
Abstract
Plant long non-coding RNA (lncRNA) plays an important role in many biological processes, mainly through its interaction with RNA binding protein (RBP). To understand the function of lncRNA, a basic step is to determine which proteins are interacted with lncRNA. Therefore, RBP can be predicted by computational approaches. However, the main challenge is that it is difficult to find interaction patterns or primitives. In this study, we propose a method based on sequences to predict plant lncRNA-protein interaction, namely PLRPI uses k-mer frequency feature for RNA and protein, stacked denoising autoencoder and gradient boosting decision tree to learn the hidden interaction between plant lncRNAs and proteins sequences. The experimental results show that PLRPI achieves good performance on the test datasets ATH948 and ZEA22133 based on lncRNA-protein interaction of Arabidopsis thaliana and Zea mays. Our method gets an accuracy of 90.4% on ATH948 and 82.6% on ZEA22133. PLRPI is also superior to other methods in some public RNA-protein interaction datasets. The result shows PLRPI has strong generalization ability and high robustness. It is an effective model for predicting plant lncRNA-protein interactions.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others

Keywords
1 Introduction
Long non-coding RNA (lncRNA) [1] is a kind of RNA molecule with specific functions in eukaryotes. Its length is generally more than 200 nt. Basically, they have no ability to encode proteins, which are large in number and are presented in the nucleus or cytoplasm. It has been found that lncRNA can participate in various levels of gene expression regulation by interacting with proteins such as chromatin-modified complexes and transcription factors. lncRNA also plays a regulatory role in many important biological processes. Their interactions are closely related to the most basic life activities of organisms [2,3,4,5]. Many key cellular processes such as signal transduction, chromosome replication, material transport, mitosis, transcription and translation, are closely related to the interaction between RNA and protein [6,7,8]. Although there is no doubt about the role of lncRNA in the regulation of gene expression, only a few functions and mechanisms of lncRNA have been studied. Since the regulatory role of lncRNA mostly requires the coordination of protein molecules, it is necessary to identify the interactions of lncRNA and protein molecules.
Research on plant lncRNA is still in its infancy compared with animals. To date, nearly 10,000 lncRNAs have been found in several plants such as Arabidopsis thaliana, wheat, corn, soybeans, and rice, accounting for 1% of total lncRNAs. They play an important role in guiding reproductive development, growth, stress response, chromosome modification, and protein interactions.
The interaction of lncRNA with protein is ubiquitous. At present, there are few structural data of protein complexes obtained by conventional methods such as X-ray diffraction, nuclear magnetic resonance, electron microscopy and neutron diffraction. This is mainly because the experimental methods have disadvantages like high cost, long time-consuming and complicated measurement process. With the development of high-throughput sequencing technology, people can quickly obtain a large amount of transcriptome and proteomic information, including a large number of potential RPI needs analysis. However, traditional experimental methods can only be studied on specific protein, RNA or protein-RNA complexes, which is far from technically sufficient. Therefore, machine learning is widely used in bioinformatics to extract features from samples and analyze them.
Traditional machine learning models require manual feature extraction, which may not be able to pinpoint hidden relationships in raw data. Deep learning provides a powerful solution to this problem. It consists of multi-layer neural network model architecture [9,10,11] that automatically extracts high-level abstractions from the data. At the same time, in the fields of image recognition [12], speech recognition, signal recognition [13], deep learning shows better performance than other commonly used machine learning methods. It has also been well applied in the field of bioinformatics [14, 15]. For example, deep learning has been successfully applied to predict RNA splicing patterns [16]. Compared with other sequence-based methods, deep learning can automatically learn the sequence characteristics of RNA and protein, discover the specific correlation between these sequences [17, 18], and reduce the influence of noise in the original data by learning the real hidden advanced features. In addition, some methods based on deep learning artificially introduce noise to reduce over-fitting, which can enhance the generalization ability and robustness of the model.
This study presents a new model, PLRPI, for predicting plant lncRNA-protein interactions based on sequence information. For a particular plant protein and lncRNA pair, PLRPI can predict whether there are interactions between them. In the experiment, we first extracted the 4-mer features of lncRNA and the 3-mer features of proteins [19]. 20 amino acids of proteins were divided into 7 groups according to their physicochemical properties [20]. They are embedded into matrices and features are extracted using stacked denoising autoencoder. Then the extracted features of lncRNAs and proteins are contacted and added into the softmax layer, which is compared with the data labels for supervised learning, the advanced features are obtained and fine-tuned. The gradient boosting descent tree classifier is used for ensemble classification, and the final result is obtained. We evaluated the performance of PLRPI on plant datasets and other RNA-protein datasets from previous studies for comparison with other advanced methods. The results show that PLRPI not only has high prediction accuracy, but also has good generalization ability and high robustness. It can effectively predict the interaction between plant lncRNAs and proteins.
