
Abstract
Higher activity and alkaline α-amylases are desired for textile desizing and detergent additive. Here, rational design was used to improve the specific activity and thermostability of the α-amylase BLA from Bacillus licheniformis. Seventeen mutants of BLA were designed based on sequence consensus analysis and folding free energy calculation, and characterized by measuring their respective activity and thermostability at pH 8.5. Among them, mutant Q360C exhibited nearly threefold improved activity than that of wild-type and retained a higher residual activity (75% vs 59% for wild-type) after preincubation at 70 ℃ for 30 min. The modeled structures and molecular dynamics simulations analysis demonstrated that the enhanced hydrophobic interaction near residue 360 and reduced disturbance to the conformation of catalytic residues are the possible reasons for the improved thermostability and activity of Q360C. The results suggest that 360th of BLA may act as a hotspot for engineering other enzymes in the GH13 superfamily.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
Amylases are one of the most important enzymes and are used broadly in biotechnology, such as textile, paper, food production, pharmaceutical industry, detergent industries, and so on. α-Amylases BLA from Bacillus licheniformis are widely used in various procedures of starch degradation in the food industry due to their ability to catalyze starch hydrolysis at high temperatures [1]. Over the past decades, there have been many researches aimed at improving the enzymatic activity and thermostability of α-amylase BLA, such as the mutants H133V [2], A209V [3], Q264S/N265Y [4], and N190F [5], which presented better thermostability. In 2003, a hyperthermostable variant that combined these five mutation sites was obtained and its half-life was improved 32 times [6]. By modifying residue near the active site, the specific activity of a mutant V286Y was fivefold than that of the wild-type [7]. In 2007, the calcium-binding site was mutated and the specific activity of the mutant N104D was improved 30% [8]. In addition to BLA, there are other α-amylases that have been modified in various ways to obtain better catalytic activity or thermal stability, which can be used as a reference program for retrofitting BLA. Multiple favorable mutations of ROAmy based on site-saturation mutagenesis on H286 were improved in the enzymatic properties and even changed the catalytic mechanism [9]. The thermal stability or activity under acid condition of ROAmy was significantly improvement by direct evolution [10]. Using computer-aided direct evolution of enzyme (CADEE), α-amylase from B. cereus GL96 was redesigned and had 20% improvement in thermal stability [11].
For industrial enzymes, thermostability and catalytic activity are the crucial factors. Therefore, enhancing the activity or thermostability of enzymes (such as α-amylases) is highly desirable to achieve better catalytic performance in the industrial processes [12]. Protein engineering strategies include rational design and random directed evolution [13]. Undoubtedly, rational design is more efficient and less labor-cost and has been successfully used to improve the stability and/or catalytic efficiency of enzymes such as α-amylase [14] and pullulanase [15]. For instance, three different algorithm predictors (FoldX, I-Mutant 3.0, and dDFIRE) were combined to improve the thermostability of pullulanase BtPul, resulting in a mutant with 2.1-fold longer half-life at 70 ℃ [15]. The thermostability of Rhizopus chinensis lipase r27RCL was improved using rational design and generated a quadruple variant, which exhibited enhanced thermostability with a 41.7-fold longer half-life at 60 ℃ compared to that of wild-type [16].
In our previous work, an α-amylase (BLA) from Bacillus licheniformis WX-02 which presented 99.6% sequence identity with widely used BLA-1BLI was over-expressed in Escherichia coli and Pichia pastoris. The recombinant BLA showed high activity and stability at 70 ℃ and pH 8.5 [17]. The optimal pH of BLA is 6.0–7.5, it retains more than 80% relative activity at pH 8–9, which presents broad pH adaption and permitted it to be used in textile industry. Alkaline amylase can be used for textile desizing, detergent additive and paper industry to reduce pulp viscosity. Together with alkaline cellulase, alkaline amylase can be used for environmental treatment. To further improve the activity and thermostability of BLA at pH 8.5, a strategy combining sequence consensus analysis and folding free energy calculation was used to design BLA mutants in this study. The 17 single BLA mutants were designed and experimentally characterized. The molecular basis of the improved activity and thermostability of the best mutant Q360C was analyzed by molecular dynamics (MD) simulations. The protein engineering strategies used in this study may be useful for engineering other microbial enzymes that have industrial applications.
Materials and methods
Primers and reagents
Primers were purchased from Sangon Biotech (Shanghai, China) in PAGE-purified grade. Soluble starch was purchased from Sigma-Aldrich (St. Louis, USA). Restriction enzymes and PrimeSTAR Max DNA polymerase were purchased from TakaRa (Dalian, China). All chemicals were of analytical grade and obtained from commercial suppliers.
