti-neutrophil cytoplasmic antibody–associated vasculitis; ESRD, end-stage renal disease
Discussion
Few reliable epidemiological data are available regarding pediatric patients with AAV, particularly MPA, due to the rarity of these diseases in children. To date, four large pediatric studies in which outcomes were described showed relatively low rates with MPA patients ranging from 13 to 58% [1,2,3,4], because of regional and ethnic differences in AAV types [21]. Therefore, the present study was performed to investigate the outcomes of this group of diseases in pediatric patients to date.
The main findings of this nationwide survey in Japanese children were as follows. (i) More than 10% of patients still do not achieve remission, and progress to ESRD. (ii) The worst renal outcomes were observed in patients with chronic histological lesions involving > 50% of all glomeruli detected on renal biopsy. (iii) The prevalence of childhood-onset AAV was estimated to be 3.41–4.28 (95% CI: 2.33–6.19) per million children.
After a median follow-up of 43 months, of 49 patients, 11 (22%) progressed to CKD stages 3–5 and seven (14%) developed ESRD. In previous studies in pediatric AAV cases, the rates of ESRD were reported to be 10–50% [2, 6, 11, 22]. ESRD was also reported to occur at 29% in a study conducted in Japan in 1999 [23]. However, we could not compare renal outcomes with these previous studies as the populations were very different. Consistent with a previous report [23], no correlation was observed between detection at school urinary screening programs and favorable outcome on multivariate analysis. Of seven patients with ESRD, four (57%) were detected by school urinary screening programs. However, the time to diagnosis was shorter and prognosis was better in the present study than in previous studies in countries with a higher proportion of MPA and without annual urine screening programs [10, 11, 24]. In addition, the rates of renal insufficiency at diagnosis were high in previous reports, ranging from 29 to 80% [10, 11, 24], and were related to later ESRD. However, in the present study, renal insufficiency was detected in only five of 38 (13%) patients at diagnosis. Early diagnosis is important to improve long-term outcome in patients with AAV, particularly in those with MPA. Renal disease, which is the primary manifestation of MPA, is seen at onset in almost all pediatric patients diagnosed with this disease [25, 26]. Although our results did not indicate a significant effect of the annual urine screening program, such screening would allow earlier intervention in childhood-onset AAV before the development of renal insufficiency.
We found that not a few patients with childhood-onset AAV still do not achieve remission and progress to ESRD because there are no specific guidelines for therapeutic management of such cases [27]. Rituximab, which has recently been shown to be as effective as cyclophosphamide, high-dose steroids, and plasmapheresis [28] as an induction agent for treatment of childhood-onset AAV, was used to treat 20% of patients in our cohort, particularly in cases with recurrent disease. This high percentage of treatment with rituximab may have improved the overall prognosis in this study. Chronic T cell activation occurring in AAV has been suggested to promote maturation of autoreactive B cells, thereby leading to ANCA production. Maintenance therapy with mycophenolate mofetil after rituximab induction was reported to significantly improve maintenance of remission and to prevent relapse in pediatric MPA [29]. Therefore, treatment regimens based on rituximab and mycophenolate mofetil may be good alternatives to cyclophosphamide-based protocols for the treatment of childhood-onset AAV.
A number of factors, including older age, female sex, elevated serum creatinine level, and chronic histological lesions, have been reported to predict poor renal outcome in adult AAV patients [30,31,32,33,34]. Here, histopathological analysis of renal biopsy specimens at diagnosis was shown to predict the risk of progression to ESRD. That is, the rates of progression to ESRD during long-term follow-up were higher in pediatric patients with chronic histological lesions involving > 50% of all glomeruli (OR, 24.20 (95% CI: 1.12–520.56), P = 0.042). Histological classification of ANCA-associated glomerulonephritis was reported to predict prognosis regarding both renal function and patient outcome in adult patients [31, 35,36,37,38,39,40,41,42]. However, limited data are available on the association between histopathology and ESRD in childhood-onset AAV, with only two studies reported to date [2, 31]. The histological chronicity of ANCA-associated glomerulonephritis indicates dysfunction of the majority of glomeruli and heavy reliance on the compensatory ability of the kidneys. Sacri et al. reported that patients with sclerotic or mixed lesions had a threefold greater risk of ESRD than those with focal or crescentic patterns [2]. Noone et al. also reported progression to ESRD in all cases of childhood-onset AAV with sclerosis at diagnosis despite early aggressive therapy [31]. Such patients may not respond to drug therapy, and therefore early diagnosis and commencement of immunomodulatory therapy would be important to improve renal outcome.
