
Abstract
Unraveling the causes and pathomechanisms of progressive disorders is essential for the development of therapeutic strategies. Here, we identified heterozygous pathogenic missense variants of LMX1A in two families of Dutch origin with progressive nonsyndromic hearing impairment (HI), using whole exome sequencing. One variant, c.721G > C (p.Val241Leu), occurred de novo and is predicted to affect the homeodomain of LMX1A, which is essential for DNA binding. The second variant, c.290G > C (p.Cys97Ser), predicted to affect a zinc-binding residue of the second LIM domain that is involved in protein–protein interactions. Bi-allelic deleterious variants of Lmx1a are associated with a complex phenotype in mice, including deafness and vestibular defects, due to arrest of inner ear development. Although Lmx1a mouse mutants demonstrate neurological, skeletal, pigmentation and reproductive system abnormalities, no syndromic features were present in the participating subjects of either family. LMX1A has previously been suggested as a candidate gene for intellectual disability, but our data do not support this, as affected subjects displayed normal cognition. Large variability was observed in the age of onset (a)symmetry, severity and progression rate of HI. About half of the affected individuals displayed vestibular dysfunction and experienced symptoms thereof. The late-onset progressive phenotype and the absence of cochleovestibular malformations on computed tomography scans indicate that heterozygous defects of LMX1A do not result in severe developmental abnormalities in humans. We propose that a single LMX1A wild-type copy is sufficient for normal development but insufficient for maintenance of cochleovestibular function. Alternatively, minor cochleovestibular developmental abnormalities could eventually lead to the progressive phenotype seen in the families.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hereditary nonsyndromic hearing impairment (NSHI, MIM: 500008) is genetically very heterogeneous. Currently, more than 100 deafness genes have been identified, and still every year novel ones are discovered (Hereditary Hearing Loss Homepage). Since 2010, whole exome sequencing (WES) and targeted next generation sequencing have enabled the rapid and cost-efficient identification of deafness genes (Vona et al. 2015). However, we need to beware of seemingly causative variants that segregate in small families or occur in several unique individuals with hearing impairment (HI) by coincidence, especially in dominant NSHI. This is illustrated by the recent disqualification of MYO1A (MIM: 601478) as a deafness gene (Eisenberger et al. 2014; Patton et al. 2016). Ideally, unique pathogenic variants are identified in several families with a similar phenotype and not in a significant number of controls. Genetic studies are preferably supported by functional and animal studies that prove the deleterious effect of a variant and demonstrate the function of a gene in hearing. As this is often very time-consuming and expensive, (homology) protein modeling or existing data on studies in mice can be supportive in the discovery of novel deafness genes, as was recently exemplified by KITLG (MIM: 184745) (Zazo Seco et al. 2015) and S1PR2 (MIM: 605111) (Santos-Cortez et al. 2016).
The identification of genetic defects underlying progressive HI and the pathomechanisms paves the way for development of therapeutic strategies. In the present study, we identified pathogenic missense variants of LMX1A (MIM: 600298), a gene associated with a complex phenotype in mice, including recessive deafness and vestibular defects (Bergstrom et al. 1999; Chizhikov et al. 2006; Steffes et al. 2012), as a cause of dominant NSHI and vestibular dysfunction in humans.
Materials and methods
Study subjects
This study was approved by the medical ethics committee of the Radboud University Medical Center and is in accordance with the principles of the World Medical Association Declaration of Helsinki. Written informed consent was obtained from all participants or their legal representatives. Medical history was obtained, using a questionnaire focusing on hearing and balance. External ear inspection and otoscopy to assess the tympanic membrane and aeration of the middle ear were performed in all subjects. Special attention was paid to possible causes of acquired HI. Pure tone audiometry was performed in a sound-treated room according to current standards. Air conduction thresholds were determined at 0.25, 0.5, 1, 2, 4, and 8 kHz in dB HL. Bone conduction thresholds were determined at 0.5, 1, 2, 4, and 8 kHz in dB HL to exclude conductive HI. HI was described according to the recommendations of the GENDEAF study group (Mazzoli et al. 2003). Individual progression of HI was calculated for each frequency with longitudinal linear regression analyses, using GraphPad Prism 6.0 (GraphPad, San Diego, CA, USA). Tympanometry was performed, and click-evoked ABR and otoacoustic emissions (OAEs) were obtained, according to current standards. Contralateral and ipsilateral acoustic reflexes were measured at 0.5, 1, 2 and 4 kHz up to loudness discomfort level. Speech perception thresholds and maximum speech recognition scores were determined using speech audiometry, which was performed in a sound-treated room with standard monosyllabic consonant–vowel–consonant Dutch word lists (Bosman and Smoorenburg 1995). Family 63136 did not participate in our clinical evaluation, but retrospective data from the affected subjects (I:2, II:2, II:3 and II:4) were analyzed.
Vestibular function was assessed in subjects II:7 and III:8 (family W15-0551) and the index case of family 63136, using electronystagmography (ENG) rotary chair stimulation and caloric irrigation testing, according to current standards. Additionally, cervical vestibular evoked myogenic potentials (cVEMP) and video head impulse tests (vHIT) were performed in subjects II:7 and III:8 of family W15-0551 to assess sacculus and vestibulo–ocular reflex (VOR) functionality, respectively.
To assess the presence of sub/infertility or neurological, cognitive or cutaneous abnormalities, subjects were screened in accordance with a predefined protocol (Supplemental Methods).