Rational design of BLA variants
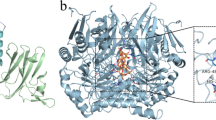
The BLA single variants were designed based on the sequence consensus analysis, folding free energy calculations, and some structural considerations according to the method in Liu et al. with minor modifications [18]. First, the amino acid sequence of BLA was used to identify the homologous sequences from non-redundant protein sequences (nr) database (https://blast.ncbi.nlm.nih.gov/Blast.cgi). As a result, 5000 protein sequences sharing a minimum of 46% sequence identity were retrieved, and the redundant sequences were removed using CD-HIT server [19]. Subsequently, the 1164 resulting sequences were aligned using MAFFT software [20], and three most frequently occurring amino acids at each position of BLA were analyzed by WebLogo 3.0 (http://weblogo.threeplusone.com/create.cgi). Secondly, the homology model of BLA (Fig. 1) was constructed based on the crystal structure of α-amylase from Bacillus licheniformis (PDB: 1BLI [21], identity 99%) using YASARA Structure v20.10.4 [22], and subjected to FoldX website (http://foldxsuite.crg.eu/) for in silico site-saturation mutagenesis of BLA. Subsequently, the stabilizing substitutions (folding free energy change ΔΔG < − 1.0 kcal/mol) were predicted.

Overall structure of wild-type BLA. The model structure of BLA was constructed through the homology modeling of YASARA. The catalytic residues D231, E261, and D328 are shown as sphere in yellow. Ca2+ are indicated as green ball, and Na+ are indicated as purple ball (colour figure online)
Site-directed mutagenesis of BLA
The expression plasmid with wild-type BLA gene pET-28a-bla was constructed in previous work [17], which was used as the template for site-directed mutagenesis. The PCR was performed under the followed conditions: initial denaturation at 98 ℃ for 5 min, followed by 20 consecutive cycles of denaturation at 98 ℃ for 10 s, anneal at 54 ℃ for 5 s, extension at 72 ℃ for 45 s, and a final extension at 72 ℃ for 10 min. The PCR products were treated by DpnI at 37 ℃ for 1 h to digest the parental methylated template DNA, then were transferred to 80 ℃ for 10 min to inactivate the DpnI. The DpnI-digested DNA was then transformed into E. coli BL21 (DE3) cells, and transformants were obtained on LB agar plate supplemented with 50 μg/mL kanamycin. Mutation was verified by sequencing (Sangon, Shanghai, China). The primers used for gene cloning and mutagenesis were listed in Table 1.
Expression and purification of wild-type and mutant enzymes
The single colonies were grown in LB medium containing 50 μg/mL kanamycin at 37 ℃ and induced by the addition of 0.1 mM IPTG when the OD600 reached 0.6–0.8, and the culture was incubated at 18 ℃ for 16–18 h. Cells were harvested by centrifugation (4000 rpm for 10 min). The cell pellet was suspended in buffer I (50 mM Tris–HCl, pH 8.5), and disrupted by sonication in an ice-water bath, and then centrifuged at 12,000 rpm for 10 min. The supernatant was applied on a Ni-Sepharose column (His60 Ni Superflow resin from Takara Biomedical Technology Co. Ltd. Dalian, China). Then the column was washed with buffer II (50 mM Tris–HCl, pH 8.5, 30 mM imidazole) and buffer III (50 mM Tris–HCl, pH 8.5, 60 mM imidazole) sequentially. At last, target protein was obtained with elution buffer (50 mM Tris–HCl, pH 8.5, 300 mM imidazole) and desalted using the buffer 50 mM Tris–HCl, pH 8.5. All the above purification steps were operated on the ice. Purified BLA and variants were analyzed on SDS-PAGE and used for enzymatic assay. The concentration of protein was determined with the Bradford Protein Assay Kit (Beyotime, China).
Amylase’s activity assays
Amylase activity was determined by a modified DNS (3,5-dinitrosalicylic acid) method [23]. The mixture including 50 μL enzyme with appropriate dilution and 500 μL 1% starch solution was incubated at 70 ℃ for 10 min, followed by adding 700 μL DNS at 100 ℃ for 10 min immediately. One unit (U) of amylase was defined as the amount of enzyme required for catalyzing starch to release 1 μmol reducing sugar (glucose) per minute under the assay conditions. The thermostability was assessed by preincubation of the enzyme at 70 ℃ for 30 min. Residual amylase activity was determined at regular time intervals of 10 min as described above.
