
Abstract
This study reported simultaneously improved thermostability and hydrolytic pattern of α-amylase from Bacillus subtilis CN7 by rationally engineering the mostly conserved central beta strands in TIM barrel fold. Nine single point mutations and a double mutation were introduced at the 2nd site of the β7 strand and 3rd site of the β5 strand to rationalize the weak interactions in the beta strands of the TIM barrel of α-amylase. All the five active mutants changed the compositions and percentages of maltooligosaccharides in final hydrolytic products compared to the product spectrum of the wild-type. A mutant Y204V produced only maltose, maltotriose, and maltopentaose without any glucose and maltotetraose, indicating a conversion from typical endo-amylase to novel maltooligosaccharide-producing amylase. A mutant V260I enhanced the thermal stability by 7.1 °C. To our best knowledge, this is the first report on the simultaneous improvement of thermostability and hydrolytic pattern of α-amylase by engineering central beta strands of TIM barrel and the novel “beta strands” strategy proposed here may be useful for the protein engineering of other TIM barrel proteins.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The TIM barrel (also called (β/α)8 barrel) is the most frequently occurring folding motif in proteins [1, 2] and is adopted by versatile enzymes in all the E. C. enzyme classes except ligase [3, 4]. The canonical TIM barrel fold consists of eight α-helices and eight parallel β strands that alternate along the peptide backbone [5]. The catalytic and substrate-binding residues locate at the “catalytic face,” which includes the C-terminal ends of β strands and the loops that extend from these strands. While the stabilizing residues are of the “stability face,” that includes the hydrophobic core, N-terminal ends of the barrel, and the αβ-loops linking the α-helices with the subsequent β strands [5]. The “division of labor” between catalytic activity and stability and the “diversity in function but similar structure” properties have made the TIM barrel an ideal object for protein engineering [4, 6].
Although various factors contribute to the formation and stability of the TIM barrel fold [7, 8], such as packing of the β strand residues in the barrel core [7, 9], amino acid clustering patterns [10], long-range interactions [11], the weak interactions [12], and protein energy networks [13], the most essential ones are attributed to the eight-stranded β sheet [6, 14]. The structurally conserved central eight β strands in TIM barrel fold are deemed to be untouchable, and are seldom taken into consideration for protein engineering of TIM barrel proteins. However, in the tightly packed hydrophobic core of TIM barrel proteins, polar residues can naturally occur by forming specific tertiary interactions [15]. For example, Silverman et al. found that the network of buried polar residues plays a crucial role in determining (β/α)8 barrel structures [16], Chothia et al. showed that polar residue in the hydrophobic interior of a protein hydrogen bond to other polar functional groups [17]. So, it seems worthy trying to protein engineer the central eight beta sheets of TIM barrel for improved specific tertiary interactions.
Alpha-amylase (E C 3.2.1.1) is a typical TIM barrel enzyme and has been extensively engineered for improved thermostability and activity due to both industrial and academic importance [18]. A number of studies have shown that the structures of α-amylases consist of domain A composing of the TIM barrel, domain B protruding between the third β strand and the third α-helix of domain A, and domain C, a C-terminal β sheet [19, 20]. Based on the “division of labor” of TIM barrel fold, various protein engineering works have been done mainly by rational design and directed evolution [21,22,23,24]. However, all the obtained beneficial mutations contributing to thermostability, pH adaptation, catalytic activity, and hydrolytic pattern are mainly located at the “stability face” and “activity face” of TIM or belong to domain B, few of them belong to the central eight beta strands of TIM. In our previous study, we exceptionally found several thermostable mutations at the beta sheets of TIM barrel of the α-amylase from Bacillus subtilis CN7 (Amy7C) using a newly developed coevolving strategy [25]. For example, both D95HT147S and G89FD95R mutants involve changes to the residue D95 of the third β strand (β3) in the TIM barrel of the domain A [25]. In this study, using the same α-amylase Amy7C as a model, we successfully and simultaneously improved the thermostability and hydrolytic pattern by rationally designing the central beta strands in TIM barrel.
Materials and Methods
Protein Modeling and Superimposition
The computational structures of Amy7C and its mutants were constructed by homology modeling via the SWISS-MODEL server (http://swissmodel.expasy.org/) [26], as previously described [25]. Experimental crystal structures of eight representative α-amylases, including 1AQH of Pseudoalteromonas haloplanktis, 3L2L of Pig pancreatic, 2TAA of Aspergillus oryzae, 1BAG of Bacillus subtilis, 3DC0 of Bacillus subtilis KR-8104, 3BH4 of Bacillus amyloliquefaciens, 1BLI of Bacillus licheniformis, and 1MWO of Pyrococcus woesei, which dramatically differed in thermostabilities and sources, were retrieved from the Protein Data Bank (PDB, http://www.rcsb.org/) [27]. Properties, sources, and PDB entries of the retrieved α-amylases were listed in supplementary Table S1. The superimposition between Amy7C and other eight α-amylases structures was done with the Swiss-PdbViewer software [28].