Genetic analyses
WES
For WES, the exome was enriched with an Agilent SureSelect kit, version 4 or 5 (Santa Clara, CA, USA), and WES was performed on an Illumina HiSeq system by BGI Europe (Denmark) (Zazo Seco et al. 2017). As a first step, variants in genes associated with HI were selected (gene list DGD20062014) and classified according to the existing guidelines from the Association for Clinical Genetic Science and the Dutch Society of Clinical Genetic Laboratory Specialists (Wallis et al. 2013), as described previously (Zazo Seco et al. 2017). Mean ≥ 20× coverage was obtained for 96% for the enriched regions.
Variants in WES, shared by individuals II:3 and II:4 of family 63136 were filtered as follows: variation reads ≥ 5 and ≥ 20% and ≤ 90%, minor allele frequency (MAF) in in-house WES database (~ 20,000 exomes) ≤ 0.05%, MAF in EXAC ≤ 0.05%, MAF in GnomAD (Exomes non-Finnish Europeans) ≤ 0.05%, and location in exons or in introns in the 20 bps flanking the exons. Missense variants were only selected for segregation analysis if at least two of four tools predicted pathogenicity. Tools employed were CADD (predicted pathogenic if ≥ 15), SIFT (predicted pathogenic ≤ 0.05), Polyphen-2 (predicted pathogenic if ≥ 0.450) and Mutation Taster. A potential effect on splicing was predicted using the tools SpliceSiteFinder-like, MaxEntScan, NNSPLICE, GeneSplicer, and Human Splicing Finder as available in Alamut Visual (v 2.10, Interactive Biosoftware).
SNP genotyping (family 63136)
All members of family 63136 were genotyped with the Infinium® Global Screening Array-24 v.1.0 (Illumina) according to protocols of the manufacturer. Genotypes were employed to exclude candidate variants derived from WES. Variants were regarded to be excluded if genotypes of SNPs located within 0.5 Mb both proximal and distal of the variant excluded the presence of the haplotype with the variant in at least one affected individual.
Sanger sequencing
PCR of fragments to be analyzed was performed using standard protocols. Primers for PCR were designed with Primer3Plus. Reference sequence identifiers, primer sequences and PCR conditions are provided in Table S3. PCR fragments were purified with ExoSAP-IT (Thermo Fisher Scientific), according to manufacturers’ protocols. For sequence analysis, the ABI PRISM BigDye Terminator Cycle Sequencing v2.0 Ready Reaction kit and the ABI PRISM 3730 DNA analyzer (Applied Biosystems, Foster City, CA, USA) were employed. Possibly deleterious effects of the identified variants were predicted with Alamut Visual version 2.7.1 (Interactive Biosoftware, Rouen, France).
Linkage analysis
Linkage analysis was performed by genotyping of all subjects of family W15-0551 with the 700 K SNP Global Screening Array (Illumina, San Diego, CA, USA), using the manufacturers’ protocols. Superlink online SNP 1.1 software was employed for approximate multipoint LOD score calculations and a window size of 20 SNPs was used (Silberstein et al. 2013). The inheritance pattern of HI was assumed to be autosomal dominant with a disease allele frequency of 0.001. The penetrance of the disease allele was set at 99%.
VNTR marker analysis
Haplotypes in the LMX1A region were determined by genotyping variable number of tandem repeat (VNTR) markers. Touchdown PCR was used to amplify the VNTR markers, and analyses were performed on an ABI Prism 3730 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). Marker heterozygosity, order, and genetic location were derived from the Marshfield genetic map. Alleles were assigned with GeneMapper v.4.0 software (Applied Biosystems, Foster City, CA, USA).
Conservation in the LIM-homeodomain protein family
All ~ 600 members of the family were extracted from the SMART domain database (http://smart.embl-heidelberg.de/) and aligned. Sequence logos for the second LIM domain and the homeodomain within this family were created with WebLogo (Crooks et al. 2004).
Results
A de novo missense variant of LMX1A cosegregates with dominant NSHI
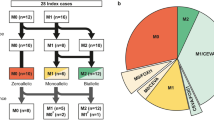
A family of Dutch origin (W15-0551, Fig. 1a) was investigated to identify the underlying genetic defect of the dominant NSHI segregating in the family. We performed WES in the index case (III:8). No (likely) pathogenic variants were identified in genes known to be associated with HI. As a next step, all WES variants were analyzed. To reduce the number of candidate variants, WES was performed in subject II:7 as well. After filtering for shared and rare (≤ 0.5%), missense, nonsense, indels, and splice site variants, 186 variants remained (Table S1). One of these variants was a heterozygous missense variant of LMX1A at g.165179962C (GRCh37/hg19), and c.721G > C (RefSeq NM_001174069.1; p.Val241Leu) (Figure S1). This variant has not been reported in gnomAD and segregated with the HI in the family (Fig. 1a).