The prevalence of childhood-onset AAV remains unclear due to the rarity of this condition in children. The results of the present study indicated a prevalence rate of childhood-onset AAV (before 16 years old) of 3.41–4.28 cases/million in Japan. Although this prevalence rate was very small compared to that in adults (46–184 cases/million) [43, 44], it may increase in the future because of increased awareness of the disease among pediatricians, and improved identification of cases due to health insurance coverage of the ANCA test.
This study had several limitations. First, this was a retrospective study using data collected over a long period, resulting in heterogeneity in the availability and accuracy of medical records. We also could not obtain information on the precise doses and durations of drugs used for therapy. In addition, all patients included in the study were Japanese. Therefore, our results may not be generalizable to non-Asian populations. Second, in this study, remission was defined as zero score in BVAS. Serum Cr and blood pressure matched to age and gender were used for BVAS in this study. However, some patients showed elevated serum Cr and proteinuria due to chronic renal scarring despite no disease activity. Such patients cannot achieve a zero score in BVAS. Therefore, the proportion of remission was lower than expected. Third, as 14 of 63 patients (22%) for whom consent was not provided were not included in the secondary survey, this may have led to overestimation of renal survival. Therefore, further studies are required to develop a prediction model and to determine the outcome of childhood-onset AAV. In conclusion, renal replacement therapy was not required in approximately 90% of the cohort at a median follow-up of 3.6 years. This observation indicated more than 10% of patients with childhood-onset did not achieve remission and progressed to ESRD. In addition, patients with chronic histological lesions involving > 50% of all glomeruli showed the best renal outcome.
References
Morishita KA, Moorthy LN, Lubieniecka JM, Twilt M, Yeung RS, Toth MB, Shenoi S, Ristic G, Nielsen SM, Luqmani RA, Li SC (2017) Early outcomes in children with antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheumatol 69:1470–1479
Sacri AS, Chambaraud T, Ranchin B, Florkin B, Sée H, Decramer S, Flodrops H, Ulinski T, Allain-Launay E, Boyer O, Dunand O (2015) Clinical characteristics and outcomes of childhood-onset ANCA-associated vasculitis: a French nationwide study. Nephrol Dial Transplant 30:i104–i112
Iudici M, Puéchal X, Pagnoux C, Quartier P, Agard C, Aouba A, Büchler M, Cevallos R, Cohen P, de Moreuil C, Guilpain P (2015) Brief report: childhood-onset systemic necrotizing vasculitides: long-term data from the french vasculitis study group registry. Arthritis Rheumatol 67:1959–1965
Cabral DA, Canter DL, Muscal E, Nanda K, Wahezi DM, Spalding SJ, Twilt M, Benseler SM, Campillo S, Charuvanij S, Dancey P, Eberhard BA, Elder ME, Hersh A, Higgins GC, Huber AM, Khubchandani R, Kim S, Klein-Gitelman M, Kostik MM, Lawson EF, Lee T, Lubieniecka JM, McCurdy D, Moorthy LN, Morishita KA, Nielsen SM, O'Neil KM, Reiff A, Ristic G, Robinson AB, Sarmiento A, Shenoi S, Toth MB, Van Mater HA, Wagner-Weiner L, Weiss JE, White AJ, Yeung RS, ARChiVe Investigators Network within the PedVas Initiative (2016) Comparing presenting clinical features in 48 children with microscopic polyangiitis to 183 children who have granulomatosis with polyangiitis (Wegener’s): an ARChiVe cohort study. Arthritis Rheumatol 68:2514–2526
Rottem M, Fauci AS, Hallahan CW, Kerr GS, Lebovics R, Leavitt RY, Hoffman GS (1993) Wegener granulomatosis in children and adolescents: clinical presentation and outcome. J Pediatr 122:26–31
Ellis EN, Wood EG, Berry P (1995) Spectrum of disease associated with anti-neutrophil cytoplasmic autoantibodies in pediatric patients. J Pediatr 126:40–43
