
Abstract
Cytoplasmic male sterility (CMS) is a maternally inherited trait resulting in failure to produce functional pollen and is widely used in the production of hybrid seed. Improper RNA editing is implicated as the molecular basis for some CMS systems. However, the mechanism of CMS in cotton is unknown. This study compared RNA editing events in eight mitochondrial genes (atp1, 4, 6, 8, 9, and cox1, 2, 3) among three lines (maintainer B, CMS A, and restorer R). These events were quantified by ultra-deep sequencing of mitochondrial transcripts and sequencing of cloned versions of these genes as cDNAs. A comparison of genomic PCR and RT-PCR products detected 72 editing sites in coding sequences in the eight genes and four partial editing sites in the 3′-untranslated region of atp6. The most frequent alteration (61.4 %) resulted in changes of hydrophilic amino acids to hydrophobic amino acids and the most common alteration was proline (P) to leucine (L) (26.7 %). In atp6, RNA editing created a stop codon from a glutamine in the genomic sequence. Statistical analysis of the frequencies of RNA editing events detected differences between mtDNA genes, but no differences between cotton cytoplasms that could account for the CMS phenotype or restoration. This study represents the first work to use next-generation sequencing to identify RNA editing positions and efficiency, and possible association with CMS and restoration in plants.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
One of the major post-transcriptional processes in plant mitochondria (mt) is RNA editing, i.e., predominantly cytidine (C) to uridine (U) modification. This process normally involves several hundred unique editing events of C-to-U conversions (Gualberto et al. 1990; Schuster et al. 1990; Salazar et al. 1991; Okuda et al. 2010) throughout the mt transcriptome to produce the evolutionary conserved functional protein sequences (Araya et al. 1992; Wei et al. 2008). RNA editing in higher plants is mainly observed in the coding region of transcripts (Hanson et al. 1996). The major editing positions are in the 1st and 2nd sites of a codon, leading to amino acid codon conversions to increase the number of hydrophobic amino acids in these mitochondrial proteins (Giegé and Brennicke 1999). RNA editing even occurs in intron or intergenic regions, but these events are much less frequent (Castandet et al. 2010). Several RNA editing sites in non-coding regions were recognized as critical factors for formation of secondary and tertiary structures of introns for splicing (Castandet et al. 2010). Alternatively, although partially edited silent sites have no effect on protein sequences, these sites may be one of the final steps in mRNA maturation (Mower and Palmer 2006).
RNA editing of mRNA in mitochondria of many different angiosperms has been reported (Covello and Gray 1989; Gualberto et al. 1989; Hiesel et al. 1989; Lamattina et al. 1989). RNA editing is a progressive process, although most of the sites are edited efficiently (Grewe et al. 2011). RNA editing sites are therefore often classified as “full editing” defined as ≥80 % of recovered sequences contain the converted sequence, or “partial editing” defined as <80 % of recovered sequences contain the converted sequence (Mower and Palmer 2006).
Numerous, mature mRNA sequences significantly differ from the genomic sequences encoding them; further, the predicted amino acid sequences of these genes in the post-edited transcripts have greater sequence similarity to the evolutionarily conserved sequences for these mt genes than the deduced genomic amino acid sequence (Schuster et al. 1991; Binder et al. 1992). In addition to amino acid conversions, RNA editing can also create start or stop codons (Bégu et al. 1990; Chapdelaine and Bonen 1991).
Occasionally, RNA editing results in the production of mature proteins that are completely distinctive in size, amino acid composition, and function from the proteins deduced to be encoded by the genomic DNA sequence (Covello and Gray 1990; Das et al. 2010). RNA editing predominantly occurs in coding regions of mRNA and the number of the editing sites differs among different tissues, ecotypes, or genes (Bentolila et al. 2008). A handful of RNA editing sites in non-coding regions, especially in introns, were reported, although their particular roles and functions are not yet known (Bonen 2008). Several mRNAs in 5′- and 3′-UTR, and intron regions are also reported to possess RNA editing sites. For example, the 5′-UTR of psbF in the chloroplast genome of Ginkgo biloba (Kudla and Bock 1999) and the 5′-UTR of maize and rice ndhG mRNAs (Corneille et al. 2000) were edited. In general, RNA editing outside of coding regions is observed less frequently than that in coding regions (Knoop et al. 1991; Binder et al. 1992).
Several studies of association between RNA editing and cytoplasmic male sterility (CMS) have been published. Howad and Kempken (1997) demonstrated that RNA editing in mt atp6 was dramatically diminished in the cytoplasmic A3Tx398 male sterile line of Sorghum bicolor in anther, although atp6 transcripts in other plant species such as wheat and several transcripts in plastid in S. bicolor indicated normal RNA editing. Their study clearly showed that the occurrence of RNA editing is highly specific to organelles and plant species. Furthermore, RNA editing efficiency of atp6 transcripts was increased in the male fertility restored line.
In CMS-S of maize, a sequence similarity between the novel chimeric gene orf77 associated with the CMS trait and atp9 was observed (Gallagher et al. 2002). Full editing of atp9 in all positions was detected in CMS-S microspores, whereas orf77 nucleotides corresponding to edited nucleotides in atp9 were either not edited or partially edited in the same CMS-S microspores. Gallagher et al. (2002) stated that the editing sites in chimeric orf77 could cause male sterility by compromising the expression of atp9.