Pedigrees, VNTR genotypes and segregation of LMX1A variants. a Genotypes of VNTR markers and segregation of the identified missense variant of LMX1A in family W15-0551. Genetic locations of the markers were derived online from the Marshfield genetic map and maker order was confirmed in the human genome assembly GRCh37/hg19. As the variant resides on an allele (depicted in red) that is shared by non-affected siblings of subject II:7, the variant was concluded to be de novo in subject II:7. It remained unclear whether subject I:2 was affected only at high age or earlier, based on conflicting subjective information provided by her family members. b Pedigree and segregation analyses of a missense variant in LMX1A in family 63136. Index cases are indicated by arrows. + Wild type (color figure online)
It was unknown whether the grandmother (subject I:2) was hearing impaired, because of conflicting subjective information provided by family members. Therefore, we investigated whether the variant of LMX1A was inherited or occurred de novo in subject II:7. Haplotypes of VNTR markers in the LMX1A region were determined. This revealed that the c.721G > C containing haplotype was also present in individuals in the second generation, who did not carry this variant (Fig. 1a). This indicates that the variant occurred de novo in subject II:7 and also that the LMX1A genomic region would have been excluded to carry the underlying genetic defect if linkage analysis would have been the primary step in our analysis.
LMX1A c.721G is highly conserved (PhyloP 5.53) and p.Val241 is fully conserved in the LIM-homeodomain protein family (Fig. 2). Also, defects of Lmx1a have been associated with HI and vestibular defects in mouse (Bergstrom et al. 1999; Chizhikov et al. 2006; Steffes et al. 2012). All together, we considered the identified LMX1A variant a promising candidate to underlie the HI in family W15-0551.

Domains and conservation of (mutated) residues in the LIM-homeodomain protein family. The presented conservation of the second LIM domain and the homeodomain shows that the mutated residues Cys97 and Val241 (indicated by arrows) are perfectly conserved. Within the second LIM domain, the residues that together bind two zinc atoms are indicated with black lines above the amino acid sequence
To exclude other candidate DNA variants (n = 185) shared by individuals II:7 and III:8, linkage analysis was performed by genotyping all subjects of family W15-0551. There were 54 regions in which linkage could not be excluded by a LOD score ≤ − 2 (Figure S2, Table S2). DNA variants in these regions were filtered and classified as described above, which resulted in seven candidate variants (Table S2). None of these segregated with the HI in the family, as determined by Sanger sequencing. As ten of the 54 regions not excluded by linkage analysis harbored known deafness genes (Table S2), coverage of all exons and exon–intron boundaries of these genes, including MYO6 (MIM: 600970), was manually checked to be at least 10×. Subsequently, Sanger sequencing was performed for regions with a lower coverage, which did not reveal any rare variants (allele frequency ≤ 0.5%). Since the HI associated with dominant defects of MYO6 is similar to that in the affected subjects of family W15-0551 with regard to (variable) age of onset and severity (Hilgert et al. 2008; Miyagawa et al. 2015), this gene was also excluded by segregation analysis of SNPs located in and flanking this gene (Figure S3).
Rare variants in the region of chromosome 6 with the strongest evidence for linkage were evaluated for a potential deleterious effect on gene function and for the genes carrying the variants (SLC17A5, PM20D1, MDN1), we searched relevant databases for indications for a role in inner ear function (Table S3). All variants were intronic and none of these were predicted to affect transcript splicing (Table S3). Although MDN1 is transcribed in the cochlea at a significant level and HI is associated with Slc17a5 defects in mouse, it is not likely that the variants of these genes are contributing to the HI phenotype in the family or to the variability of this phenotype.
The p.Cys97Ser amino acid substitution of LMX1A is associated with NSHI
We addressed further involvement of deleterious LMX1A variants in dominant NSHI. WES data of individuals with NSHI, in which (likely) pathogenic variants in all known deafness genes were excluded, were evaluated for rare variants of LMX1A. Only subjects with suspected dominant inheritance or without family history of HI were included (n = 405). This revealed a heterozygous variant at g.165218851, c.290G > C (p.Cys97Ser) in the index case of family 63136 (Figure S1), which segregated with dominantly inherited HI in the family (Fig. 1b). LMX1A c.290G is highly conserved (PhyloP 5.45) and variant c.290G > C has not been reported in the gnomAD database. Residue Cys97 is located within the second LIM domain, and is fully conserved in the LIM-homeodomain protein family (Fig. 2). Screening of WES data also revealed a heterozygous variant at g.165218765, c.376G > A (p.Gly126Lys) in the index case of family W05-233, but this variant was inherited from the normal hearing mother. As can be seen in Fig. 2, p.Gly126 is not conserved in the LIM-homeodomain protein family.
To address other potentially pathogenic variants in family 63136, in genes not (yet) associated with HI, WES was performed for an additional individual (II:4) of the family. Shared variants were identified and filtered as described in “Materials and methods” and SNP genotypes were employed to address potential co-segregation with the HI in the family. Based on these analyses, 15 candidate variants remained to be tested for segregation analysis by Sanger sequencing. Five of the 15 variants, all missense variants, were found to completely segregate with the HI in the family (Table S4a). However, no HI phenotype was reported for four of the five genes carrying the variants in mouse mutants (Table S4b) which does not support a causative nature of these variants for the HI in the family. For TTYH1, no phenotypic data were available for mouse mutants. We searched WES data of the genetically ‘unsolved’ cases with suspected dominant inheritance or without family history of HI for rare variants of this gene and did not identify any rare truncating or missense variants or variants predicted to affect splicing.
LMX1A-associated HI is nonsyndromic, sensorineural, and progressive with a variable age of onset
The affected subjects of family W15-0551 (II:7, III:8 and IV:2) were clinically examined to characterize their audiovestibular phenotype. There was no evidence of acquired causes of HI. All affected subjects had sensorineural HI except for individual II:7 of family W15-0551, who had bilateral fenestral otosclerosis and mixed HI of the left ear. We assumed that the sensorineural HI was unlikely to be caused by otosclerosis, because there were no signs of cochlear otosclerosis on computed tomography scans and her vestibular test results fit with the defect in LMX1A. There were no preoperative audiograms available before stapedectomy of the right ear in 1973 for comparison of the pre- and postoperative bone conduction thresholds of the right ear.