The optimal temperatures were determined in 50 mM Tris–HCl, pH 8.5, over the temperature range from 50 to 90 ℃. The optimal pH of enzymes was determined by assays in a wide pH range at 70 ℃, using 50 mM of four different buffers (citrate buffer pH 4.0–6.0, Na2PO4 KH2PO4 buffer pH 6.0–7.0, Tris–HCl 7.0–9.0 and Gly-NaOH pH 9.0–9.5). The pH stability was tested by diluting the purified enzyme with the different pH and incubating at 4 ℃ for 12 h. The residual activity was measured as described above. The amylase activity under optimal conditions was taken as 100% in pH stability assays. All measurements were performed in triplicate.
Homology modeling and molecular dynamics simulations
The MD simulations of BLA and its variant Q360C in explicit water were performed using YASARA Structure v20.10.4. The protein residues were treated using AMBER14 force field. The cutoff was 8 Å for Van der Waals forces, and long-range electrostatic contacts were measured by Particle-Mesh-Ewald (PME) method [24]. A cubic shape was selected for the simulation cell extending 8 Å around all atoms of the solute. Then, the enzyme was neutralized and solvated in the cell containing TIP3P water and 0.29% mass fraction of NaCl (50 mM) at pH 8.5. A two-stage geometry optimization was performed first for the water and ions, then the entire system. In each stage, the system was minimized first with steepest descent and then simulated annealing (time step of 2 fs, atom velocities scaled down by 0.9 every 10th step) starting from 98, 198, 298 and 343 K with a time averaged Berendsen thermostat until convergence was reached. The systems were heated from 0 to 343 K under NPT conditions for 10 ps. Afterwards, all MD simulations were performed using a preinstalled macro file (md_runfast.mcr). Each system was equilibrated for 10 ns, and then two independent 200 ns MD simulations (i.e., 400 ns accumulated) were performed under the NVT ensemble and 343 K.
Results and discussion
Rational design of BLA variants
In our previous work [17], the α-amylase gene bla was cloned from B. licheniformis WX-02 and expressed heterologously. The purified BLA exhibited a specific activity of 767 U/mg at pH 8.5 and 70 ℃ but lost most activity after incubation at 70 ℃ for 50 min at pH 8.5, suggesting its unsatisfied thermostability and room for activity enhancement. To enhance the activity and thermostability of BLA, the sequence consensus analysis (Fig. S1 in the supplemental material) and folding free energy calculations have been performed to design the mutation sites rationally. The sequence consensus analysis is a well-established strategy to improve the enzymatic properties using the evolutionary information encapsulated in homologous protein sequences [25, 26]. In addition, for a protein whose three-dimensional structure is available, the stabilizing mutations can be predicted through folding free energy calculations [27]. Finally, in total 17 single BLA variants were designed by taking sequence consensus, FoldX results and some structural aspects into consideration (Table 2). The mutated positions and amino acid substitutions were selected based on the following criteria: (a) the positions containing more than three stabilizing substitutions (ΔΔG < − 1 kcal/mol) predicted by FoldX were preferred; (b) the positions where there are other amino acids appearing more frequently than wild-type by consensus analysis were preferred; (c) amino acid secondary structure was considered (e.g., residues located in loops or closest to the ends of secondary structures were preferred); (d) the substitutions had to be within three most prevalent amino acids at the specific position and predicted to be stabilizing; (e) proline substitutions were preferred particularly when the mutated positions located in loop regions.
Site-directed mutagenesis of BLA
All of the mutants were constructed by site-directed mutagenesis and expressed in E. coli BL21 (DE3) as soluble protein. The enzyme activities of mutants were determined at 70 ℃ and pH 8.5 using the DNS method [23]. As shown in Fig. 2a, the catalytic activity was significantly improved for Q360C, which was 2288 U/mg against starch and about threefold higher than of the wild-type. Moreover, slightly higher catalytic activities (1–1.5-fold) were observed for other 10 single variants (H68D, G81E, D161S, W184E, T258V, V312L, T341P, T353L, A398P and A420P). However, the remaining six mutants (T139V, H140R, A426V, D430N, T453L and N473P) showed slightly decrease in catalytic activity (71, 82, 63, 96, 66, and 82%) compared with that of wild-type.