Jiang et al. (2011) cloned and sequenced atp9 from a CMS line NJCMS2A and its maintainer line NJCMS2B in soybeans. They detected two RNA editing sites in atp9 in the maintainer line, which resulted in the conversion of the codon for serine into the codon for leucine. However, no RNA editing was identified in atp9 transcripts in the CMS line. In rice, RNA editing in the atp9 transcript in the maintainer line Yingxiang B caused a conversion of arginine to a stop codon, producing a conserved size protein for this gene product. However, in the CMS line, Yingxiang A with cytoplasm from Yunnan purple rice, no RNA editing was detected at this position, so that the absence of a stop codon resulted in production of a non-functional ATP9 subunit (Wei et al. 2008).
In wheat, comparisons between euplasmic and alloplasmic CMS lines have detected RNA editing of atp6 at 12 codons in the fertile euplasmic Triticum timopheevi, while 17 % of the clones in the CMS lines were only partially edited. In atp9 transcripts, full RNA editing occurred at eight codons in different tissues such as embryos, roots, shoots, and anthers in the euplasmic wheat lines, while only 19 % of the clones from the CMS lines were partially edited (Kurek et al. 1997). Thus, differences in RNA editing efficiency were observed between CMS plants and their corresponding male fertile plants with different nuclear backgrounds.
The reports cited above suggest a correlation between RNA editing and CMS in the mitochondria of higher plants. However, in previous studies, RNA editing efficiency was assessed based on traditional cloning and only limited numbers of clones were sequenced that did not allow identification of rare partial RNA editing sites. RNA editing in mtDNA genes in cotton (Gossypium) is currently unknown. Based on RFLP analysis, Wang et al. (2010) showed that atp1 and atp6 in CMS-D8 and these two genes together with cox1 and cox2 in CMS-D2 might be involved in CMS in cotton. The objectives of the present study were to detect full and partial RNA editing positions in selected mtDNA genes in cotton and to study their possible relationship with CMS in the CMS-D8 system using both the traditional cloning and the next-generation sequencing methods. Eight mitochondrial genes, atp1, 4, 6, 8 and 9, and cox 1, 2 and 3, were selected for the analysis of RNA editing events based on their associations with CMS in cotton (Wang et al. 2010) and other plant species (Schnable and Wise 1998; Hanson and Bentolila 2004; Chase 2007). The results raise interesting questions about the differences in RNA editing in cotton lines and illustrate the power of next-generation ultra-deep sequencing for these types of analyses.
Materials and methods
Plant materials
Cotton plants from the three lines, maintainer B line (8518), CMS A line (D8CMS8518) and restorer R line (D8R8518) (Zhang and Stewart 2001) were grown in the greenhouse, New Mexico State University, Las Cruces, NM, USA. Young leaves were harvested and pooled for both DNA and RNA extraction. Information on the nuclear and cytoplasmic genotypes and phenotypes of the plant materials used in this study is listed in Table 1. The maintainer line is fertile with normal fertile Upland cotton AD1 cytoplasm and possesses homozygous recessive non-functional fertility restorer genotype (rf 2 rf 2 ). The CMS line is sterile with the sterile CMS-D8 cytoplasm and possesses homozygous recessive fertility restorer genotype (rf 2 rf 2 ). The restorer line is fertile with the sterile CMS-D8 cytoplasm but possesses homozygous dominant fertility restorer genotype (Rf 2 Rf 2 ) to restore the male fertility of the CMS plants.
Total DNA and RNA extraction
Total DNA was isolated using a quick CTAB method (Zhang and JMcD 2000) from at least five folded or newly unfolded young leaves in each genotype. Both CMS-D8 and restorer Rf2 genes express in both sporophytic and gametophytic tissues and it is extremely difficult to harvest developing sufficient anthers for RNA isolation. Total RNA from young leaves was isolated using the hot borate method (Wan and Wilkins 1994; Wilkins and Smart 1996) with modifications in micro-preparation, convenience and quickness (Pang et al. 2011).