The reported age of onset varied from congenital to 35 years (Table 1). Affected subjects had mild to profound HI with overall a downsloping audiogram configuration (Fig. 3). Subject IV:2 of family W15-0551 and subject II:2 of family 63136 displayed asymmetric HI, whereas the other affected individuals demonstrated symmetric HI. Longitudinal linear regression analysis of hearing thresholds revealed significant progression in all affected subjects of both families, except for subject IV:2 of family W15-0551, probably because follow-up time was too short (3 years) to measure significant deterioration of hearing in this individual. Progression of HI was not analyzed in subject II:7 of family W15-0551, as we cannot exclude deterioration of hearing due to her otosclerosis. The progression rate and severity of HI varied widely among the affected subjects (Fig. 3, Table 1).

Audiometric characterization of families affected by deleterious LMX1A variants. Air conduction thresholds of both ears of the affected individuals are shown of first-visit and last-visit audiometry. The 95th percentile threshold values of presbyacusis (p95) were calculated for the last-visit audiogram, and matched to the individual’s sex and age, according to the ISO 7029 standard. a Family W15-0551. For subject II:7, also bone conduction thresholds of the left ear are depicted, because of mixed HI due to fenestral otosclerosis (conductive HI) and the LMX1A variant (sensorineural HI). b Family 63136. Subjects did not participate in our clinical evaluation; only retrospective data were used for analysis. R right ear, L left ear
Speech reception thresholds were lower than or comparable to pure tone average thresholds at 0.5, 1 and 2 kHz for the better hearing ear, indicating the absence of retrocochlear pathology (Table 1). This is confirmed by results from acoustic reflex measurements in all affected subjects and brainstem evoked response audiometry in subject II:2 of family 63136, which showed no indications of retrocochlear pathology (data not shown). CT scans in subject II:7 of family W15-0551 and the index case (II:3) of family 63136 did not show cochleovestibular malformations, except for known fenestral otosclerosis in the former individual.
Four out of seven affected individuals reported sudden episodes of vertigo in the past that lasted minutes to days, with or without tinnitus, but without simultaneous deterioration of hearing (Table 1). Individuals could not identify triggers for the vertigo episodes. Vestibular function was assessed in subjects II:7 and III:8 (family W15-0551) and the index case of family 63136. The vestibular tests revealed bilateral symmetric hyporeflexia in subject III:8 of family W15-0551 (42-year-old), asymmetric severe hyporeflexia to areflexia to the detriment of the left vestibulum in subject II:3 of family 63136 (52-year-old), and bilateral areflexia in subject II:7 of family W15-0551 (73-year-old). Subjects II:7 and III:8 of family W15-0551 also underwent a video head impulse test (vHIT) and cervical vestibular evoked myogenic potential (cVEMP) recordings. The vHIT displayed bilateral weakness of the posterior semicircular canals in individual II:7 and normal function in individual III:8. cVEMPs showed no responses up to 100 dBnHL in both subjects, indicating dysfunction of the saccule. An overview of vestibular test results is presented in Table S6. Besides standard vestibular tests, subjects also underwent oculomotor testing, consisting of smooth pursuit, gaze (frontal/right/left), saccade, optokinetic and spontaneous nystagmus tests. Oculomotor testing revealed no abnormalities (data not shown). Since vestibular abnormalities are more severe in the older individuals, we hypothesize that there is progressive deterioration of vestibular function. There were, however, no longitudinal data available to confirm this.
As mice with bi-allelic loss-of-function variants of Lmx1a display neurological, skeletal, pigmentation, and reproductive system abnormalities, besides HI and vestibular dysfunction (Bergstrom et al. 1999; Chizhikov et al. 2006; Steffes et al. 2012), the affected subjects of families W15-0551 and 63136 were screened for syndromic abnormalities (Supplemental Methods). However, none of the affected individuals of families W15-0551 and 63136 displayed any cutaneous abnormalities, signs of cognitive dysfunction, or peripheral or central nervous system involvement. History taking did not indicate fertility problems.
Discussion
This study provides evidence that LMX1A is involved in hearing and vestibular function. In two families of Dutch origin, mono-allelic deleterious variants of Lmx1a were found to be associated with a progressive phenotype. HI started between birth and the age of 35 years, and severity and progression rate varied widely between affected individuals. About half of the affected individuals displayed vestibular dysfunction and experienced symptoms thereof. Apart from the vestibular abnormalities, the observed HI phenotype is similar to that of DFNA7, for which the underlying defect has been localized to chromosome 1q21–q23 encompassing LMX1A (Fagerheim et al. 1996). Therefore, LMX1A is a candidate gene for DFNA7. Evaluation of rare variants segregating with the HI in family 63136 revealed no strong candidates for being causative of the HI other than the described LMX1A variant.