2 Materials and Methods
2.1 Datasets
To test the performance of PLRPI, we created the datasets ATH948 and ZEA22133 based on Arabidopsis thaliana and Zea mays. Firstly, we downloaded Arabidopsis thaliana and Zea mays lncRNA-protein datasets from Ming Chen’s bioinformatics group (http://bis.zju.edu.cn/PlncRNADB/index.php?p=network&spe=Zea%20mays). In order to reduce the bias of sequence homology, the redundant sequences with sequence similarity greater than 90% for both protein and lncRNA sequences were excluded by using CD-HIT [21]. For constructing non-interaction pairs, the same number of negative pairs were generated through randomly pairing proteins with lncRNAs and further removing the existing positive pairs [19]. After redundancy removal, ATH948 dataset, including 948 interactive pairs and 948 non-interactive pairs, was obtained consisting of 35 protein chains and 109 lncRNA chains. Similarly, ZEA22133 dataset, including 22133 interactive pairs and 22133 non-interactive pairs, was obtained consisting of 42 protein chains and 1704 lncRNA chains. It should be pointed out that compared with other datasets, it is more difficult to extract features from plant lncRNA-protein interaction datasets. This is due to the poor homology of plant lncRNA and the fact that a larger number of interactions require only a smaller number of lncRNAs and proteins. It may increase the noise which is more evident in ZEA22133. The details are shown in Table 1.
To test the robustness of PLRPI, we also collected other RNA-protein datasets from previous studies, such as RPI1807 [22], RPI369 [19], RPI2241 [19] and RPI488 [23], which were all extracted based on structure-based experimental complexes. RPI1807, RPI369 and RPI2241 datasets are RNA-protein interactions from many species, including human, animals and plants. Only RPI488 dataset is lncRNA-protein interaction.
2.2 Methods
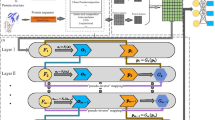
We first extracted 4-mer features of lncRNAs and 3-mer features of proteins, and then put them into stacked denoising autoencoder models, respectively. The results are fine-tuned using label information from RNA-protein pairs. After high-level features were fine-tuned and they were classified using gradient boosting decision tree to get the output. The detailed process is shown in Fig. 1.

The flowchart of proposed PLRPI.
The datasets and python code supporting the findings of this study are available at https://github.com/zhr818789/PLRPI. The source code for the experiments was written in python 3.5.2 using Keras 2.2.2 with Tensorflow 1.10.0 backend.
Sequence Information Processing
In order to obtain the raw features of autoencoder, we extracted simple sequence component composition features from both RNAs and proteins. For RNA sequences, 4-mer frequency features of RNA sequences (A, C, G, T) are extracted, we got 4 × 4 × 4 × 4 = 256 dimensional features. Each feature value is the normalized frequency of 4-mer nucleotides in RNA sequences, which is AAAA…CATC…TTTT. For protein sequences, analysis by existing studies indicates that RNA-binding residues are prone to amino acids with certain properties. According to the physicochemical properties of amino acids and the effects of interactions, the 20 amino acids are divided into 7 categories. They include: {Val, Gly, Ala}, {Phe, Pro, Leu, Ile}, {Ser, Tyr, Met, Thr}, {His, Asn, Tpr, Gln}, {Arg, Lys}, {Glu, Asp} and {Cys}. We divided the protein sequences into 7 groups according to the rules above. Since the conjoint triad (3-mer) of protein is composed by 3 amino acids, we extracted the 3-mer features of protein trimer and got 7 × 7 × 7 = 343 dimensional features.
Stacked Denoising Autoencoder (SDAE)
Autoencoder (AE)
Autoencoder belongs to unsupervised learning and does not need to label training samples. It is composed of two parts. The first part is an encoding network consisting of input layer and middle layer which is used to compress the signal. The second part is a decoding network consisting of middle layer and output layer which is used to restore the compressed signal.