Stegmayr BG, Gothefors L, Malmer B, Müller DE, Nilsson K, Sundelin B (2000) Wegener granulomatosis in children and young adults. A case study of ten patients. Pediatr Nephrol 14:208–213
Deshpande PV, Gilbert R, Alton H, Milford DV (2000) Microscopic polyarteritis with renal and cerebral involvement. Pediatr Nephrol 15:134–135
Belostotsky VM, Shah V, Dillon MJ (2002) Clinical features in 17 paediatric patients with Wegener granulomatosis. Pediatr Nephrol 17:754–761
Yu F, Huang JP, Zou WZ, Zhao MH (2006) The clinical features of anti-neutrophil cytoplasmic antibody-associated systemic vasculitis in Chinese children. Pediatr Nephrol 21:497–502
Peco-Antic A, Bonaci-Nikolic B, Basta-Jovanovic G, Kostic M, Markovic-Lipkovski J, Nikolic M, Spasojevic B (2006) Childhood microscopic polyangiitis associated with MPO-ANCA. Pediatr Nephrol 21:46–53
Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, Flores-Suarez LF, Gross WL, Guillevin L, Hagen EC, Hoffman GS (2013) 2012 revised International Chapel Hill Consensus Conference nomenclature of vasculitides. Arthritis Rheum 65:1–11
Ozen S, Ruperto N, Dillon MJ, Bagga A, Barron K, Davin JC, Kawasaki T, Lindsley C, Petty RE, Prieur AM, Ravelli A (2006) EULAR/PReS endorsed consensus criteria for the classification of childhood vasculitides. Ann Rheum Dis 65:936–941
Ozen S, Pistorio A, Iusan SM, Bakkaloglu A, Herlin T, Brik R, Buoncompagni A, Lazar C, Bilge I, Uziel Y, Rigante D (2010) EULAR/PRINTO/PRES criteria for Henoch- Schönlein purpura, childhood polyarteritis nodosa, childhood Wegener granulomatosis and childhood Takayasu arteritis: Ankara 2008. Part II: final classification criteria. Ann Rheum Dis 69:798–806
Schwartz GJ, Muñoz A, Schneider MF, Mak RH, Kaskel F, Warady BA, Furth SL (2009) New equations to estimate GFR in children with CKD. J Am Soc Nephrol 20:629–637
Mukhtyar C, Lee R, Brown D, Carruthers D, Dasgupta B, Dubey S, Flossmann O, Hall C, Hollywood J, Jayne D, Jones R (2009) Modification and validation of the Birmingham Vasculitis Activity Score (version 3). Ann Rheum Dis 68:1827–1832
Dolezalova P, Price-Kuehne FE, Özen S, Benseler SM, Cabral DA, Anton J, Brunner J, Cimaz R, O'Neil KM, Wallace CA, Wilkinson N, Eleftheriou D, Demirkaya E, Böhm M, Krol P, Luqmani RA, Brogan PA (2013) Disease activity assessment in childhood vasculitis: development and preliminary validation of the Paediatric Vasculitis Activity Score (PVAS). Ann Rheum Dis 72:1628–1633
Hashimoto S, Fukutomi K, Nagai M, Nakamura Y, Yanagawa H, Sasaki R, Ohno Y (1990) A note on methods for estimating the number of patients in the nationwide epidemiological survey on intractable diseases. Nihon Koshu Eisei Zasshi 37:768–774 (in Japanese)
Little RJ, Vartivarian S (2003) On weighting the rates in non-response weights. Stat Med 22:1589–1599
The Statistics Bureau of the Ministry of Internal Affairs and Communications of Japan http://www.stat.go.jp/english/index.html. Accessed 12 May 2018
Savage CO, Winearls CG, Evans DJ, Rees AJ, Lockwood CM (1985) Microscopic polyarteritis: presentation, pathology and prognosis. Q J Med 56:467–483
Siomou E, Tramma D, Bowen C, Milford DV (2012) ANCA-associated glomerulonephritis/systemic vasculitis in childhood: clinical features-outcome. Pediatr Nephrol 27:1911–1920
Hattori M, Kurayama H, Koitabashi Y (2001) Antineutrophil cytoplasmic autoantibody- associated glomerulonephritis in children. J Am Soc Nephrol 12:1493–1500
Sun L, Wang H, Jiang X, Mo Y, Yue Z, Huang L, Liu T (2014) Clinical and pathological features of microscopic polyangiitis in 20 children. J Rheumatol 41:1712–1719
Yamato K, Ishii T, Kawamura T (2012) Microscopic polyangiitis in a girl with severe anemia and no respiratory symptoms. Pediatr Int 54:541–543
Chen M, Yu F, Zhang Y, Zhao MH (2005) Clinical [corrected] and pathological characteristics of Chinese patients with antineutrophil cytoplasmic autoantibody associated systemic vasculitides: a study of 426 patients from a single centre. Postgrad Med J 81:723–727