Identification of Weak Interactions
The canonical hydrogen bonds in the eight strands of the TIM barrel were identified using the Pymol software (The PyMOL Molecular Graphics System, Version 1.2r3pre, Schrödinger, LLC., Cambridge, USA). The other weak interactions termed non-canonical interactions (NCI), such as C-H…O, C-H…π, O-H…π, and N-H…π interactions, were analyzed by the program Hydrogen Bond Analysis Tool (HBAT, edition 1.1, Hyderabad, Telangana State, India) [29]. All the NCI analyses of TIM barrel were executed using the default parameter values of the program. The cation–π interactions were calculated using the online web-based program CAPTURE (available at http://capture.caltech.edu) [30]. For the hydrophobic-hydrophilic binary pattern analysis of beta strands, the C, F, I, L, M, V, W, and Y were considered to be hydrophobic amino acids according to Sweet and Eisenberg [31]. The Van Der Waals (VDW) contacts were identified using a software package-Molecular Operating Environment (MOE) (Chemical Computing Group Inc., Montreal, Quebec, Canada). The salt bridge was considered to exist only between the anionic carboxylate (RCOO−) of either aspartic acid or glutamic acid and cationic ammonium (RNH3+) from lysine or the guanidinium (RH(CH2)2+) of arginine.
Rational Design of Beta Strands in TIM Barrel
The beta strands in TIM barrel of Amy7C were rationally designed from two apparently contradictory directions, i.e., strengthening and eliminating the weak interactions between central β strands in three steps. Step 1, the mutation sites were chosen based on three considerations: (1) most conserved and deeply buried in the interior core of the β sheet; (2) the side chains participating in specific weak interactions; (3) convenient to engineer specific tertiary structures for design objectives. Step 2, the hydrogen bonding between the β strands was optimized by substituting the wild-type amino acid residues at the chosen sites by residues with comparable chemical and physical properties but totally different hydrogen bonding ability, and so did the salt bridge and VDW contacts between β strands. Step 3, the hydrophobic-hydrophilic binary pattern of beta strand was designed by changing the hydrophobic amino acid residues with hydrophilic ones or with more hydrophobic ones. The involved amino acids in this study and their properties were listed in supplementary Table S2.
Gene Clone and Site-Directed Mutagenesis
The gene coding for the α-amylase (Amy7C) from B. subtilis CN7 was cloned into the expression vector pSE380 as described previously [25]. The recombinant plasmid was then used as template to introduce the site-directed mutations by one-step PCR protocol, using the mutagenic primers listed in supplementary Table S3 according to the procedures previously described [24, 25]. The PCR conditions were as follows: 95 °C for 3 min; 30 cycles of 98 °C for 15 s, 68 °C for 5 min 40 s; 72 °C for 10 min. Each reaction system contained 10 ng of plasmid DNA, 1 × PrimeSTAR buffer, 0.2 mM of dNTPs, 0.2 μM of mutagenic primers, and 1 U PrimeSTAR DNA polymerase (Takara Bio Company, Dalian, China) in a total volume of 25 μL. The PCR products were digested with 10 U of Dpn I (Fermentas Company, Massachusetts, USA) enzyme to digest template DNA, and then the Dpn I was inactivated at 80 °C for 30 min. The digested PCR mixture was transformed directly into competent E. coli XL1-Blue cells (Stratagene company of Agilent Technologies Inc., Shanghai, China) following the standard transformation procedure [32]. The transformants were selected on the basis of their ampicillin resistance and the ability to hydrolyze starch, and further sequenced to confirm the mutations (Sangon Company, Shanghai, China).
Enzyme Expression and Purification
All the recombinant enzymes were produced by culturing the above-mentioned E. coli XL1-Blue cells containing corresponding recombinant plasmids in 1 L LB medium (Amresco Inc., Solon, Ohio, USA) supplemented with 50 μg/mL of ampicillin. The cultures were grown at 37 °C and 200 rpm for 8 h to mid-log phase (OD600 = 0.6), and then the proteins were expressed by induction with 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG, Amresco Inc., Solon, Ohio, USA) for further 18 h under the same conditions. After which, the cells were harvested by centrifugation at 5000 rpm for 10 min. The obtained pellets were resuspended in 50 mM Na2HPO4-citrate acid buffer (pH 6.5) at 2–5 mL per wet weight. The suspension was sonicated on ice using a sonicator equipped with a microtip using 40 cycles of 8 s bursts at 200–300 W with an 8-s cooling period between each burst. The cell debris was removed by centrifugation at 12,000 rpm for 30 min at 4 °C. The his6-tagged proteins were purified by Ni-NTA chromatography (Ni-NTA, Qiagen, Hilden, Germany) and Sephacyl S300 chromatography according to the manufacturers’ instructions. Then, the solution buffer was exchanged to 50 mM Na2HPO4-citrate acid buffer (pH 6.5) by a standard Amicon® Ultra centrifugal filter with cutoff of 10,000 Da (Merck KGaA, Darmstadt, Germany). The homogeneity of the purified proteins was analyzed with SDS-PAGE. Protein concentrations of purified enzymes were determined by the method of Bradford using BSA as standard [33].