Relative activity (a) and thermostability (b) of 17 mutants and wild-type. Soluble starch (1%) was used as substrate for amylases activity assay. The wild-type activity under 70 ℃ and pH 8.5 was taken as 100%. The thermostability was determined by preincubation enzyme at 70 ℃ for 30 min and residual activity were tested at pH 8.5 and 70 ℃
The thermostability of BLA and variants were determined after preincubated at 70 ℃ for 30 min. As shown in Fig. 2b, in contrast to only 59% of residual activity of wild-type, two of 17 mutants (T139V and T258V) exhibited significantly higher residual activities (95% and 96%), and six mutants (H68D, G81E, H140R, Q360C, D430N and T453L) showed slight to moderate increase in residual activity (71, 64, 68, 78, 65 and 63%). Nevertheless, the remaining nine mutants showed varying degrees of decrease in residual activity, with seven mutants (V312L, T341P, T353L, A398P, A420P, A426V and N473P) showing minor decrease (55, 44, 47, 35, 48, 50 and 46%) and two mutants (D161S and W184E) showing drastically decrease (1.4% and 12%).
In the previous studies [3,4,5], α-amylase BLA was usually engineered using the strategy of random mutations, which were time consuming and costly, and often only a few favorable mutations were obtained out of thousands of mutants, with a low success rate. Besides, the mutants with improved thermal-stability often had no change in hydrolytic activity, and some of them even decreased. Recently, using multiple sequence alignment and PoPMuSiC algorithm had obtained a mutant which had improvement in half-life and catalytic efficiency under acidic conditions [11]. Therefore, the rational design was used in this study, resulting in 11 mutants with improved catalytic activity and eight mutants with improved thermal stability among all 17 mutants. Importantly, four mutants with both improved stability and activity were obtained, which demonstrated the feasibility and efficiency of rational design and could provide reference for other enzymes of GH13 family engineering to simultaneously improve the specific activity and thermostability.
Combinational mutation for further improvement
The beneficial amino acid mutations may have an additive effect on the thermostability and activity of an enzyme [28]. Meanwhile, it has also been reported that combing two mutations of structurally distant residues may result in superposition effect [29]. By analyzing the location of beneficial mutations on the BLA structure, two double mutants H68D/G81E and T258V/Q360C were designed and constructed. As shown in Fig. 3, the relative activity of H68D/G81E was 1.2-fold higher than that of wild-type, which was higher than G81E but lower than H68D. And the residual activity of H68D/G81E increased to 66%, which was only 7% improvement over the wild-type. For the double mutant T258V/Q360C, its residual activity was 67% and the activity was 1.8-fold higher than wild-type, which was significantly lower than the threefold of single mutant Q360C. In short, the mutants H68D/G81E and T258V/Q360C didn’t have an overwhelming advantage compared to the single mutant Q360C in terms of activity and thermostability. The results in our work are not uncommon. Several previous studies have shown that beneficial mutations would be antagonistic for some proteins [30].

Relative activity (a) and thermostability (b) of combination mutants and wild-type. Soluble starch (1%) was used as substrate for amylases activity assay. The wild-type activity under 70 ℃ and pH 8.5 was taken as 100%. The thermostability was determined by preincubation enzyme at 70 ℃ for 30 min and residual activity were tested at pH 8.5 and 70 ℃
Temperature and pH dependency of the mutant Q360C
The effect of temperature on Q360C activity was measured at various temperatures ranging from 40 to 90 ℃, under standard assay conditions. Results showed that the optimal temperature of Q360C was 80 ℃, same to that of the wild-type (Fig. 4a) [17]. The pH dependence of the mutant Q360C activity was investigated in various buffers at pH values ranging from 4 to 9.5, under standard assay conditions. Results showed that the optimal pH of Q360C was 6.5, and kept nearly 80% activity at pH 8.5, similar to that of the BLA [17] (Fig. 4b). Therefore, the substitution of the 360th residue had no effect on either optimal pH or temperature. Additionally, when Q360C was incubated in 70 ℃ for 30 min, the residual activity of Q360C dropped to 75% (Fig. 4c), which indicated that the Q360C has stronger thermostability. And, when pH is over 9, the relative activity of Q360C was still retain above 50%, showing the Q360C has good resistance to strong alkali conditions (Fig. 4d).

Effects of temperature and pH on activity (a, b) and stability (c, d) of Q360C. Soluble starch (1%) was used as substrate for activity assay. The highest activity was taken as 100%. The thermostability was tested by preincubation the enzyme at 70 ℃ for 60 min. The pH stability was tested by diluting the purified enzyme with the different pH buffer and incubating at 4 ℃ for 12 h. Then the residual activity was measured at pH 8.5 and 70 ℃
The enzyme activity of the purified Q360C was 2288 U/mg against starch, whereas that of BLA was 767 U/mg at 70 ℃, pH 8.5 against starch. The specific activity of Q360C was about threefold higher than that of the wide type, which indicated that the specific activity of Q360C had been improved significantly. Also, its residual activity reduced to 75% after preincubation at 70 ℃ for 30 min when the BLA reduced to 59%, which indicated the higher thermostability of Q360C at pH 8.5. Some BLA mutants with high heat resistance were reported while their optimal pH were weakly acidic [6], which is not suitable for the textile industry, where the amylase needs to be highly active at pH values above 8.0. Many amylases can function in alkaline conditions, but the activity is poor. For instance, the specific activity of α-amylase (N-Amy) was only 170 U/mg at pH 8.5 [31], and the specific activity of α-amylase (TfAmy48) was 1500 U/mg using soluble starch as substrate at 80 ℃, pH 8 [32]. Therefore, the higher specific activity and thermostability of Q360C in alkaline conditions make it more advantage to be used in the textile industry.