Vanoni F, Bettinelli A, Keller F, Bianchetti MG, Simonetti GD (2010) Vasculitides associated with IgG antineutrophil cytoplasmic autoantibodies in childhood. Pediatr Nephrol 25:205–212
Geetha D, Specks U, Stone JH, Merkel PA, Seo P, Spiera R, Langford CA, Hoffman GS, Kallenberg CG, Clair EW, Fessler BJ (2015) Rituximab versus cyclophosphamide for ANCA-associated vasculitis with renal involvement. J Am Soc Nephrol 26:976–985
Basu B, Mahapatra TK, Mondal N (2015) Favourable renal survival in paediatric microscopic polyangiitis: efficacy of a novel treatment algorithm. Nephrol Dial Transplant 30:i113–i118
Sinico RA, Di Toma L, Radice A (2013) Renal involvement in anti-neutrophil cytoplasmic autoantibody associated vasculitis. Autoimmun Rev 4:477–482
Noone DG, Twilt M, Hayes WN, Thorner PS, Benseler S, Laxer RM, Parekh RS, Hebert D (2014) The new histopathologic classification of ANCA-associated GN and its association with renal outcomes in childhood. Clin J Am Soc Nephrol 7:1684–1691
Lee T, Gasim A, Derebail VK, Chung Y, McGregor JG, Lionaki S, Poulton CJ, Hogan SL, Jennette JC, Falk RJ, Nachman PH (2014) Predictors of treatment outcomes in ANCA-associated vasculitis with severe kidney failure. Clin J Am Soc Nephrol 7:905–913
Chen Y, Bao H, Liu Z, Liu X, Gao E, Zeng C, Zhang H, Liu Z, Hu W (2017) Risk factors for renal survival in Chinese patients with myeloperoxidase-ANCA-associated GN. Clin J Am Soc Nephrol 12:417–425
Vega LE, Espinoza LR (2016) Predictors of poor outcome in ANCA-associated vasculitis (AAV). Curr Rheumatol Rep 18:70
Berden AE, Ferrario F, Hagen EC, Jayne DR, Jennette JC, Joh K, Neumann I, Noël LH, Pusey CD, Waldherr R, Bruijn JA (2010) Histopathologic classification of ANCA- associated glomerulonephritis. J Am Soc Nephrol 21:1628–1636
Hilhorst M, Wilde B, van Breda Vriesman P, van Paassen P, Tervaert JW (2013) Estimating renal survival using the ANCA-associated GN classification. J Am Soc Nephrol 24:1371–1375
Chang DY, Wu LH, Liu G, Chen M, Kallenberg CG, Zhao MH (2012) Re-evaluation of the histopathologic classification of ANCA-associated glomerulonephritis: a study of 121 patients in a single center. Nephrol Dial Transplant 27:2343–2349
Tanna A, Guarino L, Tam FW, Rodriquez-Cubillo B, Levy JB, Cairns TD, Griffith M, Tarzi RM, Caplin B, Salama AD, Cook T (2015) Long-term outcome of anti-neutrophil cytoplasm antibody-associated glomerulonephritis: evaluation of the international histological classification and other prognostic factors. Nephrol Dial Transplant 30:1185–1192
Ford SL, Polkinghorne KR, Longano A, Dowling J, Dayan S, Kerr PG, Holdsworth SR, Kitching AR, Summers SA (2014) Histopathologic and clinical predictors of kidney outcomes in ANCA-associated vasculitis. Am J Kidney Dis 63:227–235
Iwakiri T, Fujimoto S, Kitagawa K, Furuichi K, Yamahana J, Matsuura Y, Yamashita A, Uezono S, Shimao Y, Hisanaga S, Tokura T (2013) Validation of a newly proposed histopathological classification in Japanese patients with anti-neutrophil cytoplasmic antibody-associated glomerulonephritis. BMC Nephrol 17:125
Togashi M, Komatsuda A, Nara M, Omokawa A, Okuyama S, Sawada K, Wakui H (2014) Validation of the 2010 histopathological classification of ANCA-associated glomerulonephritis in a Japanese single-center cohort. Mod Rheumatol 24:300–303
Muso E, Endo T, Itabashi M, Kakita H, Iwasaki Y, Tateishi Y, Komiya T, Ihara T, Yumura W, Sugiyama T, Joh K, Suzuki K (2013) Evaluation of the newly proposed simplified histological classification in Japanese cohorts of myeloperoxidase-anti- neutrophil cytoplasmic antibody-associated glomerulonephritis in comparison with other Asian and European cohorts. Clin Exp Nephrol 17:659–662
Ormerod AS, Cook MC (2008) Epidemiology of primary systemic vasculitis in the Australian Capital Territory and South-Eastern New South Wales. Intern Med J 38:816–823
Reinhold-Keller E, Zeidler A, Gutfleisch J, Peter HH, Raspe HH, Gross WL (2000) Giant cell arteritis is more prevalent in urban than in rural populations: results of an epidemiological study of primary systemic vasculitides in Germany. Rheumatology 39:1396–1402