LMX1A is a transcription factor that belongs to the highly conserved LIM-homeodomain protein family. LIM-homeodomain proteins are characterized by two cysteine-rich zinc-binding LIM motifs that are known to be involved in protein–protein interactions (Kadrmas and Beckerle 2004), and a homeodomain that is known to bind DNA (Hobert and Westphal 2000). The mutated LMX1A residues identified in this study are located within the second LIM domain and the homeodomain. The p.Cys97Ser affects one of the two zinc-binding residues, which is highly likely to be deleterious for protein folding and function. LIM-homeodomain transcription factors play a pivotal role in various developmental processes, and can be divided into subgroups based on sequence similarities. The vertebrate Lmx subgroup consists of paralogs LMX1A and LMX1B that have identical homeodomain sequences (Figure S4) (Hobert and Westphal 2000).
Bi-allelic loss-of-function variants of the murine Lmx1a gene cause congenital deafness, vestibular defects, and neurological, skeletal, pigmentation, and reproductive system abnormalities (Bergstrom et al. 1999; Chizhikov et al. 2006; Steffes et al. 2012). However, no abnormalities in addition to inner ear dysfunction were observed in the studied families. In contrast to humans, there is no auditory phenotype in heterozygous Lmx1a mouse mutants, but occasionally mild pigmentation abnormalities are seen (Patrylo et al. 1990; Steffes et al. 2012). Normal hearing thresholds have been measured up to 16 weeks (Steffes et al. 2012) and 3 months (unpublished data, kindly provided by Dr. Johnson, Jackson Laboratory, Bar Harbor, Maine, concerning heterozygous Lmx1adr-J mice). Interestingly, the genetic defect in the well-studied mouse model Lmx1adr-J is p.Cys82Tyr and affects one of the zinc-binding cysteine residues of the LIM1 domain (Millonig et al. 2000), which supports the pathogenicity of the identified p.Cys97Ser variant.
Heterozygous loss-of-function variants of LMX1B (MIM: 602575), a paralog of LMX1A, have been associated with nail–patella syndrome in human (NPS, MIM: 161200) (Dreyer et al. 1998; McIntosh et al. 1998; Vollrath et al. 1998). NPS is characterized by dysplastic nails, absent or hypoplastic patellae, elbow dysplasia, and iliac horns, and may be accompanied by nephropathy and/or glaucoma (Mimiwati et al. 2006; Sweeney et al. 2003). A study by Bongers et al. (2005) indicated that late-onset sensorineural HI can also be associated with defects of LMX1B (Bongers et al. 2005). There are striking similarities between the identified genetic defects of LMX1A and those of LMX1B. The mutated residues Cys97 and Val241 of LMX1A are in the sequence alignments at the same positions as the amino acids Cys118 and Val265 of LMX1B, respectively (Figure S4). Amino acid variant p.Val265Leu of LMX1B is known to be disease-causing and displays the same substitution as the LMX1A p.Val241Leu variant (Dunston et al. 2004). The equivalent of LMX1A variant p.Cys97Ser has not been identified in LMX1B, but other substitutions at this position that would also impair the binding of a zinc atom, p.Cys118Phe (Vollrath et al. 1998) and p.Cys118Tyr (Clough et al. 1999), and the equivalent cysteine–serine substitution in LIM domain 1 (p.Cys59Ser), have been associated with NPS. Resemblance of the identified LMX1A variants with disease-causing variants in LMX1B supports the former’s pathogenicity.
We hypothesize that the identified deleterious LMX1A variants lead to haploinsufficiency, which is also the proposed pathogenic mechanism of NPS, caused by variants of LMX1B (Bongers et al. 2008; Dreyer et al. 2000; Sato et al. 2005). A number of LMX1B variants have been tested for a dominant-negative effect, including p.Val265Leu, the equivalent of LMX1A p.Val241Leu, but no inhibitory effect on wild-type protein function was found (Dreyer et al. 2000; Sato et al. 2005). Haploinsufficiency of LMX1A might also explain the sensorineural HI in a subject with a heterozygous interstitial 1q23.3q24.1 deletion encompassing LMX1A (patient 9 in Chatron et al. 2015). However, the absence of an HI phenotype in mice heterozygous for loss of function variants of Lmx1a is not supportive of haploinsufficiency as the disease mechanism.
Our data do not support the suggested involvement of LMX1A in ID (Chatron et al. 2015; Mackenroth et al. 2016), as affected subjects of families W15-0551 and 63136 displayed normal cognition. Based on our findings and those of Chatron et al. (2015) and Mackenroth et al. (2016), we recommend screening for HI in subjects with mono-allelic loss of LMX1A.
In mouse, Lmx1a expression in the inner ear starts at the otocyst stage (from E10.5 onwards) and is later restricted to non-sensory epithelia of the developing cochlea and vestibular system (Huang et al. 2008; Koo et al. 2009; Nichols et al. 2008; Steffes et al. 2012). Bi-allelic Lmx1a defects lead to disorganization of the inner ear and lack of differentiation and separation of sensory, non-sensory and neurogenic domains of the vestibulocochlear system. As a result, Lmx1a mouse mutants have a short and malformed cochlear duct, no endolymphatic duct and sac, no semicircular canals, and the sacculus and utriculus remain rudimentary (Koo et al. 2009; Nichols et al. 2008; Steffes et al. 2012). Hair cells in the basal part of the cochlea display severe disorganization, whereas hair cells in the apical turn are only mildly disorganized (Nichols et al. 2008). Although expression of Lmx1a in the inner ear is reduced from E16.5 onwards (Huang et al. 2008), Lmx1a has been suggested to function in the maintenance of hair cells, as there is progressive hair cell loss in adult mutant mice (Nichols et al. 2008).