Structural interpretation of the improved thermostability
In recent years, homology modeling has become an important auxiliary tool in structural biology, which is significantly beneficial to narrowing the distance between known protein sequences and experimentally determined structures. In our work, to reveal the possible molecular basis behind the enhanced activity and thermostability of Q360C, the structure of Q360C and wild-type were modeled and analyzed. The overall structures of BLA and Q360C were almost identical, and they had a typical TIM barrel consisting of eight α-helices and eight parallel β strands (Fig. 1).
As shown in Fig. 5a, the Q360 was located in the middle of the TIM barrel, which was distant from the active sites (> 5.5 Å). Since the majority residues near Q360 (within 4 Å) were hydrophobic (e.g., L7, F284, V321, F323, I351, V361, and F362), the highly polar residue Q360 was unfavorable for the structural stability of BLA. Thus, when Q360 was mutated to Cys, the hydrophobic interaction near position 360 was significantly enhanced, which may explain for the increased thermostability of Q360C. The enhanced hydrophobic interaction that may contribute to the enzymatic thermostability has been widely reported. For instance, Dessy et al. reported that enhancing the hydrophobic interaction between two domains could significantly improve the residual activity of Sfamy01, which made it suitable for raw-starch degradation in the rice and cassava fermentation [33]. Silverman et al. suggested that the buried polar residues may have a crucial role in determining (α/β)8 barrel structures [34]. The thermostability and hydrolytic pattern of α-amylase Amy7C were simultaneously improved by rational engineering of the mostly conserved central beta strands in TIM barrel fold [35]. Therefore, engineering the central eight beta sheets of TIM barrel may provide a valuable strategy for improving stability and activity of GH13 family. Apart from Q360C, the mutant T139V and T258V also exhibited improved thermostability even better than Q360C. We assumed that an interior polar residue replaced by a more hydrophobic residue contributed to increase the stability. T139 was surrounded by many hydrophobic amino acids, including half of aromatic amino acids, such as A109, A111, V115, A137, F141, W155, W157, F160, F177 and A199 (Fig. S2a). Interestingly, the sequence consensus analysis showed 86% occurrence for T and only 8% for V at the residue 139th, which perhaps explained the improved thermostability but the decreased catalytic activity of T139V. Similarly, T258 located at the interior of the TIM barrel was also surrounded by hydrophobic amino acids, including F228, L230, W244, V245, M256, and A260 (Fig. S2b). The sequence consensus analysis indicated the 258th residue always be hydrophobic.

Structural interpretation of the improved thermostability of Q360C. a Residues around position 360 in BLA. The green stick is Q360, light-blue sticks are residues around Q360, and the yellow sticks represented the active sites. b The sequence consensus analysis of α‐amylases. The amino acid sequence of BLA and 1163 homologous sequences sharing a minimum of 46% sequence identity were aligned using MAFFT software, and the occurring residues at each position were analyzed by WebLogo 3.0. The occurring probability of several key residues are given in main text (colour figure online)
Interestingly, our sequence consensus analysis showed that a high percentage of Cys (82%) appeared at position 360, whereas < 1% of Gln was occurred, suggesting the importance and benefit of Cys at the position 360 for the structure and function of α‐amylases (Fig. 5b). Additionally, three catalytic residues (D231, E261 and D328), as well as some key amino acids (Q9, R229, N326 and H327) near the 360th residue were highly conserved and occurred more than 99% in the consensus analysis result (Fig. 5b). Noteworthy, since BLA wild-type contains no Cys residues, it is unable to form a disulfide bond for variant Q360C. From an evolutionary perspective, the probability of Cys occurring at position 360 in homologous sequences with BLA is much higher than that of Gln, suggesting that Cys may let enzyme easier adapt to the complex and various external environment.