Activity Assays
The enzymatic activity of α-amylase was determined by dinitrosalicylic acid (DNS, Sinopharm Chemical Reagent Co., Ltd, Beijing, China) method, in which reducing sugars in the starch hydrolyzates were assayed and represented in terms of glucose [34]. The assay solution was comprised of 0.5% soluble starch (S9765, Sigma, USA), and appropriate dilutions of the purified α-amylase (up to 2 μg/mL) in 50 mM Na2HPO4-citrate acid buffer (pH 6.5). One unit (U) of α-amylase was defined as the amount of enzyme required to of releasing one micromolar reducing sugar equivalent per minute under the assay conditions. All the values were averaged from at least three parallels. The kinetic parameters were assayed at pH 6.5 and 65 °C and determined using the Lineweaver-Burk plot method [35].
Thermostability Assays
The thermostability of α-amylase was characterized by two parameters: the optimum temperature and the half-inactivation temperature (\( {T}_{50}^{30} \)) as described previously [25, 36]. The \( {T}_{50}^{30} \) value denotes the temperature at which α-amylase lost 50% activity after incubation for 30 min and was determined by measuring residual activity at pH 6.5 and 65 °C after incubation at various temperatures (50–80 °C) for 30 min.
Hydrolytic Pattern Assays
The hydrolysis of soluble starch of α-amylase was performed at 50 °C for 24 h in 50 mM Na2HPO4-citrate acid buffer (pH 6.5) containing 1% potato soluble starch and 1 U/mL α-amylase. The reaction was terminated by heat for 10 min in boiling water and the inactivated enzymes were removed by centrifugation at 12,000 rpm for 10 min. The supernatant was filtered with 0.22 μm pore size membrane; the permeate solution was analyzed on HPLC in an Aminex HPX-87H column (Bio-Rad Laboratories, Hercules, CA, USA). The detection conditions were RI temperature of 50 °C, mobile phase of 5 mM H2SO4, and velocity of 0.5 mL/min. The standard high purity sugars—glucose (G1), maltose (G2), maltotriose (G3), maltotetraose (G4), and maltopentaose (G5) were purchased from Sigma, Chemical Co. (St Louis, Mo., USA).
Results
In this study, we identified the weak interactions preserving the structural integrity of the central eight beta strands in the TIM barrel of Amy7C, rationally designed ten mutations at the 3rd amino acid site of β5 (Y204) and the 2nd amino acid site of β7 (V260) to reform the local interaction network, and identified five mutants with improved thermostability and altered hydrolytic pattern.
Weak Interactions in the Central Beta Strands of Amy7C
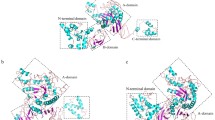
Although the eight representative α-amylases and Amy7C come from different bacteria, archaea, and animal domains and show different thermostabilities ranging from 25 to 95 °C in terms of optimum temperature (see supplementary Table S1 for details), their TIM barrels superimposed very well, as illustrated by the average values of 2.78 Å of the overall RMSD value of Cα atoms of all TIM barrels and 0.13 Å of the eight beta strands (Fig. 1a, b, see supplementary Table S4 for pair-wise RMSD values of all the superimpositions). In spite of different lengths and compositions, all the β strands showed a similar “hydrophobic-hydrophilic” binary pattern, in which the hydrophobic and hydrophilic amino acid residues arranged alternatively. The β3, β4, β5, and β7 strands were the four best structurally conserved regions, which pertain closely to the active sites of the α-amylase family (Fig. 1c) [37]. So, the Amy7C should be a good representative of the whole α-amylase family.