Suppose that we input an n-dimensional signal x (x < [0, 1]) through the input layer to the middle layer, the signal becomes y, which is expressed by the following formula:
where s is a non-linear function, such as sigmoid. W is the link weight from input layer to middle layer, and b is the bias of middle layer. Signal y is decoded by decoding layer and output to output layer with n neurons, and then the signal becomes z. The following formula is used:
where s is a non-linear function, such as sigmoid. W′ is the link weight from the middle layer to the output layer, b′ is the bias of the output layer, and z is regarded as the prediction of x. Then the network parameters are adjusted to make the final output z as close to the original input signal x as possible.
Denoising Autoencoder
Due to the complexity of the model, the amount of training data and the noise of data, the initial model obtained by autoencoder often has the risk of over-fitting. In order to prevent overfitting of the input data (input layer network), noise is added, so as to enhance the generalization ability of the model.
As shown in Fig. 2, x is the original input data, and the denoising autoencoder sets the value of the input layer node to 0 with a certain probability, so as to get the model input xˆ with noise. This is similar to dropout, except that dropout sets the neurons in the hidden layer to 0. By calculating y and z with the corrupted data x′ and iterating errors with z and the original x, the network learns the corrupted data.

The flowchart of denoising autoencoder.
Through the comparison with non-corrupted data training, the weight noise of corrupted data is relatively small. This is because the input noise is accidentally removed, and the corrupted data alleviates the generation gap between training data and test data to a certain extent. Because part of the data is removed, the corrupted data is close to the test data to a certain extent.
Stacked Denoising Autoencoder (SDAE)
The idea of SDAE is to stack multiple DAEs together to form a deep architecture [24]. Noise is added to the input when training the model. A SDAE with two hidden layers is shown in Fig. 3.

A SDAE with two hidden layers.
Each encoding layer carries out unsupervised training separately. The training objective is to minimize the error between input (input is the hidden output of the previous layer) and reconstruction results. The output of layer K is obtained through forward propagation, and then layer K + 1 is trained with the output of layer K as the input.
Once SDAE training is completed, its high-level features are used as input of traditional supervised algorithms. A layer of logistic regression layer can be added at the top level, and then the network can be fine-tuned with labeled data.
Gradient Boosting Decision Tree (GBDT)
GBDT is one of the best algorithms to fit the real distribution in traditional machine learning algorithms. Its effect is good and it is used for classification and regression.
GBDT uses multiple iterations, and each iteration produces a weak classifier. Each classifier is trained on the basis of the residual of the previous one. The requirement for weak classifiers is usually simple enough with low variance and high deviation, because the training process is to improve the accuracy of the final classifier by reducing the deviation. The weak classifier will generally choose CART (classification and regression tree). Because of the above high deviation and simple requirement, the depth of each classification regression tree will not be very deep. The final total classifier is the sum of weighted weak classifiers obtained from each round of training (that is the additive model).
Evaluation Criteria
In this study, we classify protein and lncRNA pairs to be interacting or not. We follow the widely used evaluation measure by means of the classification accuracy, precision, sensitivity, specificity and MCC defined respectively as follows:
where TP, TN, FP, FN represents true positive, true negative, false positive, and false negative, respectively. To guarantee unbiased comparison, the testing and training datasets do not overlap with each other.
3 Results and Discussion
3.1 Results
In this study, PLRPI method is tested on ATH948 and ZEA22133 datasets which are the interactions between lncRNA and protein. The test results are shown in Table 2.
Through the experimental results, we find that our method not only has high accuracy, but also has excellent sensitivity and precision. This indicates that PLRPI has a strong ability to recognize negative samples, and the proportion of actual positive set samples in the predicted positive set is large. Although deep learning models generally require enough data as support, the larger amount of data, does not yield higher accuracy. The data of ZEA22133 is more, however, its accuracy is not as good as that of ATH948.
3.2 Comparing with Other Methods
We compared PLRPI with other sequence-based methods IPMiner [23], RPISeq [19] and lncPro [25] on our datasets. In study [19], the authors proposed RPISeq-RF and RPISeq-SVM for predicting RNA-protein interaction, and RPISeq-RF performed better than RPISeq-SVM on most datasets. Accordingly, here we only compared PLRPI with RPISeq-RF. As shown in Table 3, on data ATH488 and ZEA22133, PLRPI achieved the best performance. On dataset ATH488 it increased the accuracy with 10% over IPMiner. Compared with other methods, it obtained the best performance in other indexes with a little advantage over IPMiner, RPISeq-RF and lncPro. On dataset ZEA22133, PLRPI achieved a prediction accuracy of 82.6% with an increase of about 20% over other methods. It achieved a precision of 99.9% and a specificity of 99.6% with an increase of about 50% over other methods. This shows that our model performs well in plant lncRNA-protein interactions datasets, and can effectively extract advanced features and make predictions.