cDNA synthesis, PCR, RT-PCR, cloning, and sequencing
RNA (45 μg) was digested with 9 units of RNase-free DNase I (New England Biolabs, Ipswich, MA, USA) at 37 °C for 10 min. Total RNA was extracted with RNeasy MinElute kit (Qiagen, Hilden, Germany); cDNA synthesis was then performed with 200 units of Superscript III RT (Invitrogen, Carlsbad, CA, USA) and 250 ng of random hexamers (Qiagen, Hilden, Germany) as primers. The primer sequences for both PCR and RT-PCR amplifications of mt genes atp1, 4, 6, 8 and 9, and cox 1, 2 and 3 genes are listed in Table 2. High fidelity amplification reactions for both PCR and RT-PCR were performed in 25 μL: 25 ng of template DNA, 1 unit of AccuPrime Taq high fidelity DNA polymerase (Invitrogen, Carlsbad, CA, USA), 0.2 μmol of each primer, 10× AccPrimer PCR buffer mixture I for cDNA or II for genomic DNA templates, as per the manufacturer’s protocol (Invitrogen, Carlsbad, CA, USA). The PCR cycling program was: 94 °C for 1 min and 40 s, 50–55 °C for 30 s, 68 °C for 1 min, and a final extension at 68 °C for 7 min. Agarose gel resolved amplicons were photographed under ultraviolet light using Gel Logic 200 (CareStream Health, Inc., Rochester, NY, USA), the amplicons were purified using the Agencourt® AMPure® XP kit (Beckman Coulter, Brea, California, USA). RT-PCR products were stored for the subsequent ultra-deep sequencing using an Ion Torrent Personal Genome Machine (PGM™) sequencer (see below). The amplicons were cloned with the pGEM-T Easy Vector as recommended by the manufacturer (Promega, Madison, WI, USA). Sequencing of the cloned products was performed by ElimBiopharm (Hayward, CA, USA; http://www.elimbio.com/dna_sequencing.htm). The thermal asymmetric interlaced (TAIL) PCR method (Mazers et al. 1991; Liu et al. 1995; Liu and Whittier 1995; Terauchi and Kahl 2000; Liu and Chen 2007; Tan and Singh 2011) was utilized for full sequencing of atp6. The percentage of editing efficiency was determined by sequencing an average of 15 (8–30) cDNA clones from each of the three genotypes (maintainer, CMS line, and restorer) for each gene and utilizing the multiple sequence alignment software termed ClustalW (http://www.ebi.ac.uk/Tools/msa/clustalw2/). For consideration as a site of RNA editing, the threshold was set so that at least two C-to-U conversions at a position should be detectable to be regarded as an authentic RNA editing (Grewe et al. 2011). Therefore, single C-to-U conversion at one position was not recorded as RNA editing due to the possibility of PCR amplification and DNA sequencing errors. Chi-square tests were performed to compare differences in RNA editing efficiencies among the three genotypes.
cDNA library preparation
For library preparation, two independent samples of RT-PCR products for eight mitochondrial genes in each genotype were pooled in an equimolar ratio to produce the pooled concentration of 30 nM per genotype.
Non-barcoded cDNA library was generated using the Ion Xpress™ Plus Fragment Library kit (Life Technologies, Foster City, CA) as described by the manufacturer’s protocol. Equimolar pooled RT-PCR amplicons (100 ng) per genotype were prepared to be enzymatically fragmented by incubation at 37 °C for 25 min using Ion Shear™ Plus Reagents, and subsequently purified using the Agencourt® AMPure® XP kit (Beckman Coulter, Brea, CA, USA). The fragmented amplicon sizes were assessed using Agilent High Sensitivity DNA kit (Agilent Technologies, Santa Clara, CA, USA) with Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). Adapter ligation and nick repair to the fragmented amplicons were performed and followed by the purification of adapter ligated amplicons employing the Agencourt® AMPure® XP kit. The final cDNA library was eluted in 20 μL of low TE buffer and stored at −20 °C. DNA concentration was determined utilizing the Qubit® dsDNA HS Assay kit (Life technologies, Foster City, CA, USA) with the Qubit® 2.0 Fluorometer (Life technologies, Foster City, CA, USA). The final fragment size of the adapter ligated libraries for each genotype was evaluated using Agilent High Sensitivity DNA kit with Agilent 2100 Bioanalyzer.
Emulsion PCR and ultra-deep sequencing
Emulsion PCR was performed in a 1-mL reaction volume to clonally amplify cDNA libraries attached onto Ion Sphere Particles (ISP) for sequencing using the Ion Template Preparation kit (Life Technologies, Foster City, CA, USA). Briefly, PCR master mix, ISP, and diluted library template were combined thoroughly. Emulsion oil was added to an Ion Template Preparation tube. All components were mixed together using the Ultra-Turrax® Tube Drive (Life Technologies, Foster City, CA, USA) to generate the emulsion. The mixed emulsion was then transferred to a 96-well PCR plate and amplified using a BIO-RAD T100 Thermal Cycler (BIO-RAD, Hercules, CA, USA) for 45 cycles. ISP was recovered using reagents supplied in the Ion Xpress Template kit (Life Technologies, Foster City, CA, USA) according to manufacturer’s protocol. The cDNA libraries were washed with a recovery solution and then enriched by eliminating any ISP without cDNA fragments. The Ion Torrent PGM™ sequencer equipped at the University of New Mexico Health Sciences Center (Albuquerque, NM, USA) was operated for sequencing all clonally amplified cDNA libraries with Ion Torrent 318 chips. Sequence raw data were submitted to the Sequence Read Archive (http://trace.ncbi.nlm.nih.gov/Traces/sra/) under SRA accession number SRA063971.