There is large phenotypic variability within families W15-0551 and 63136, regarding age of onset (a)symmetry, severity and progression rate of HI. Large intrafamilial phenotypic variation has also been reported for NPS (Ghoumid et al. 2016). The variable HI phenotype suggests involvement of environmental and/or genetic factors in the expression of the phenotype. The expression level of the wild-type LMX1A allele might well be one of the genetic factors. The overall downsloping audiogram configuration observed in the affected subjects corresponds to the abnormal development of the sensory epithelium in the organ of Corti in homozygous Lmx1a mouse mutants, displaying severe abnormalities of this epithelium in the basal part of the cochlea compared to mild defects in the apical regions (Koo et al. 2009).
The onset of HI caused by LMX1A defects was in the second or third decade in most of the cases, which suggests that one LMX1A copy is sufficient for normal cochlear development. As homozygous Lmx1a mutant mice displayed loss of cochlear hair cells in the adult stage, the gene has been suggested to be essential for long-term maintenance of hair cells (Nichols et al. 2008). It is therefore tempting to speculate that in the families the deterioration of HI over time is due to progressive loss of hair cells. Since vestibular complaints occurred during adulthood and vestibular dysfunction seemed progressive, we speculate that LMX1A is also critical for maintenance of the adult vestibular organs. Interestingly, it has been demonstrated that both Lmx1a and Lmx1b not only function in the developing mouse brain, as expression of both genes was found to be sustained in the adult midbrain and to be essential for survival of adult dopaminergic neurons in the midbrain (Doucet-Beaupre et al. 2016). A role in the regulation of genes with a mitochondrial function was indicated. It remains to be determined whether LMX1A, possibly in concert with LMX1B, has a similar function in the adult inner ear. So far, there are no indications for LMX1A/Lmx1a expression in the adult inner ear (Huang et al. 2008; Schrauwen et al. 2016). However, the expression might well be at a very low level and/or in a specific cell type. An alternative explanation for the HI in the presented families is that a single copy of LMX1A causes minor cochleovestibular developmental abnormalities that eventually lead to a progressive disease-phenotype. The phenotype of bi-allelic deleterious LMX1A variants in humans is still elusive. As suggested by Steffes et al. (2012), bi-allelic loss-of-function variants of LMX1A might well be lethal in human (Steffes et al. 2012).
One of the two LMX1A variants identified in this study occurred de novo. Also, we identified de novo variants with a dominant effect to explain four of 67 cases for whom a genetic diagnosis was established by testing a deafness gene panel and Kasakura-Kimura et al. (2017) identified de novo variants as the cause of dominantly inherited HI in two of 74 ‘solved’ cases (Kasakura-Kimura et al. 2017; Zazo Seco et al. 2015). Interestingly, also for retinitis pigmentosa that displays very high genetic heterogeneity as does HI, the underlying genetic defect was found to be a de novo variant in 3 of 28 individuals without family history of the disease (Neveling et al. 2012). These data demonstrate that in families with an isolated case of a genetically highly heterogeneous disorder for which reproductive fitness is not reduced, the scenario of an underlying de novo variant is not uncommon.
In conclusion, mono-allelic missense variants of LMX1A were found to underlie nonsyndromic HI and vestibular defects. The HI phenotype has a variable age of onset, severity and progression rate. Haploinsufficiency is the most likely pathogenic mechanism. As in LMX1B, we expect that both truncating and missense variants can lead to the identified phenotype, with a preferential presence of missense variants in the conserved residues of the LIM domains and homeodomain of LMX1A.
Web resources
The URLs for data presented herein are as follows:
Alamut Visual http://www.interactive-biosoftware.com/alamut-visual/.
ExAC Browser http://exac.broadinstitute.org/.
GnomAD browser http://gnomad.broadinstitute.org/.
Hereditary Hearing loss Homepage http://hereditaryhearingloss.org/.
Marshfield Genetic Maps http://research.marshfieldclinic.org/genetics/GeneticResearch/compMaps.asp.
OMIM http://www.omim.org/.
OMIM Phenotypic Series http://www.omim.org/phenotypicSeriesTitle/all.
Primer3Plus http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi.
SMART http://smart.embl-heidelberg.de/.
Superlink Online SNP http://cbl-hap.cs.technion.ac.il/superlink-snp/.
UCSC Genome Browser https://genome.ucsc.edu.
WebLogo http://weblogo.berkeley.edu/.