PLRPI outperforms other models on ATH948 and ZEA22133 datasets is because it uses GBDT as a classifier. For GBDT, trees are not a multi-training average relationship. They are interrelated, hierarchical, and the variance must be relatively large. However, because its learning ability is relatively strong, its deviation is very small, and the more trees there are, the stronger the learning ability and the smaller the deviation. Thus, as long as the number of trees for learning is enough, the predicted mean will be infinitely close to the target.
3.3 Testing the Robustness of PLRPI
To test the robustness of PLRPI, we also compared it with other sequence-based methods on other published ncRNA-protein and RNA-protein datasets. On dataset RPI2241 and RPI369, the proposed method achieved higher performance than the other methods. This shows that our method has strong robustness (Table 4).
On dataset RPI488 and RPI1807, PLRPI has not achieved the best performance but its indicators are almost the same as other methods. The reason is that the datasets are mixed with samples of different organisms, and our model is better at dealing with the plant lncRNA with poor homology, that is, our single species dataset.
PLRPI achieves good results on public datasets, mainly because of the use of stacked denoising autoencoder. When the amount of training data is small, if we use the traditional autoencoder to build the learning network, after passing the first few layers, the error is extremely small. In addition, the training becomes invalid, and the learning speed is slow. SDAE first performs unsupervised pre-training on each single hidden layer of the denoising autoencoder, then stacks them, and finally performs overall fine-tuning training to avoid the above problems and obtain better results.
In the process of training, the early stop method is used, which means that training is stopped when the performance of the model begins to decline on the verification set, thus avoiding the problem of over-fitting caused by continued training. PLRPI stops training when the generalization loss exceeds the threshold, which reduces the impact of over-fitting and save time. To further reduce the impact of over-fitting, we set dropout to 0.5 [26], which is a common setting.
It can be found that PLRPI is not strict with the requirement of data quantity. From hundreds to tens of thousands of sequences, the performance is excellent, but if the number of interaction between lncRNA and protein is large and the number of their respective sequences is relatively small (which is common in plant data), other general models do not perform well, and our model still maintains a good performance. This proves that PLRPI can adapt well to the data of plant lncRNA-protein interaction and obtain higher performance.
4 Conclusion
In this study, we propose a computational method PLRPI based on stacked denoising autoencoder and gradient boosting decision tree to predict plant lncRNA-protein interactions. It achieved a better performance on our constructed lncRNA-protein datasets ATH948 and ZEA22133. The comprehensive experimental results of other previously published datasets also show the effectiveness of PLRPI. In dataset ZEA22133, it improves the performance of the model by about 20% compared with other existing sequence-based methods. The results show that stacked denoising autoencoder extracts discriminant high-level features, which is very important for building deep learning model. The high-level features are the features automatically learned from multiple layers of neural network. PLRPI has shown good performance in plant lncRNA-protein, which is better than other advanced methods. In future work, we will apply different methods for sequence information of lncRNA and protein such as OPT, PSSM, One-hot, and adjust the network structure according to different datasets. We hope that we can use this model to construct network for plant lncRNAs and proteins, which can be used to infer the functions of plant lncRNAs.