Structural alignments of the central eight beta strands in TIM barrel of nine representative α-amylases. a A schematic drawing of the superimposed central eight strands in TIM barrel from a top view, in which the C-terminal side of the β strands is toward the reader. The 1AQH, 2TAA, 1BAG, 3DC0, 3BH4, 1BLI, 3L2L, 1MWO, and the computational structure of Amy7C are shown in purple, pink, red, brown, green, cyan, dark blue, blue, and yellow, respectively. The three catalytic triad residues are shown in ball and sticks. b A bottom view in which the N-terminal side of the β strands is toward the reader. c The structure-based sequence alignments of β3, β4, β5, and β7 strands in the TIM barrel of nine α-amylases, which correspond to the four most conserved regions of the α-amylase family. The hydrophobic and hydrophilic amino acid residues of beta strands are colored in yellow and blue, respectively. The three catalytic residues (red) are framed and marked by asterisks. The mutational sites in this study are shown by triangles
The eight beta strands of Amy7C are β1 of 9-ILH-11, β2 of 34-AIQT-38, β3 of 91-KVIVDA-97, β4 of 170-GFRF-174, β5 of 202-FQYG-206, β6 of 223-NVT-226, β7 of 259-LVT-261, and β8 of 298-TPLFF-303. These 32 amino acids together with 3 interaction were identified (Table 1). Among the 122 weak interactions, 103 interactions were mediated by main-chain atoms, while only 10 NCI and 2 canonical hydrogen bonds were of side chains atoms. There were totally 28 canonical hydrogen bonds, 5 of which were participated by the side chains of the β strand amino acids (supplementary Table S5). On average, each β strand was connected laterally by about 3 backbone hydrogen bonds with the nearest neighboring β stands, except that the β3 formed 5 hydrogen bonds, β5 6 hydrogen bonds, and β7 only one hydrogen bond. All the 28 canonical H-bonds were formed between neighboring β strands except one H-bond between the Tyr 204 OH of β5 and Gln36 NE2 of β2, which was formed by two spatially opposite β strands and spread the hydrophobic TIM barrel core.
Rational Design of Central Beta Strands in TIM Barrel
Based on the above structural analysis, the β5 and β7 strands were chosen to design nine single point mutations and one double mutation. For the β5 strand (202-FQYG-206), five mutations were designed to substitute the hydrophobic Y204 at the 2nd site. The substitution by a Phe was envisaged to eliminate the hydrogen bonding formed by the side chain. The substitution by a Val was envisaged to weaken the hydrophobicity of the β5 strand, and so did the substitution of an Ile. The substitution by an Arg was expected to strengthen the electrostatic interactions and form more H-bonds. The double substitutions of Y204 by an arginine and Q36 by a glutamate were designed to introduce a new salt bridge into the hydrophobic core, which will replace the original H-bond between the side chains of Y204 and Q36. Meanwhile, the binary pattern of β5 strand was changed from “HPHP” to “HPPP” form by the Y204R and Y204RQ36R mutants.
Five single mutants were designed on the V260 site (the 2nd site) of β7 strand (259-LVT-261) of Amy7C. The substitution by an Ile was expected to strengthen the weak interactions by introducing more VDW contacts. Two hydrophilic substitutions by Ser and Thr were devised to introduce extra NCI and hydrogen bonds, and to change the binary pattern of β7 (LVT) from “NNP” to “NPP.” Meanwhile, two counterpart amino acid residues at the 2nd site of β7 of other α-amylases, Gly and Leu (Fig. 1c), were also introduced at the V260 site of Amy7C. Both the substitutions by Gly and Leu were supposed to increase the hydrogen bonds but decrease the VDW contacts (Tables 1 and S5).
Improvement of Thermostability by Rational Design
All the rationally designed α-amylase variants were created by site-directed mutagenesis, and then the wild-type and mutant α-amylases were over-expressed, purified, and characterized. Compared to the wild-type, five out of the ten variants, Y204F, Y204I, Y204V, V260I, and V260L retained decreased but considerable α-amylase activity (Table 2). The turnover number (kcat) values of Y204F, Y204I, Y204V, V260I, and V260L reduced by 63%, 40%, 81%, 52%, and 38% to 466.40 s−1, 756.33 s−1, 239.50 s−1, 605.06 s−1, and 781.54 s−1, respectively. The substrate affinity for soluble starch (Km) values of Y204F, Y204I, and Y204V showed a moderate increase by 8%, 25%, and 5% to 3.57 g/L, 4.14 g/L, and 3.48 g/L, while V260I and V260L decreased by 18% and 12% to 2.71 g/L and 2.91 g/L, respectively. As a result, the catalytic efficiency values of Y204F, Y204I, Y204V, V260I, and V260L were decreased by 66%, 52%, 82%, 41%, and 30% to 130.47 L.(g.s)−1, 182.80 L.(g.s)−1, 68.91 L.(g.s)−1, 222.93 L.(g.s)−1, and 268.31 L.(g.s)−1, respectively. Generally speaking, all the three active variants at Y204 site, Y204F, Y204I, and Y204V reduced the affinity for the substrate of soluble starch, the other two active variants at V260 site, V260I and V260L, enhanced the affinity (Table 2). The substitutions at V260 site showed higher catalytic efficiency than that at Y204 site, although the Y204I showed very comparable catalytic number to the variants at the V260 site.