References
Bergstrom DE, Gagnon LH, Eicher EM (1999) Genetic and physical mapping of the dreher locus on mouse chromosome 1. Genomics 59:291–299. https://doi.org/10.1006/geno.1999.5873
Bongers EM et al (2005) Genotype–phenotype studies in nail-patella syndrome show that LMX1B mutation location is involved in the risk of developing nephropathy. Eur J Hum Genet EJHG 13:935–946. https://doi.org/10.1038/sj.ejhg.5201446
Bongers EM, de Wijs IJ, Marcelis C, Hoefsloot LH, Knoers NV (2008) Identification of entire LMX1B gene deletions in nail patella syndrome: evidence for haploinsufficiency as the main pathogenic mechanism underlying dominant inheritance in man. Eur J Hum Genet EJHG 16:1240–1244. https://doi.org/10.1038/ejhg.2008.83
Bosman AJ, Smoorenburg GF (1995) Intelligibility of Dutch CVC syllables and sentences for listeners with normal hearing and with three types of hearing impairment. Audiol Off Organ Int Soc Audiol 34:260–284
Chatron N et al (2015) Refinement of genotype-phenotype correlation in 18 patients carrying a 1q24q25 deletion. Am J Med Genet Part A 167A:1008–1017. https://doi.org/10.1002/ajmg.a.36856
Chizhikov V, Steshina E, Roberts R, Ilkin Y, Washburn L, Millen KJ (2006) Molecular definition of an allelic series of mutations disrupting the mouse Lmx1a (dreher) gene. Mamm Genome Off J Int Mamm Genome Soc 17:1025–1032. https://doi.org/10.1007/s00335-006-0033-7
Clough MV, Hamlington JD, McIntosh I (1999) Restricted distribution of loss-of-function mutations within the LMX1B genes of nail-patella syndrome patients. Hum Mutat 14:459–465. https://doi.org/10.1002/(sici)1098-1004(199912)14:6<459::aid-humu3>3.0.co;2-9
Crooks GE, Hon G, Chandonia JM, Brenner SE (2004) WebLogo: a sequence logo generator. Genome Res 14:1188–1190. https://doi.org/10.1101/gr.849004
Doucet-Beaupre H et al (2016) Lmx1a and Lmx1b regulate mitochondrial functions and survival of adult midbrain dopaminergic neurons. Proc Natl Acad Sci USA 113:E4387–E4396. https://doi.org/10.1073/pnas.1520387113
Dreyer SD et al (1998) Mutations in LMX1B cause abnormal skeletal patterning and renal dysplasia in nail patella syndrome. Nat Genet 19:47–50. https://doi.org/10.1038/ng0598-47
Dreyer SD et al (2000) LMX1B transactivation and expression in nail-patella syndrome. Hum Mol Genet 9:1067–1074
Dunston JA et al (2004) The human LMX1B gene: transcription unit, promoter, and pathogenic mutations. Genomics 84:565–576. https://doi.org/10.1016/j.ygeno.2004.06.002
Eisenberger T et al (2014) Targeted and genomewide NGS data disqualify mutations in MYO1A, the “DFNA48 gene”, as a cause of deafness. Hum Mutat 35:565–570. https://doi.org/10.1002/humu.22532
Fagerheim T et al (1996) Identification of a new locus for autosomal dominant non-syndromic hearing impairment (DFNA7) in a large Norwegian family. Hum Mol Genet 5:1187–1191
Ghoumid J et al (2016) Nail-Patella Syndrome: clinical and molecular data in 55 families raising the hypothesis of a genetic heterogeneity. Eur J Hum Genet EJHG 24:44–50. https://doi.org/10.1038/ejhg.2015.77
Hilgert N, Topsakal V, van Dinther J, Offeciers E, Van de Heyning P, Van Camp G (2008) A splice-site mutation and overexpression of MYO6 cause a similar phenotype in two families with autosomal dominant hearing loss. Eur J Hum Genet EJHG 16:593–602. https://doi.org/10.1038/sj.ejhg.5202000
Hobert O, Westphal H (2000) Functions of LIM-homeobox genes. Trends Genet 16:75–83
Huang M, Sage C, Li H, Xiang M, Heller S, Chen ZY (2008) Diverse expression patterns of LIM-homeodomain transcription factors (LIM-HDs) in mammalian inner ear development. Dev Dyn Off Publ Am Assoc Anatomists 237:3305–3312. https://doi.org/10.1002/dvdy.21735
Kadrmas JL, Beckerle MC (2004) The LIM domain: from the cytoskeleton to the nucleus. Nat Rev Mol Cell Biol 5:920–931. https://doi.org/10.1038/nrm1499
Kasakura-Kimura N et al (2017) WFS1 and GJB2 mutations in patients with bilateral low-frequency sensorineural hearing loss. Laryngoscope 127:E324–E329. https://doi.org/10.1002/lary.26528
Koo SK, Hill JK, Hwang CH, Lin ZS, Millen KJ, Wu DK (2009) Lmx1a maintains proper neurogenic, sensory, and non-sensory domains in the mammalian inner ear. Dev Biol 333:14–25. https://doi.org/10.1016/j.ydbio.2009.06.016
Mackenroth L, Hackmann K, Klink B, Weber JS, Mayer B, Schrock E, Tzschach A (2016) Interstitial 1q23.3q24.1 deletion in a patient with renal malformation, congenital heart disease, and mild intellectual disability. Am J Med Genet Part A 170:2394–2399. https://doi.org/10.1002/ajmg.a.37785
Mazzoli M, Van Camp G, Newton V, Giarbini N, Declau F, Parving A (2003) Recommendations for the description of genetic and audiological data for families with nonsyndromic hereditary hearing impairment. Audiol Med 1:148–150
McIntosh I et al (1998) Mutation analysis of LMX1B gene in nail-patella syndrome patients. Am J Hum Genet 63:1651–1658. https://doi.org/10.1086/302165
Millonig JH, Millen KJ, Hatten ME (2000) The mouse Dreher gene Lmx1a controls formation of the roof plate in the vertebrate CNS. Nature 403:764–769. https://doi.org/10.1038/35001573