Structural interpretation of the improved enzymatic activity
In an attempt to reveal how the 360th residue affect the activity of BLA at the molecular level, we next performed MD simulation of BLA wild-type or variant Q360C in explicit water. The interactions (e.g., hydrogen bonds, salt-bridge, hydrophobic interaction) near the 360th residue were analyzed, and the results showed that Q360 may interact with catalytic residues (D231, E261 and D328) by the hydrogen bond network. As shown in Fig. 6a, for BLA wild-type, the side-chain carbonyl group of Q360 was highly polar and participated in several hydrogen bond interactions, such as with Q9, which in turn interacted with one of the active sites, E261. In another representative structure (Fig. 6b), Q360 interacted with Q9 via a water-mediated hydrogen bond network, and then affected the conformations of N326 and active site D328. Besides, it was also observed that Q360 could form additional hydrogen bonds with surrounding water or protein residues (Fig. 6a, b). Thus, we speculated that the polar Q360 may affect the conformation of the catalytic residues and restrict their flexibility via hydrogen bond network, and then alter the enzyme activity.

Hydrogen bond networks surrounding the 360th residue in representative structures of BLA wild-type (a, b) and variant Q360C (c, d). W1, W2 and W3 are water molecules. The black dashed lines indicate hydrogen bonds (colour figure online)
In contrast to BLA wild-type, the side-chain of C360 for variant Q360C often had no polar contacts with surrounding residues (Fig. 6c), or only formed a weak hydrogen bond with water molecule (Fig. 6d). Thus, the hydrogen bond network involving the side-chain of Q360 in wild-type disappeared, resulting in no interference for the conformation of catalytic residues. On the other hand, in the resolved crystal structure of the α-amylase (PDB ID: 1BLI) [21], no interactions were formed between the side-chain of Q9 and other residues, suggesting the reduced contacts of Q9 with the catalytic residues may make the conformation of active sites more flexible to act with the large substrate, thus improving the enzymatic activity. A number of studies had demonstrated that eliminating or weakening the hydrogen bond network near the active sites could improve the catalytic efficiency. For example, the loss of a hydrogen bond between D285 near the proton donor (E261) with S320 led to the improved catalytic activity of mutant S320A of BLA [36]. The catalytic activity of the isoamylase had been improved significantly when G608 near the catalytic residue E609 was replaced by a more hydrophobic residue V, which eliminated the hydrogen bonds network with G608 [37]. The protein engineering of pullulanase showed that decreased catalytic activity of mutant S622R was owing to R622 forming two new hydrogen bonds with Q664 and T667, leading to weak interaction between catalytic loop 5 with substrate [38].
Conclusions
In this study, the activity and thermostability of α‐amylase BLA in alkaline conditions were simultaneously enhanced through a sequence and structure-guided rational design approach. By combining sequence consensus analysis and free energy calculations, 17 single BLA mutations were designed. Subsequently, their relative activities and residual activities after incubation at 70 ℃ were measured, and a mutant Q360C improved nearly threefold in the specific activity at pH 8.5 than wild-type and retained 75% residual activity after preincubation at 70 ℃ for 30 min. MD simulations showed that when polar Q360 was mutated to the highly conserved Cys, the hydrophobic interaction near position 360 was significantly enhanced, contributing to the enzyme stability. Additionally, the polar Q360 can affect the conformation of the catalytic residues and restrict their flexibility via hydrogen bond network, and may be non-favorable for the enzyme activity. In conclusion, the present study provides a deeper understanding of TIM barrel on the function and stability of BLA and could help design other enzymes particularly with the similar structure.