As shown in Table 2, all the five active mutants showed the same optimum temperature of 63.2 °C as that of the wild-type, except the V260I that performed the maximum activity at 70.3 °C. However, the half-inactivation temperature (\( {T}_{50}^{30} \)) values of Y204I, Y204V, and V260I increased by 1.6 °C, 1 °C, and 4.8 °C to 63.2 °C, 62.4 °C, and 67.2 °C, that of Y204F and V260L decreased by 2.5 °C and 6.3 °C, respectively. To a certain extent, the thermostability of α-amylase enhances as the weak interactions between the central β strands increase. For example, the V260I variant obtained extra 8 VDW contacts and increased the thermostability by 4.9 °C, the Y204F variant eliminated 2 H-bonds and 2 VDW contacts and decreased the thermostability by 3.5 °C in comparison to the wild-type (Fig. 2 and Table 1). But, it is hampered to further decompose the specific contributions of each weak interaction. For example, the Y204V variant eliminated 10 NCI, 2 H-bonds, and 1 cation-π interaction, but still kept nearly the same thermostability as the wild-type.

The calculated weak interactions between central β strands in the TIM barrels of the wild-type and mutant α-amylases. All the amino acid residues consisting of the β strands in the (β/α)8-barrel are shown in line representations, except that the amino acid residues at 204 site of β5 and 260 site of β3, the catalytic triad residues of aspartates (D174 and D267) and glutamate (E206), and the residues participating in VDW contacts (red dashed lines) are shown in ball and stick from. The canonical hydrogen bonds are shown as black suspension points, except for the hydrogen bonds formed by side chains of amino acid residues at 36 and 204 sites in green. a Hydrogen bonding network in the wild-type. b VDW contacts in the wild-type. c Hydrogen bonding network in Y204F. d VDW contacts in Y204F. e hydrogen bonding network in V260I. f VDW contacts in V260I
In contrast, the other five variants, V260S, V260T, V260G, Y204R, and Y204RQ36E completely lost the catalytic activity. The substitutions of hydrophobic V260 by hydrophilic Ser and Thr, which changed the binary pattern of β7 (LVT) from “NNP” to “NPP,” completely inactivated the α-amylase. What was surprising, the substitution by a naturally occurring hydrophilic amino acid residue Gly, which resulted in the same β7 strand as in 2TAA (Fig. 1c), also inactivated the α-amylase. The substitution of Y204 by an Arg changed the “HPHP” pattern of wild-type β5 strand to “HPPP” pattern, which also resulted in an inactivated variant. The double mutant Y204RQ36E showed no activity at all and did not seem to adopt the expected orientation to introduce a salt bridge into the hydrophobic core.
Alteration of Hydrolytic Pattern by Rational Design
All the five active mutants altered the hydrolytic pattern on the soluble starch, and changed the class and composition of oligosaccharides in the final hydrolytic product in comparison to the wild-type (Table 3, Fig. S1). The two major components of the final hydrolytic products of all the wild-type and mutants except Y204V were G1 and G2. Although the percentages of the oligosaccharides were different, all the five active variants except Y204V produced more G1 than G2, whereas the wild-type Amy7C yielded more G2. Y204V produced only G2, G3, and G5 without any G1 and G4 in the final hydrolytic products, indicating a conversion from typical endo-amylase to novel maltooligosaccharides-producing amylase, which is especially appealing for the production of maltooligosaccharides from starch and shows great promise in various industrial applications like the bread industry [38].
Discussion
The “Binary Pattern” and Weak Interactions of Central β Strands
All the substitutions of hydrophobic amino acid residues at β5 and β7 strands by hydrophilic ones (V260S, V260T, V260G, Y204R, and Y204RQ36E) totally inactivated the α-amylases, but the replacements by other hydrophobic residues (Y204F, Y204I, Y204V, V260I, and V260L) retained the activity and stability, even the V260I markedly enhanced the thermostability (Table 2). The results are supported by previous theoretical and experimental studies, which showed that the hydrophobic pattern is one of the most conserved features of specific fold proteins [39] and specifies secondary and tertiary structure [40, 41]. Our results also suggest the indispensability of hydrophobic-hydrophilic binary pattern of beta sheets to the structural integrity of TIM barrel for stability and activity, while the binary pattern can be further optimized by protein engineering the component amino acid residues as shown by V260I.
It is well established that the weak interactions, such as H-bond, NCI and VDW contacts, are the fundamental forces to maintain TIM barrel proteins, and the contributions of which have also been extensively investigated [11, 12]. However, to the best of our knowledge, no attention has been paid on the rational design of the weak interactions between the central β strands in the TIM barrel. Here, we provided the first example of rationally reconstructing the weak interaction network between the central β strands of α-amylase. As expected, the thermostability of α-amylase enhances as the weak interactions between the central β strands increase as shown by the wild-type, V260I, Y204F, and Y204I but Y204V (Fig. 2 and Table 1). The exception of Y204V might be attributed to subtle and intricate structural reorientation to compensate the negative influence by the elimination of weak interactions, which matches well with the alteration of hydrolytic pattern.