Acknowledgments
The authors would like to thank all the institutions that participated in the surveys and Takayuki Okamoto (Hokkaido University Graduate School of Medicine), Kazushi Tsuruga (Hirosaki University), Naonori Kumagai (Tohoku University School of Medicine), Shigeo Suzuki (Ohara General Hospital), Kazuhide Suyama (Fukushima Medical University Hospital), Yasuo Oyake (Hitachi General Hospital), Yoko Ohwada (Dokkyo Medical University), Isamu Kamimaki (National Hospital Organization Saitama National Hospital), Koji Sakuraya (Saitama Children’s Medical Center), Yoshihiro Aoki (Asahi Hospital), Shosuke Sunami (Japanese Red Cross Narita Hospital), Shinsuke Matsumoto (Matsudo City General Hospital), Takashi Sato (Funabashi Futawa Hospital), Mamiko Suehiro (Chiba Children’s Hospital), Yuji Tomii (Tokyo Women’s Medical University Hospital), Tae Omori (Tokyo Metropolitan Bokutoh Hospital), Shojiro Okamoto (Tokai University Hachioji Hospital), Riku Hamada (Tokyo Metropolitan Children’s Medical Center), Takeshi Yanagihara (Nippon Medical School Musashi Kosugi Hospital), Hisashi Kaneda (Toyama City Hospital), Shoko Iwata (Ogaki Municipal Hospital), Masayoshi Yamada (Shizuoka Children’s Hospital), Naoya Fujita (Aichi Children’s Health and Medical Center), Nami Okamoto (Osaka Medical College Hospital), Kosuke Shabana (Osaka Medical College Hospital), Rika Fujimaru (Osaka City General Hospital), Takahisa Kimata (Kansai Medical University Hospital), Takeshi Ninchoji (Kobe University Graduate School of Medicine), Shingo Ishimori (Kakogawa City Hospital), Toshiyuki Ohta (Hiroshima Prefectural Hospital), Masafumi Teramachi (Fukuoka University Hospital), Kei Nishiyama (Kyushu University Hospital), Toshihiko Shirakawa (Nagasaki University Hospital), Hitoshi Nakazato (Kumamoto University Hospital), Akio Furuse (Japanese Red Cross Kumamoto Hospital), Hideaki Imamura (Faculty of Medicine, University of Miyazaki Hospital), and Tomoo Kise (Nanbu Medical Center/Nanbu Child Medical Center) for their contributions to the study. The authors would also like to thank Mr. Masanori Nimura of Yamate Information Processing Center Ltd. (Tokyo, Japan) for his participation in data management.
Funding
This work was supported by a Health and Labour Sciences Research Grant for Research in Rare and Intractable Diseases from the Ministry of Health, Labour, and Welfare, Japan (H26-nanchitou(nan)-ippan-036).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
This study was conducted in accordance with the ethical principles of the Declaration of Helsinki and with the ethical guidelines for epidemiological studies issued by the Ministry of Health, Labour and Welfare, Japan. The study was approved by the Ethics Review Committee of Graduate School of Medicine, Yokohama City University (institution of the principal investigator, SI, ID: B151201009) before commencement. Informed consent was not deemed necessary because the data were obtained retrospectively from the patient charts.
Conflict of interest
S.I. has received consulting fees from Novartis Pharma and Loche. S.I. has also received research grants from Asahi Kasei Pharma, Astellas Pharma, and Chugai Pharmaceutical. T.S. has received consulting fees from Takeda Pharmaceutical Company Limited. T.S. has also received lecture fees from Takeda Pharmaceutical Company Limited and Statcom Company Limited.
Additional information
The results presented in this paper have not been published previously in whole or part, except in abstract form.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Hirano, D., Ishikawa, T., Inaba, A. et al. Epidemiology and clinical features of childhood-onset anti-neutrophil cytoplasmic antibody–associated vasculitis: a clinicopathological analysis. Pediatr Nephrol 34, 1425–1433 (2019). https://doi.org/10.1007/s00467-019-04228-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-019-04228-4