Abbreviations
- PCR:
-
Polymerase chain reaction
- IPTG:
-
Isopropyl-β-d-thiogalactopyranoside
- OD:
-
Optical density
- SDS-PAGE:
-
Sodium dodecyl sulfate polyacrylamide gel electrophoresis
References
Lee S, Oneda H, Minoda M et al (2006) Comparison of starch hydrolysis activity and thermal stability of two Bacillus licheniformis α-amylases and insights into engineering α-amylase variants active under acidic conditions. J Biochem 139:997–1005. https://doi.org/10.1093/jb/mvj113
Declerck N, Joyet P, Gaillardin C, Masson JM (1990) Use of amber suppressors to investigate the thermostability of Bacillus licheniformis alpha-amylase. Amino acid replacements at 6 histidine residues reveal a critical position at His-133. J Biol Chem 265:15481–15488. https://doi.org/10.1016/S0021-9258(18)55421-1
Declerck N, Joyet P, Trosset JY et al (1995) Hyperthermostable mutants of Bacillus licheniformis alpha-amylase: multiple amino acid replacements and molecular modelling. Protein Eng 8:1029–1037. https://doi.org/10.1093/protein/8.10.1029
Declerck N, Machius M, Joyet P et al (2003) Hyperthermostabilization of Bacillus licheniformis α-amylase and modulation of its stability over a 50 °C temperature range. Protein Eng Des Sel 16:287–293. https://doi.org/10.1093/proeng/gzg032
Declerck N, Machius M, Wiegand G et al (2000) Probing structural determinants specifying high thermostability in Bacillus licheniformis α-amylase. J Mol Biol 301:1041–1057. https://doi.org/10.1006/jmbi.2000.4025
Machius M, Declerck N, Huber R, Wiegand G (2003) Kinetic stabilization of Bacillus licheniformis α-amylase through introduction of hydrophobic residues at the surface. J Biol Chem 278:11546–11553. https://doi.org/10.1074/jbc.M212618200
Rivera MH, Lopez-Munguia A, Soberon X, Saab-Rincon G (2003) α-Amylase from Bacillus licheniformis mutants near to the catalytic site: effects on hydrolytic and transglycosylation activity. Protein Eng Des Sel 16:505–514. https://doi.org/10.1093/protein/gzg060
Priyadharshini R, Gunasekaran P (2007) Site-directed mutagenesis of the calcium-binding site of α-amylase of Bacillus licheniformis. Biotechnol Lett 29:1493–1499. https://doi.org/10.1007/s10529-007-9428-0
Li S, Yang Q, Tang B, Chen A (2018) Improvement of enzymatic properties of Rhizopus oryzae α-amylase by site-saturation mutagenesis of histidine 286. Enzyme Microb Technol 117:96–102. https://doi.org/10.1016/j.enzmictec.2018.06.012
Li S, Yang Q, Tang B (2020) Improving the thermostability and acid resistance of Rhizopus oryzae α-amylase by using multiple sequence alignment based site-directed mutagenesis. Biotechnol Appl Biochem 67:677–684. https://doi.org/10.1002/bab.1907
Pouyan S, Lagzian M, Sangtarash MH (2022) Enhancing thermostabilization of a newly discovered α-amylase from Bacillus cereus GL96 by combining computer-aided directed evolution and site-directed mutagenesis. Int J Biol Macromol 197:12–22. https://doi.org/10.1016/j.ijbiomac.2021.12.057
Guo J, Wang Y, Zhang X et al (2021) Improvement of the catalytic activity of chitosanase BsCsn46A from Bacillus subtilis by site-saturation mutagenesis of proline121. J Agric Food Chem 69:11835–11846. https://doi.org/10.1021/acs.jafc.1c04206
Terashima M, Katoh S (1996) Modification of alpha-amylase functions by protein engineering. Ann NY Acad Sci 799:65–69. https://doi.org/10.1111/j.1749-6632.1996.tb33179.x
Lim SJ, Oslan SN (2021) Native to designed: microbial α-amylases for industrial applications. PeerJ 9:e11315. https://doi.org/10.7717/peerj.11315
Bi J, Chen S, Zhao X et al (2020) Computation-aided engineering of starch-debranching pullulanase from Bacillus thermoleovorans for enhanced thermostability. Appl Microbiol Biotechnol 104:7551–7562. https://doi.org/10.1007/s00253-020-10764-z
Wang R, Wang S, Xu Y, Yu X (2020) Enhancing the thermostability of Rhizopus chinensis lipase by rational design and MD simulations. Int J Biol Macromol 160:1189–1200. https://doi.org/10.1016/j.ijbiomac.2020.05.243
Hu X, Yuan X, He N et al (2019) Expression of Bacillus licheniformis α-amylase in Pichia pastoris without antibiotics-resistant gene and effects of glycosylation on the enzymic thermostability. 3 Biotech 9:427. https://doi.org/10.1007/s13205-019-1943-x
Liu C, Zhao J, Liu J et al (2019) Simultaneously improving the activity and thermostability of a new proline 4-hydroxylase by loop grafting and site-directed mutagenesis. Appl Microbiol Biotechnol 103:265–277. https://doi.org/10.1007/s00253-018-9410-x
Huang Y, Niu B, Gao Y et al (2010) CD-HIT suite: a web server for clustering and comparing biological sequences. Bioinformatics 26:680–682. https://doi.org/10.1093/bioinformatics/btq003