Central β Strands and “Division of Labor” Within TIM Barrel
The mutations at the central β strands in TIM barrel achieved a clear simultaneous alteration of hydrolytic pattern and improvement of thermostability (Table 3, Fig. S1). Such simultaneous changes of stability and activity can probably be attributed to structural reorientation of active site residues brought by the rational engineering of the central β strands, considering that the catalytic amino acid residue D267 and the transition state stabilizing residue E206 locate nearby the C-termini of β7 and β5 strands (Figs. 1 and 2). This speculation is further supported by the fact that all the five active mutations are naturally occurring residues in central beta strands of TIM barrel of α-amylases (Fig. 1). The results bring an amendment to two widely accepted views on TIM barrel fold: one is the “division of labor” between the “activity” and “stability” and the independence between “catalytic face” and “stability face” within the TIM barrel [3, 6], the other is that the central β strands in the hydrophobic core of the TIM barrel stabilize the whole protein structure and are untouchable [16, 42].
Comparison Between “Beta Strands” Strategy and Traditional Strategies
By protein engineering the “stability face,” the thermostability of α-amylase from B. licheniformis (BLA) has been greatly enhanced to the extent that it can be used for almost any high-temperature application [43]. By introducing up to seven mutations, BLA increased the half-inactivation temperature by over 20 °C. However, no single mutation or even double mutations increases by over 5 °C in terms of \( {T}_{50}^{30} \) value, the increase of 4.9 °C in this study by single point mutation (V260I) at β7 strand of TIM barrel is of the top-class level in the protein engineering of α-amylase [25]. In considering the simultaneous alteration of hydrolytic pattern, the “beta strands” strategy here seems even more appealing in engineering the α-amylase.
In contrast to the “beta strands” strategy here and traditional “division of labor” ones focusing on the highly conserved TIM barrel (domain A), there also exist some strategies aiming at the highly variable domain B to improve the stability and activity of α-amylases [24, 25, 44,45,46]. For example, the thermostability of α-amylases of Bacillus amyloliquefaciens (BAA), Bacillus KSM-1378 (LAMY), and Bacillus stearothermophilus (AmyS) were significantly increased by deleting an Arg-Gly peptide belonging to the domain B, and the deletion also decreased the requirement for calcium to maintain activity of AmyS [44,45,46]. So, it should be promising to combine the “beta strands” strategy here and traditional ones for a synergistic effect in the future protein engineering of α-amylases.
An interesting question is why have not much attentions been paid on the seemly simple but effective “beta strands” strategy so far? One possible reason may be that the central eight β strands are buried deeply in the hydrophobic core, and usually are safe from the evolution pressure. As shown in the Amy7C, the Tyr204 and Val260 are 100% buried, and the relative mutability values of Tyr204 and Val260 are 41% and 74%, respectively (Table S2). We artificially introduced rational mutations at the Tyr204 and Val260 of Amy7C by site-directed mutagenesis here and firstly proved the effectiveness of “beta strands” strategy here. By applying the time-tested enzyme engineering methods of directed evolution and rational design, the novel “beta strands” strategy should play a greater role in improving the catalytic activity and stability of α-amylase and other TIM enzymes and proteins.
In conclusion, we have demonstrated for the first time that the thermostability and hydrolytic pattern of B. subtilis α-amylase can be simultaneously improved by rational engineering of the central β strands in TIM barrel. One substitution (V260I) at the β7 strand increased the thermal stability by 7.1 °C in comparison to the wild-type α-amylase. All the five active mutants introduced substitutions at either β7 or β5 strands altered the hydrolytic pattern. The new “beta strands” strategy to improve stability and activity by engineering the central beta strands in the TIM barrel proposed here may benefit other TIM barrel proteins.
References
Farber, G. K., & Petsko, G. A. (1990). The evolution of [alpha]/[beta] barrel enzymes. Trends in Biochemical Sciences, 15(6), 228–234.
Brändén, R. I. (1991). The TIM barrel—The most frequently occurring folding motif in proteis. Current Opinion in Structural Biology, 1, 978–983.
Petsko, G. A. (2000). Enzyme evolution. Design by necessity. Nature, 403(6770), 606–607.
Wierenga, R. K. (2001). The TIM-barrel fold: A versatile framework for efficient enzymes. FEBS Letters, 492(3), 193–198.
Hocker, B., Jurgens, C., Wilmanns, M., & Sterner, R. (2001). Stability, catalytic versatility and evolution of the (beta alpha)(8)-barrel fold. Current Opinion in Biotechnology, 12(4), 376–381.