Ultra-deep sequence data analysis
Transcriptome sequencing data were analyzed using CLC GenomicWorkbench 5.5.1 (Cambridge, MA, USA). Quality control for elimination of undesirable fragment reads was arbitrarily adjusted using mapping parameters, such as length fraction (0.8) and similarity (0.9) according to the CLC GenomicWorkbench 5.5.1. Sequence reads were mapped back to the cotton mitochondrial genomic DNA sequences, which were retrieved from NCBI GenBank (KC149532–KC149552) (Suzuki et al. 2013). The number of sequence reads for each gene was compared among the three genotypes using a t test. Each C-to-U RNA editing position, total read count and coverage depth were identified by CLC GenomicWorkbench 5.5.1, and the baseline threshold for consideration of C-to-U RNA editing was arbitrarily set as above 3 % of thymine detection at specific positions in at least one of the genotypes. Therefore, less than 3 % of thymine in any genotype at a particular position was not recorded as C-to-U RNA editing site due to the possibility of PCR amplification and sequencing error.
The fraction between C and U was calculated by the numbers of identified C and T at each RNA editing position. The calculated ratio between C and U at each RNA editing site was converted into percentages. An unpaired, two-tailed t test was conducted to determine if RNA editing efficiency differences were significant on the three distinct comparisons between the maintainer B line and the CMS A line and between maintainer B and restorer R lines due to the cytoplasmic effect, and between CMS A and restorer R lines due to the restorer gene’s effect. P values below 0.05 were considered statistically significant.
Results
Transcript abundance of mtDNA genes
The gene expression between the three genotypes was quantified for each gene based on sequencing reads from ultra-deep sequencing (Table 3). Sequence reads for each gene varied from 17,772–26,525 for atp9 to 400,589–528,128 for atp6. Expression of the eight genes, with the exception of atp1, was the same in all three genotypes. For atp1, however, both A and R lines, with the CMS-D8 cytoplasm, had similar sequence reads (273,988 and 254,990, respectively), which were significantly (P < 0.10 or 0.05) lower (with 41.4 and 45.7 % reduction, respectively) than that (469,211) in B line with the AD1 cytoplasm. Expression of atp1 in the CMS line was significantly decreased and the restorer gene Rf 2 did not mitigate the negative effect of the CMS-D8 cytoplasm in the Upland cotton nuclear background. As shown below, the difference due to the introduction of the D8 cytoplasm into Upland cotton was not associated with RNA editing in atp1 gene.
Comparison between traditional cloning and ultra-deep sequencing for detection of RNA editing events
To obtain information on efficiency and distribution of RNA editing events in mt genes in cotton, RNA editing sites were determined for eight genes, including atp1, 4, 6, 8 and 9 and cox 1, 2 and 3. A total of 76 RNA editing sites were detected from either the traditional cloning or ultra-deep sequencing (supplementary Table 1). For all the full RNA editing sites in all the mtRNA sequenced except for atp6, the two methods yielded the same results. This high consistency between the two sequencing methods supports a sampling method of sequencing 15 clones to detect full RNA editing sites. Specific analysis of atp6 editing events is presented below.
For partial editing, 11 sites were commonly detected by both sequencing methods. However, the traditional cloning detected five different sites, while the ultra-deep sequencing detected eight additional sites. The results indicated that the ultra-deep sequencing is more effective in detecting partial RNA editing sites and the traditional cloning method is complementary. The reason why the ultra-deep sequencing did not detect the five RNA editing sites was unknown, given its extremely high genome coverage. However, the possibility that the five sites detected by the traditional cloning were sequencing errors should be minimal because the C-to-U conversion was detected in each of the five sites in at least two cDNA clones (see “Materials and methods”).
RNA editing events identified in cotton
RNA editing events were detected in each of the eight genes. Of a total of 76 editing sites found across the eight genes, 45 were full editing, 27 were partial editing in coding regions, and 4 were partial editing in the 3′-UTR of atp6 (Table 4). The editing efficiency of the eight genes was determined by the ultra-deep sequencing at an average coverage of 59,000 (range 15,000–113,000) for each mtDNA gene for each cotton genotype to obtain precise estimates of editing efficiency (supplementary Table 1).
The general characteristics of alteration of the biochemical predispositions of the edited sites of codons in the coding region of the mitochondrial respiratory transport chain in cotton were investigated by combining the data from the three genotypes. The most frequent alteration (61.4 %) of codons observed in the set of full RNA editing events resulted in the change of hydrophilic amino acids to hydrophobic amino acids (Fig. 1). The second most frequent alteration changed an amino acid but both were hydrophobic amino acids (31.8 %). A small percentage of alterations (6.8 %) resulted in hydrophilic amino acids replacing hydrophobic amino acids. No changes between hydrophilic amino acids were identified (Fig. 1). This increase in hydrophobic amino acids following RNA editing has been described in other plant mitochondrial systems (Yura and Go 2008) and is expected, given that these mtDNA genes encode membrane-bound and membrane-associated proteins.

Distribution of editing events across changes that resulted in different hydrophilic/hydrophobic amino acid replacements; full (filled bars) or partial (open bars) editing events; n = 45 for full editing, n = 22 for partial editing
Interestingly, within the set of partially edited sites, changes replacing between hydrophobic amino acids were the most abundant (54.5 %), followed by changes replacing hydrophilic amino acids with hydrophobic amino acids (36.4 %). Again, editing events replacing hydrophobic amino acids with hydrophilic amino acids were the least frequent (9.1 %). All of the partial editing events at numerous silent editing sites may have characterized an inefficiency of editing at these sites.