Katoh K, Standley DM (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780. https://doi.org/10.1093/molbev/mst010
Machius M, Wiegand G, Huber R (1995) Crystal structure of calcium-depleted Bacillus licheniformis α-amylase at 2.2 Å resolution. J Mol Biol 246:545–559. https://doi.org/10.1006/jmbi.1994.0106
Krieger E, Vriend G (2014) YASARA view—molecular graphics for all devices—from smartphones to workstations. Bioinformatics 30:2981–2982. https://doi.org/10.1093/bioinformatics/btu426
Xiao Z, Storms R, Tsang A (2006) A quantitative starch–iodine method for measuring alpha-amylase and glucoamylase activities. Anal Biochem 351:146–148. https://doi.org/10.1016/j.ab.2006.01.036
Essmann U, Perera L, Berkowitz ML et al (1995) A smooth particle mesh Ewald method. J Chem Phys 103:8577–8593. https://doi.org/10.1063/1.470117
Sternke M, Tripp KW, Barrick D (2019) Consensus sequence design as a general strategy to create hyperstable, biologically active proteins. Proc Natl Acad Sci USA 116:11275–11284. https://doi.org/10.1073/pnas.1816707116
Bai X, Li D, Ma F et al (2020) Improved thermostability of creatinase from Alcaligenes Faecalis through non-biased phylogenetic consensus-guided mutagenesis. Microb Cell Fact 19:194. https://doi.org/10.1186/s12934-020-01451-9
Huang J, Xie D-F, Feng Y (2017) Engineering thermostable (R)-selective amine transaminase from Aspergillus terreus through in silico design employing B-factor and folding free energy calculations. Biochem Biophys Res Commun 483:397–402. https://doi.org/10.1016/j.bbrc.2016.12.131
Zhao H, Arnold FH (1999) Directed evolution converts subtilisin E into a functional equivalent of thermitase. Protein Eng Des Sel 12:47–53. https://doi.org/10.1093/protein/12.1.47
Wen S, Tan T, Zhao H (2013) Improving the thermostability of lipase Lip2 from Yarrowia lipolytica. J Biotechnol 164:248–253. https://doi.org/10.1016/j.jbiotec.2012.08.023
Deng Z, Yang H, Shin H et al (2014) Structure-based rational design and introduction of arginines on the surface of an alkaline α-amylase from Alkalimonas amylolytica for improved thermostability. Appl Microbiol Biotechnol 98:8937–8945. https://doi.org/10.1007/s00253-014-5790-8
Lu Z, Wang Q, Jiang S et al (2016) Truncation of the unique N-terminal domain improved the thermos-stability and specific activity of alkaline α-amylase Amy703. Sci Rep 6:22465. https://doi.org/10.1038/srep22465
Allala F, Bouacem K, Boucherba N et al (2019) Purification, biochemical, and molecular characterization of a novel extracellular thermostable and alkaline α-amylase from Tepidimonas fonticaldi strain HB23. Int J Biol Macromol 132:558–574. https://doi.org/10.1016/j.ijbiomac.2019.03.201
Natalia D, Vidilaseris K, Ismaya WT et al (2015) Effect of introducing a disulphide bond between the A and C domains on the activity and stability of Saccharomycopsis fibuligera R64 α-amylase. J Biotechnol 195:8–14. https://doi.org/10.1016/j.jbiotec.2014.12.002
Silverman JA, Balakrishnan R, Harbury PB (2001) Reverse engineering the (α/β)8 barrel fold. Proc Natl Acad Sci USA 98:3092–3097
Wang C-H, Lu L-H, Huang C et al (2020) Simultaneously improved thermostability and hydrolytic pattern of alpha-amylase by engineering central beta strands of TIM barrel. Appl Biochem Biotechnol 192:57–70. https://doi.org/10.1007/s12010-020-03308-8
Liu Y, Lu F, Li Y et al (2008) Acid stabilization of Bacillus licheniformis alpha amylase through introduction of mutations. Appl Microbiol Biotechnol 80:795–803. https://doi.org/10.1007/s00253-008-1580-5
Li Y, Zhang L, Ding Z et al (2016) Engineering of isoamylase: improvement of protein stability and catalytic efficiency through semi-rational design. J Ind Microbiol Biotechnol 43:3–12. https://doi.org/10.1007/s10295-015-1708-4
Chen A, Li Y, Nie J et al (2015) Protein engineering of Bacillus acidopullulyticus pullulanase for enhanced thermostability using in silico data driven rational design methods. Enzyme Microb Technol 78:74–83. https://doi.org/10.1016/j.enzmictec.2015.06.013
Acknowledgements
This work was supported by National Key R and D Program of China (NO.2018YFA0901100), National Natural Science Foundation of China (31970059) and Fundamental Research Funds for the Central Universities (buctrc202131).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Cui, X., Yuan, X., Li, S. et al. Simultaneously improving the specific activity and thermostability of α-amylase BLA by rational design. Bioprocess Biosyst Eng 45, 1839–1848 (2022). https://doi.org/10.1007/s00449-022-02790-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00449-022-02790-0