References
Okazaki, Y., Furuno, M., Kasukawa, T., Adachi, J., Bono, H., Kondo, S., et al.: Analysis of the mouse transcriptome based on functional annotation of 60,770 full-length cDNAs. Nature 420(6915), 563–573 (2002)
Chen, Y., Varani, G.: Protein families and RNA recognition. FEBS J. 272(9), 2088–2097 (2005)
Cooper, T.A., Wan, L., Dreyfuss, G.: RNA and disease. Cell 136(4), 777–793 (2012)
Lukong, K.E., Chang, K.W., Khandjian, E.W., Richard, S.: RNA-binding proteins in human genetic disease. Trends Genet. 24(8), 416–425 (2008)
Chen, X., Sun, Y.Z., Guan, N.N., et al.: Computational models for lncRNA function prediction and functional similarity calculation. Brief. Funct. Genomics 18(1), 58–82 (2018)
Lunde, B.M., Moore, C., Varani, G.: RNA-binding proteins: modular design for efficient function. Nat. Rev. Mol. Cell Biol. 8(6), 479–490 (2007)
Zhang, L., Zhang, C., Gao, R., Yang, R., Song, Q.: Prediction of aptamer-protein interacting pairs using an ensemble classifier in combination with various protein sequence attributes. BMC Bioinform. 17(1), 225–238 (2016)
Gawronski, A.R., Uhl, M., Zhang, Y., et al.: MechRNA: prediction of lncRNA mechanisms from RNA-RNA and RNA-protein interactions. Bioinformatics 34(18), 3101–3110 (2018)
Bengio, Y., Courville, A., Vincent, P.: Representation learning: a review and new perspectives. IEEE Trans. Pattern Anal. Mach. Intell. 35(8), 1798–1828 (2012)
Hinton, G.E., Salakhutdinov, R.R.: Reducing the dimensionality of data with neural networks. Science 313, 504–507 (2006)
Lecun, Y., Bengio, Y., Hinton, G.: Deep learning. Nature 521(7553), 436–444 (2015)
Litjens, G., Kooi, T., Bejnordi, B.E., et al.: A survey on deep learning in medical image analysis. Med. Image Anal. 42(9), 60–88 (2017)
Deng, L., Yu, D.: Deep learning: methods and applications. Found. Trends® Sig. Process. 7(3), 197–387 (2014)
Alipanahi, B., Delong, A., Weirauch, M.T., Frey, B.J.: Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat. Biotechnol. 33(8), 831–838 (2015)
Zhou, J., Troyanskaya, O.G.: Predicting effects of noncoding variants with deep learning–based sequence model. Nat. Methods 12(10), 931–934 (2015)
Leung, M.K.K., Xiong, H.Y., Lee, L.J., Frey, B.J.: Deep learning of the tissue-regulated splicing code. Bioinformatics 30(12), i121–i129 (2014)
Ray, D., Kazan, H., Cook, K.B., Weirauch, M.T., et al.: A compendium of RNA-binding motifs for decoding gene regulation. Nature 499(7457), 172–177 (2013)
Cook, K.B., Hughes, T.R., Morris, Q.D.: High-throughput characterization of protein–RNA interactions. Brief. Funct. Genomics 14(1), 74–89 (2015)
Muppirala, U.K., Honavar, V.G., Dobbs, D.: Predicting RNA-protein interactions using only sequence information. BMC Bioinform. 12(1), 489–500 (2011)
Pan, X.Y., Zhang, Y.N., Shen, H.B.: Large-scale prediction of human protein-protein interactions from amino acid sequence based on latent topic features. J. Proteome Res. 9(10), 4992–5001 (2010)
Huang, Y., Niu, B., Gao, Y., Fu, L., Li, W.: CD-HIT Suite: a web server for clustering and comparing biological sequences. Bioinformatics 26(5), 680–682 (2010)
Suresh, V., Liu, L., Adjeroh, D., Zhou, X.: RPI-Pred: predicting ncRNA-protein interaction using sequence and structural information. Nucleic Acids Res. 43(3), 1370–1379 (2015)
Pan, X., Fan, Y.X., Yan, J., Shen, H.B.: IPMiner: hidden ncRNA-protein interaction sequential pattern mining with stacked autoencoder for accurate computational prediction. BMC Genom. 17(1), 582–596 (2016)
Vincent, P., Larochelle, H., Lajoie, I., Bengio, Y., Manzagol, P.A.: Stacked denoising autoencoders: learning useful representations in a deep network with a local denoising criterion. J. Mach. Learn. Res. 11(12), 3371–3408 (2010)
Lu, Q., Ren, S., Lu, M., Zhang, Y., Zhu, D., Zhang, X., et al.: Computational prediction of associations between long non-coding RNAs and proteins. BMC Genom. 14(1), 651–661 (2013)
Dahl, G.E., Sainath, T.N., Hinton, G.E.: Improving deep neural networks for LVCSR using rectified linear units and dropout. In: International Conference on Acoustics, Speech and Signal Processing, pp. 8609–8613. IEEE, Vancouver (2013)
Acknowledgment
The current study was supported by the National Natural Science Foundation of China (Nos. 61872055 and 31872116).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this paper

Cite this paper
Zhou, H., Luan, Y., Wekesa, J.S., Meng, J. (2019). Prediction of Plant lncRNA-Protein Interactions Using Sequence Information Based on Deep Learning. In: Huang, DS., Huang, ZK., Hussain, A. (eds) Intelligent Computing Methodologies. ICIC 2019. Lecture Notes in Computer Science(), vol 11645. Springer, Cham. https://doi.org/10.1007/978-3-030-26766-7_33
Download citation
DOI: https://doi.org/10.1007/978-3-030-26766-7_33
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-26765-0
Online ISBN: 978-3-030-26766-7
eBook Packages: Computer ScienceComputer Science (R0)