Sterner, R., & Hocker, B. (2005). Catalytic versatility, stability, and evolution of the (betaalpha)8-barrel enzyme fold. Chemical Reviews, 105(11), 4038–4055.
Jaenicke, R., & Bohm, G. (1998). The stability of proteins in extreme environments. Current Opinion in Structural Biology, 8(6), 738–748.
Vieille, C., & Zeikus, G. J. (2001). Hyperthermophilic enzymes: Sources, uses, and molecular mechanisms for thermostability. Microbiology and Molecular Biology Reviews, 65(1), 1–43.
Lesk, A. M., Branden, C. I., & Chothia, C. (1989). Structural principles of alpha/beta barrel proteins: The packing of the interior of the sheet. Proteins, 5(2), 139–148.
Selvaraj, S., & Gromiha, M. M. (1998). An analysis of the amino acid clustering pattern in (alpha/beta)8 barrel proteins. Journal of Protein Chemistry, 17(5), 407–415.
Selvaraj, S., & Gromiha, M. M. (1998). Importance of long-range interactions in (alpha/beta)8 barrel fold. Journal of Protein Chemistry, 17(7), 691–697.
Chakkaravarthi, S., Babu, M. M., Gromiha, M. M., Jayaraman, G., & Sethumadhavan, R. (2006). Exploring the environmental preference of weak interactions in (alpha/beta)8 barrel proteins. Proteins, 65(1), 75–86.
Vijayabaskar, M. S., & Vishveshwara, S. (2012). Insights into the fold organization of TIM barrel from interaction energy based structure networks. PLoS Computational Biology, 8(5), e1002505.
Gromiha, M. M., Pujadas, G., Magyar, C., Selvaraj, S., & Simon, I. (2004). Locating the stabilizing residues in (alpha/beta)8 barrel proteins based on hydrophobicity, long-range interactions, and sequence conservation. Proteins, 55(2), 316–329.
Ponder, J. W., & Richards, F. M. (1987). Tertiary templates for proteins. Use of packing criteria in the enumeration of allowed sequences for different structural classes. Journal of Molecular Biology, 193(4), 775–791.
Silverman, J. A., Balakrishnan, R., & Harbury, P. B. (2001). Reverse engineering the (beta/alpha)8 barrel fold. Proceedings of the National Academy of Sciences of the United States of America, 98(6), 3092–3097.
Chothia, C. (1976). The nature of the accessible and buried surfaces in proteins. Journal of Molecular Biology, 105(1), 1–12.
MacGregor, E. A. (1988). Alpha-amylase structure and activity. Journal of Protein Chemistry, 7(4), 399–415.
Buisson, G., Duee, E., Haser, R., & Payan, F. (1987). Three dimensional structure of porcine pancreatic alpha-amylase at 2.9 A resolution. Role of calcium in structure and activity. The EMBO Journal, 6(13), 3909–3916.
Nielsen, J. E., & Borchert, T. V. (2000). Protein engineering of bacterial alpha-amylases. Biochimica et Biophysica Acta, 1543(2), 253–274.
Bessler, C., Schmitt, J., Maurer, K. H., & Schmid, R. D. (2003). Directed evolution of a bacterial alpha-amylase: Toward enhanced pH-performance and higher specific activity. Protein Science, 12(10), 2141–2149.
Yang, H., Liu, L., Shin, H. D., Chen, R. R., Li, J., Du, G., & Chen, J. (2013). Structure-based engineering of histidine residues in the catalytic domain of alpha-amylase from Bacillus subtilis for improved protein stability and catalytic efficiency under acidic conditions. Journal of Biotechnology, 164(1), 59–66.
Liu, Y., Fan, S., Liu, X., Zhang, Z., Wang, J., Wang, Z., & Lu, F. (2014). A highly active alpha amylase from Bacillus licheniformis: Directed evolution, enzyme characterization and structural analysis. Journal of Microbiology and Biotechnology, 24(7), 898–904.
Wang, C. H., Liu, X. L., Huang, R. B., He, B. F., & Zhao, M. M. (2018). Enhanced acidic adaptation of Bacillus subtilis Ca-independent alpha-amylase by rational engineering of pKa values. Biochemical Engineering Journal, 139, 146–153.
Wang, C., Huang, R., He, B., & Du, Q. (2012). Improving the thermostability of alpha-amylase by combinatorial coevolving-site saturation mutagenesis. BMC Bioinformatics, 13, 263.
Peitsch, M. C. (1995). Protein modeling by E-mail. Nature Biotechnology, 13(7), 658–660.
Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., Shindyalov, I. N., & Bourne, P. E. (2000). The protein data bank. Nucleic Acids Research, 28(1), 235–242.
Guex, N., & Peitsch, M. C. (1997). SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis, 18(15), 2714–2723.