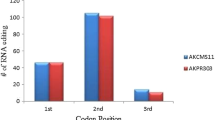
The distribution of editing events by position within the codon, 1st, 2nd, and 3rd positions, was counted to detect the positional tendency of RNA editing (Fig. 2). In full editing events, 97.8 % of those changes occurred at the 1st and 2nd position of the codons, while 77.3 % of partial editing events occurred at the 1st and 2nd position, with an increased frequency of RNA editing in the 3rd position (22.7 %). Editing events at the 3rd position in the codons usually result in silent editing due to the wobble degeneracy of this position in the codons.

Distribution of editing events at each position with a codon for full (filled bars) or partial (open bars) editing events; n = 45 for full editing, n = 22 for partial editing
The specific amino acid changes as a result of editing were compared between those sites classified as full editing and partial editing (Fig. 3). In full RNA editing, the most frequent change was proline (P) > leucine (L) (26.7 %), followed by serine (S) > leucine (L) (22.2 %), serine (S) > phenylalanine (F) (15.6 %), and arginine (R) > tryptophan (W) (11.1 %). In partial editing sites S > L (22.7 %) and P > L (20 %) were the most frequent, followed by P > S (9.1 %), L > L (9.1 %), alanine (A) to valine (V) (9.1 %), isoleucine (I) > I (9.1 %) and P > P (9.1 %) (Fig. 3). Full editing caused only 2.2 % of silent RNA editing (i.e., the encoded amino acid was unchanged), whereas partial editing gave rise to 31.8 % of silent editing.

Amino acid conversions as a result of full (filled bars) or partial (open bars) editing events; n = 45 for full editing, n = 22 for partial editing
Comparison of RNA editing frequencies in mtDNA genes in cotton and three other dicot plants
The number of RNA editing sites in each of the eight mtDNA genes reported for Beta vulgaris subsp. vulgaris (NC_002511), Brassica napus (NC_008285) and Arabidopsis thaliana (NC_001284) was compared with the number of editing sites in cotton detected in this study (Table 5). The number of RNA editing sites was relatively conserved across this set of plant taxa, for six of the eight genes examined: cox2, cox3, atp1, atp4, atp8, and atp9.
In cotton, atp6 and cox1 appeared to have more RNA editing sites than those found in these genes of other plant species (Table 5). In cox1, there are 15 edited sites in cotton, which is considerably higher than the rare editing events in this gene in B. napus (1), A. thaliana (0) and B. vulgaris (0). In atp6, B. vulgaris and cotton have substantially higher numbers of RNA editing sites, 12 and 15, respectively, compared with B. napus and A. thaliana, each of which possesses only one RNA editing site. In atp9, B.napus, A. thaliana and B. vulgaris have four or five RNA editing sites, which are higher than the single RNA editing site in cotton (Table 5). The number of edited sites in atp9 in cotton could be underestimated since only a partial genomic sequence was available for this gene (170 bp), considerably shorter than the full-length versions of this gene of other plant species, in B. napus (225 bp), A. thaliana (258 bp) and B. vulgaris (225 bp). A comparison of the specific location of the edited sites and specific amino acid changes in these eight genes was conducted (supplementary Figs. 1–8).
Comparison of RNA editing events in eight mtDNA genes in CMS, maintainer and restorer cytoplasms in cotton
The ultra-deep sequencing approach had sufficient sample size for editing events for each gene in the three genotypes, to allow statistical analysis to test for genotypic differences in RNA editing events. These results are provided and compared with the results of the traditional cloning approach (Table 4; supplementary Table 2). With the large sample set for statistical analysis, differences in RNA editing frequencies were detected between cytoplasms that were not observed in the traditional cloning approach, and further in most cases, these differences were very small values, i.e., 98.24 vs. 98.74 %. Therefore, we further refined the analysis such that RNA editing events needed to be both statistically significant and at biologically relevant rates for the site to be identified as a distinct RNA editing event associated with cytoplasmic genetic differences.
- atp1:
-
Five full editing (by both sequencing methods) and two partial editing (by ultra-deep sequencing) were detected and no statistical differences in RNA editing were detected between any of the cytoplasms, CMS A, maintainer B, or restorer R, at any of these sites
- atp4:
-
Six full editing and three (including one from ultra-deep sequencing) partial editing were identified. No biologically relevant, statistical differences in RNA editing were detected between any of the cytoplasms, CMS A, maintainer B, or restorer R, at any of these sites
- atp6:
-
One full editing of glutamine in the last codon of atp6 resulted in the alteration into a stop codon, producing 993 bp coding region in the maintainer line with AD1 cytoplasm and 1,002 bp coding region in the CMS and restorer lines with D8 cytoplasm (Tables 4, 5; supplementary Fig. 3). The length variation between AD1 and D8 cytoplasms in atp6 is ascribed to a nine nt insertion–deletion (InDel) sequence (AATTGTTTT) at the 59–67 bp positions of atp6, which is present in the CMS and restorer lines with the D8 cytoplasm but absent in the maintainer line with the AD1 cytoplasm
A total of 19 RNA editing sites including 4 in the 3′-UTR were detected for this gene. The ultra-deep sequencing detected 4 additional partial editing sites. In the intron region at the 1,333 nt cDNA position from the start codon, the CMS A line had significantly higher RNA editing efficiency than the restorer R line (35.19 vs. 21.53 %) based on ultra-deep sequencing and a similar trend was seen from the traditional cloning (Table 4). The editing efficiency differences may explain the outcome of the restorer gene’s effect. Even though numerical differences in RNA editing efficiency were noted at four sites (i.e., 29th, 34th, 166th and 184th amino acids from the start codon) among the three genotypes based on the traditional cloning, these differences were not statistically significant due perhaps to lower numbers of cDNA clones sequenced. The differences were also not confirmed by the ultra-deep sequencing.