Mimiwati Z et al (2006) Nail-patella syndrome and its association with glaucoma: a review of eight families. Br J Ophthalmol 90:1505–1509. https://doi.org/10.1136/bjo.2006.092619
Miyagawa M, Nishio SY, Kumakawa K, Usami S (2015) Massively parallel DNA sequencing successfully identified seven families with deafness-associated MYO6 mutations: the mutational spectrum and clinical characteristics. Ann Otol Rhinol Laryngol 124(Suppl 1):148S–157S. https://doi.org/10.1177/0003489415575055
Neveling K et al (2012) Next-generation genetic testing for retinitis pigmentosa. Hum Mutat 33:963–972. https://doi.org/10.1002/humu.22045
Nichols DH, Pauley S, Jahan I, Beisel KW, Millen KJ, Fritzsch B (2008) Lmx1a is required for segregation of sensory epithelia and normal ear histogenesis and morphogenesis. Cell Tissue Res 334:339–358. https://doi.org/10.1007/s00441-008-0709-2
Patrylo PR, Sekiguchi M, Nowakowski RS (1990) Heterozygote effects in dreher mice. J Neurogenet 6:173–181
Patton J, Brewer C, Chien W, Johnston JJ, Griffith AJ, Biesecker LG (2016) A genotypic ascertainment approach to refute the association of MYO1A variants with non-syndromic deafness. Eur J Hum Genet EJHG 25:147–149. https://doi.org/10.1038/ejhg.2016.140
Santos-Cortez RL et al (2016) Autosomal-recessive hearing impairment due to rare missense variants within S1PR2. Am J Hum Genet 98:331–338. https://doi.org/10.1016/j.ajhg.2015.12.004
Sato U, Kitanaka S, Sekine T, Takahashi S, Ashida A, Igarashi T (2005) Functional characterization of LMX1B mutations associated with nail-patella syndrome. Pediatr Res 57:783–788. https://doi.org/10.1203/01.PDR.0000157674.63621.2C
Schrauwen I et al (2016) A comprehensive catalogue of the coding and non-coding transcripts of the human inner ear. Hear Res 333:266–274. https://doi.org/10.1016/j.heares.2015.08.013
Silberstein M et al (2013) A system for exact and approximate genetic linkage analysis of SNP data in large pedigrees. Bioinformatics 29:197–205. https://doi.org/10.1093/bioinformatics/bts658
Steffes G et al (2012) Mutanlallemand (mtl) and Belly Spot and Deafness (bsd) are two new mutations of Lmx1a causing severe cochlear and vestibular defects. PLoS One 7:e51065. https://doi.org/10.1371/journal.pone.0051065
Sweeney E, Fryer A, Mountford R, Green A, McIntosh I (2003) Nail patella syndrome: a review of the phenotype aided by developmental biology. J Med Genet 40:153–162
Vollrath D, Jaramillo-Babb VL, Clough MV, McIntosh I, Scott KM, Lichter PR, Richards JE (1998) Loss-of-function mutations in the LIM-homeodomain gene, LMX1B, in nail-patella syndrome. Hum Mol Genet 7:1091–1098
Vona B, Nanda I, Hofrichter MA, Shehata-Dieler W, Haaf T (2015) Non-syndromic hearing loss gene identification: a brief history and glimpse into the future. Mol Cell Probes 29:260–270. https://doi.org/10.1016/j.mcp.2015.03.008
Wallis Y et al (2013) Practice guidelines for the evaluation of pathogenicity and the reporting of sequence variants in clinical molecular genetics. ACGS/VGKL. Available at http://www.acgs.uk.com/media/774853/evaluation_and_reporting_of_sequence_variants_bpgs_june_2013_-_finalpdf.pdf. Accessed 23 Mar 2018
Zazo Seco C et al (2015) Allelic mutations of KITLG, encoding KIT ligand, cause asymmetric and unilateral hearing loss and Waardenburg syndrome type 2. Am J Hum Genet 97:647–660. https://doi.org/10.1016/j.ajhg.2015.09.011
Zazo Seco C et al (2017) The diagnostic yield of whole-exome sequencing targeting a gene panel for hearing impairment in The Netherlands. Eur J Hum Genet 25:308–314. https://doi.org/10.1038/ejhg.2016.182
Acknowledgements
We are grateful to the participating subjects and their families. We thank Laura Tomas Roca for her contribution to the WES analysis. This work was financially supported by a Grant from the Heinsius Houbolt Foundation (to H. K., R. J. E. P. and H. P. M. K.). The DOOFNL consortium consists of M.F. van Dooren, H.H.W. de Gier, E.H. Hoefsloot, M.P. van der Schroeff (ErasmusMC, Rotterdam, The Netherlands), S.G. Kant, L.J.C. Rotteveel (LUMC, Leiden, The Netherlands), S.G.M. Frints, J.R. Hof, R.J. Stokroos, E.K. Vanhoutte (MUMC+, Maastricht, The Netherlands), R.J.C. Admiraal, I. Feenstra, H. Kremer, H.P.M. Kunst, R.J.E. Pennings, H.G. Yntema (Radboudumc, Nijmegen, The Netherlands) A.J. van Essen, R.H. Free and J.S. Klein-Wassink (UMCG, Groningen, The Netherlands).
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflict of interest to declare.
Additional information
The members of the DOOFNL Consortium group are listed in the “Acknowledgements” section.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Wesdorp, M., de Koning Gans, P.A.M., Schraders, M. et al. Heterozygous missense variants of LMX1A lead to nonsyndromic hearing impairment and vestibular dysfunction. Hum Genet 137, 389–400 (2018). https://doi.org/10.1007/s00439-018-1880-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-018-1880-5