Tiwari, A., & Panigrahi, S. K. (2007). HBAT: A complete package for analysing strong and weak hydrogen bonds in macromolecular crystal structures. In Silico Biology, 7(6), 651–661.
Gallivan, J. P., & Dougherty, D. A. (1999). Cation-pi interactions in structural biology. Proceedings of the National Academy of Sciences of the United States of America, 96(17), 9459–9464.
Sweet, R. M., & Eisenberg, D. (1983). Correlation of sequence hydrophobicities measures similarity in three-dimensional protein structure. Journal of Molecular Biology, 171(4), 479–488.
Sambrook, J., & Russell, D. (2001). Molecular cloning: A laboratory manual (third ed.). New York: Cold Spring Harbor Laboratory Press.
Bradford, M. M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical Biochemistry, 72, 248–254.
Miller, G. L. (1959). Use of dinitrosalicylic acid reagent for determination of reducing sugar. Analytical Chemistry, 31(3), 426–428.
Lineweaver, H., & Burk, D. (1934). The determination of enzyme dissociation constants. Journal of the American Chemical Society, 56(3), 658–666.
Wang, C., Wang, Q., Liao, S., He, B., & Huang, R. (2014). Thermal stability and activity improvements of a Ca-independent alpha-amylase from Bacillus subtilis CN7 by C-terminal truncation and hexahistidine-tag fusion. Biotechnology and Applied Biochemistry, 61(2), 93–100.
van der Maarel, M. J., van der Veen, B., Uitdehaag, J. C., Leemhuis, H., & Dijkhuizen, L. (2002). Properties and applications of starch-converting enzymes of the alpha-amylase family. Journal of Biotechnology, 94(2), 137–155.
Pan, S., Ding, N., Ren, J., Gu, Z., Li, C., Hong, Y., Cheng, L., Holler, T. P., & Li, Z. (2017). Maltooligosaccharide-forming amylase: Characteristics, preparation, and application. Biotechnology Advances, 35(5), 619–632.
Bowie, J. U., Reidhaar-Olson, J. F., Lim, W. A., & Sauer, R. T. (1990). Deciphering the message in protein sequences: Tolerance to amino acid substitutions. Science, 247(4948), 1306–1310.
Skolnick, J., Kolinski, A., & Yaris, R. (1989). Monte Carlo studies on equilibrium globular protein folding. II. Beta-barrel globular protein models. Biopolymers, 28(6), 1059–1095.
Yue, K., & Dill, K. A. (1992). Inverse protein folding problem: Designing polymer sequences. Proceedings of the National Academy of Sciences of the United States of America, 89(9), 4163–4167.
Hocker, B., Beismann-Driemeyer, S., Hettwer, S., Lustig, A., & Sterner, R. (2001). Dissection of a (betaalpha)8-barrel enzyme into two folded halves. Nature Structural Biology, 8(1), 32–36.
Declerck, N., Machius, M., Joyet, P., Wiegand, G., Huber, R., & Gaillardin, C. (2002). Engineering the thermostability of Bacillus licheniformis a-amylase. Biologia, Bratislava, 57(Suppl. 11), 9.
Suzuki, Y., Ito, N., Yuuki, T., Yamagata, H., & Udaka, S. (1989). Amino acid residues stabilizing a Bacillus alpha-amylase against irreversible thermoinactivation. The Journal of Biological Chemistry, 264(32), 18933–18938.
Igarashi, K., Hatada, Y., Ikawa, K., Araki, H., Ozawa, T., Kobayashi, T., Ozaki, K., & Ito, S. (1998). Improved thermostability of a Bacillus alpha-amylase by deletion of an arginine-glycine residue is caused by enhanced calcium binding. Biochemical and Biophysical Research Communications, 248(2), 372–377.
Gai, Y., Cheng, J., Zhang, S., Zhu, B., & Zhang, D. (2018). Property improvement of α-amylase from Bacillus stearothermophilus by deletion of amino acid residues arginine 179 and glycine 180. Food Technology and Biotechnology, 56(1), 58–64.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China [grant number 21868003]; the Chinese National Basic Research Program (“973”) [grant number 2009CB724703]; the Major Program of Natural Science Foundation of Guangxi [grant number 2016GXNSFEA380003]; and the Guangxi BaGui Scholars of Guangxi Zhuang Autonomous Region of China.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
ESM 1
(DOCX 738 kb)
Rights and permissions
About this article

Cite this article
Wang, CH., Lu, LH., Huang, C. et al. Simultaneously Improved Thermostability and Hydrolytic Pattern of Alpha-Amylase by Engineering Central Beta Strands of TIM Barrel. Appl Biochem Biotechnol 192, 57–70 (2020). https://doi.org/10.1007/s12010-020-03308-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-020-03308-8