At 179th/182th, 313th/316th (histidine to tyrosine), 315th/318th (serine to phenylalanine), and 331th/334th (glutamine to a stop codon) in maintainer B/CMS A lines (3 of which were full editing), RNA editing efficiency differences between B line with AD1 cytoplasm and A line with D8 cytoplasm were not detected based on the traditional cloning. However, at these four sites based on the ultra-deep sequencing, the maintainer B line had significantly lower RNA editing efficiencies than the CMS A line (53.80 vs. 59.58 %, 62.90 vs. 74.39 %, 51.70 vs. 67.50 %, and 78.47 vs. 89.68 %, respectively). A similar trend was also noted between maintainer B line and the restorer R line. Thus, the D8 cytoplasm appeared to have higher levels of partial RNA editing efficiency than the AD1 cytoplasm in these four positions. However, the restorer gene Rf 2 did not affect RNA editing because CMS line without Rf 2 and restorer line with Rf 2 gene in the D8 cytoplasm had similar RNA editing efficiencies.
- atp8:
-
Two partial RNA editing sites were identified by both sequencing methods. At the 1st (proline to serine) and the 2nd (proline to leucine) positions of the 13th amino acid from the 5′-primer end, the CMS line had significantly higher editing efficiencies than the R restorer line (33.51 vs. 27.44 %, and 28.56 vs. 21.29 %, respectively) (Table 4). The same was true for the difference between the B maintainer line and the R restorer line (25.58 vs. 21.29 %) at the 2nd position of the 13th amino acid. The ultra-deep sequencing results may indicate that the restorer gene could decrease RNA editing efficiency in these two positions. However, the trends were reversed based on the results from the traditional cloning. Therefore, differences in RNA editing among the three genotypes at these two positions were not biologically significant
- atp9:
-
One full editing site and 1 partial editing site were detected for this gene. There was no significant difference detected among the three genotypes based on the traditional cloning results. While the ultra-deep sequencing detected significantly lower RNA editing efficiency in the B maintainer than the R restorer line (98.47 vs. 98.91 %) (Table 4), the difference was numerically too small and not biologically relevant
- cox1:
-
Both sequencing methods detected 15 full editing sites and the traditional cloning also detected 1 partial editing site in the CMS line. No statistical differences were detected between AD1 (B line) and D8 cytoplasms (A or R line) or between CMS (A) without Rf 2 and restorer (R) line with Rf 2 in the D8 cytoplasm, except for the 181th amino acid position from the 5′-primer end. At this position, the CMS line had slightly lower but significant editing efficiency than the restorer line (97.16 vs. 97.44 %) based on ultra-deep sequencing. However, this difference was very small and not confirmed by the traditional cloning, and therefore did not have any biological significance
- cox2:
-
Both sequencing methods identified 13 full editing sites and 1 partial editing site and the traditional cloning further detected 3 more partial editing sites (Table 4). No biologically relevant, statistical differences in RNA editing were detected between any of the cytoplasms, CMS A, maintainer B, or restorer R, at any of these sites
However, notably, a consistent difference in RNA editing was detected at the 118th amino acid position (isoleucine remained unchanged due to silent editing) from the start codon (Table 4). The CMS line A had significantly higher editing efficiency than the R restorer line based on both the ultra-deep sequencing (16.64 vs. 1.29 %) and the traditional cloning methods (32.4 vs. 0 %).
- cox3:
-
Three full RNA editing and 2 partial editing sites were detected in this gene. The ultra-deep sequencing detected two differences at two sites (Table 4). At the 75th amino acid position from the 5′-primer end where isoleucine remained unchanged, the maintainer line had significantly lower partial editing efficiency than the restorer line (2.10 vs. 3.42 %). At the 89th amino acid position where serine was altered to phenylalanine, the maintainer B line also had significantly lower editing efficiency than the CMS A line (88.87 vs. 91.06 %) (Table 4). However, the differences were very small and the traditional cloning did not confirm the significant differences in these two sites. Therefore, the differences in these two editing sites were biologically insignificant
Discussion
Although associations between CMS phenotypes and RNA editing have been reported in a few CMS systems, the molecular mechanism of CMS and its correlation with RNA editing in cotton is unknown. This present study is the first report in cotton to identify RNA editing sites in mitochondrial genes responsible for ATP synthase subunits 1, 4, 6, 8, and 9, and cytochrome c oxidase subunits 1, 2, and 3 using an isogenic “three line” (A, B and R) system (Table 1). This is also the first report of the full-length sequences of atp6 and cox2 in the three cotton genotypes. Further, this study is one of the first reports to use an ultra-deep sequencing platform to identify RNA editing positions and efficiency, and possible association with effects from CMS cytoplasm or a restorer gene in higher plants. This approach has been used in grape to detect 401 mRNA editing sites in a single genotype (Picardi et al. 2010). Picardi et al. (2010) also detected 314 non-significant RNA editing sites, implying that many sites may be undiscovered in other plants using the traditional cloning method. In this present study, more RNA editing sites were detected in atp6 and cox1 genes than other three dicot species (Table 5). However, the partial RNA editing events in the eight mtDNA genes (1/3 of all detected C-to-U changes) in our study were much lower than that (76 %) detected by Picardi et al. (2010). Partial RNA editing sites may reflect true differential editing efficiencies in different positions or may be due to incompletion of the RNA editing process (therefore immature transcripts).
Our study showed that both the traditional cloning approach, using on average of 15 sequenced clones per gene, and the ultra-deep sequencing gave virtually identical results. RNA editing sites classified as full editing sites were detected by both methods and almost half of the partial editing sites were detected by both methods. We demonstrated that the ultra-deep sequencing approach detected more partial editing sites, whereas the traditional cloning detected a few new partial editing. There was only one significant discrepancy between the two methods. Four full RNA editing sites were detected in atp6 based on the traditional cloning, but these sites were scored as partially edited (53.8–78.5 %) based on the ultra-deep sequencing. The reason for this inconsistency is not known. Picardi et al. (2010) also showed minor discrepancies in detecting RNA editing sites using two next-generation sequencing platforms (Illumina vs. ABI SOLiD) and supported a combined approach to reduce false positives.
Based on RFLP analysis, Wang et al. (2010) indicated that atp1 and atp6 might be the candidate genes for CMS in the cotton CMS-D8 system used in this study. This present study has ruled out a possible association between CMS and RNA editing in atp1 or other seven mtDNA genes which are CMS causal genes in various plant species. However, whether the reduction in expression of atp1 in A and R lines is associated with CMS remains to be elucidated. In atp6, the glutamine was altered into the stop codon, resulting in the production of a 331-aa product in the B maintainer line and 334-aa product in CMS A and R restorer lines.
Most of the fertility restorer genes cloned and sequenced so far encode pentatricopeptide repeat (PPR) proteins (Hanson and Bentolila 2004; Chase 2007; Hu et al. 2012) and many other PPR proteins are required for RNA editing for mitochondrial or plastid transcripts (Fujii and Small 2011; Okuda et al. 2010; Sosso et al. 2012). However, the roles of PPR-coding restorer genes in RNA editing are currently unknown. The D8 cytoplasm together with the nuclear D8 genome functions normally in its original G. trilobum (i.e., D8 genome) species, giving rise to fertile pollen, while CMS-D8 cytoplasm, when transferred into another species Upland cotton nuclear background, causes male sterility. Unlike natural CMS mutations in other crops such as CMS-T and CMS-S in maize, the D8 mitochondrial DNA genes when transferred to Upland cotton were not changed, as demonstrated by RFLP analysis (Wang et al. 2010). However, certain mitochondrial transcripts in CMS-D8 may need species-specific PPR protein(s) for editing before being functional. We speculate that the Rf 2 gene from the D8 genome may serve this purpose for the restoration of male fertility in CMS-D8. The CMS-D8 and its restorer provide an excellent biological system for such studies. However, in this present study, based on both the traditional cloning and deep-sequencing methods, the restorer R line with Rf2 had significantly lower RNA editing efficiencies in a position of the intron of atp6 and a position (with a silent editing) in cox2 than the CMS A line without Rf2. The reduction in editing efficiencies in both positions by the restorer gene appeared to be unrelated to restoration of male fertility. The role of the restorer gene in RNA editing needs further studies.
Male sterility in the CMS-D8 occurs with no pollen production due to the failure of meiosis in the anthers, a typical of sporophytic CMS system. However, its fertility restoration by Rf2 is a gametophytic system because two pollen grains (sterile and fertile in an equal ratio) exist after meiosis in the restored F1 (CMS line × its restorer line) plants. Zhang et al. (2008) indicated that the CMS-associated gene(s) are also expressed in sporophytic issues and the Rf2 is also not an anther- or pollen-specific gene. Considering that both the CMS and restorer genes are expressed in both somatic and reproductive tissues and the difficulty in harvesting sufficient amount of anther tissues from young flower buds for RNA extraction, this study used leaf samples instead of immature anthers as the source for RNA editing analysis of candidate CMS-causal genes. This is a limitation in the present study. Picardi et al. (2010) were able to detect tissue-specific patterns of RNA editing (28 % of 401 C-to-U conversions) using RNA-Seq approach in grape stem, leaf, root and callus samples. If cotton RNA editing is similarly tissue specific and a role for RNA editing in the cotton CMS-D8 cytoplasm is possible, it will require mt transcriptome analyses of these genotypes using immature floral organs. Immature anthers from both CMS and fertile cotton plants may be necessary to identify differences in editing efficiency between these lines since the anther is organ with the dysfunctional development in cotton CMS.
References
Araya A, Domec C, Bégu D, Litvak S (1992) An in vitro system for the editing of ATP synthase subunit 9 mRNA using wheat mitochondrial extracts. Proc Natl Acad Sci USA 89:1040–1044
Bégu D, Graves PV, Domec C, Arselin G, Litvak S, Araya A (1990) RNA editing of wheat mitochondrial ATP synthase subunit 9: direct protein and cDNA sequencing. Plant Cell 2:1283–1290
Bentolila S, Elliott LE, Hanson MR (2008) Genetic architecture of mitochondrial editing in Arabidopsis thaliana. Genet 178:1693–1708
Binder S, Marchfelder A, Brennicke A, Wissinger B (1992) RNA editing in trans-splicing intron sequences of nad2 mRNAs in Oenothera. Mitochondria 267:7615–7623
Bonen L (2008) Cis- and trans-splicing of group II introns in plant mitochondria. Mitochondrion 8:26–34
Castandet B, Choury D, Begu D, Jordana X, Araya A (2010) Intron RNA editing is essential for splicing in plant mitochondria. Nucleic Acids Res 38:7112–7121
Chapdelaine Y, Bonen L (1991) The wheat mitochondrial gene for subunit I of the NADH dehydrogenase complex: a f/ww-splicing model for this gene-in-pieces. Cell 65:465–472
Chase CD (2007) Cytoplasmic male sterility: a window to the world of plant mitochondrial–nuclear interactions. Trends Genet 23:81–90
Corneille S, Lutz K, Maliga P (2000) Conservation of RNA editing between rice and maize plastids are most editing events dispensable? Mol Gen Genet 264:419–424
Covello PS, Gray MW (1989) RNA editing in plant mitochondria. Nature 341:662–666
Covello PS, Gray MW (1990) Differences in editing at homologous sites in messenger RNAs from angiosperm mitochondria. Nucleic Acids Res 18:5189–5196
Das S, Sen S, Chakraborty A, Chakraborti P, Maiti MK, Basu A, Basu D, Sen SK (2010) An unedited 1.1 kb mitochondrial orfB gene transcript in the wild abortive cytoplasmic male sterility (WA-CMS) system of Oryza sativa L. subsp. Indica. BMC Plant Biol 10:39
Fujii S, Small I (2011) The evolution of RNA editing and pentatricopeptide repeat genes. New Phytol 191:37–47
Gallagher LJ, Betz SK, Chase CD (2002) Mitochondrial RNA editing truncates a chimeric open reading frame associated with S male-sterility in maize. Curr Genet 42:179–184
Giegé P, Brennicke A (1999) RNA editing in Arabidopsis mitochondria effects 441 C to U changes in ORFs. Proc Natl Acad Sci USA 96:15324–15329
Grewe F, Herres S, Viehöver P, Polsakiewicz M, Weisshaar B, Knoop V (2011) A unique transcriptome: 1782 positions of RNA editing alter 1406 codon identities in mitochondrial mRNAs of the lycophyte Isoetes engelmannii. Nucleic Acids Res 39:2890–2902
Gualberto JM, Lamattina L, Bonnard G, Weil JH, Grienenberger JM (1989) RNA editing in wheat mitochondria results in the conservation of protein sequences. Nature 341:660–662
Gualberto JM, Weil JH, Grienenberger JM (1990) Editing of the wheat coxIII transcript: evidence for twelve C to U and one U to C conversions and for sequence similarities around editing sites. Nucleic Acids Res 18:3771–3776
Handa H (2003) The complete nucleotide sequence and RNA editing content of the mitochondrial genome of rapeseed (Brassica napus L.): comparative analysis of the mitochondrial genomes of rapeseed and Arabidopsis thaliana. Nucleic Acids Res 31:5907–5916
Hanson MR, Bentolila S (2004) Interactions of mitochondrial and nuclear genes that affect male gametophyte development. Plant Cell 16:S154–S169
Hanson MR, Sutton CA, Lu BW (1996) Plant organelle gene expression: altered by RNA editing. Trends Plant Sci 1:57–64
Hiesel R, Wissinger B, Schuster W, Brennicke A (1989) RNA editing in plant mitochondria. Science 246:1632–1634
Howad W, Kempken F (1997) Cell type-specific loss of atp6 RNA editing in cytoplasmic male sterile Sorghum bicolor. Proc Natl Acad Sci USA 94:11090–11095
Hu J, Wang K, Huang W, Liu G, Gao Y, Wang J, Huang Q, Ji Y, Qin X, Wan L, Zhu R, Li S, Yang D, Zhu Y (2012) The rice pentatricopeptide repeat protein RF5 restores fertility in Hong-Lian cytoplasmic male-sterile lines via a complex with the glycine-rich protein GRP162. Plant Cell 24:109–122
Jiang W, Yang S, Yu D, Gai J (2011) A comparative study of ATPase subunit 9 (Atp9) gene between cytoplasmic male sterile line and its maintainer line in soybeans. Afr J Biotech 10:10387–10392
Knoop V, Schuster W, Wissinger B, Brennicke A (1991) Trans splicing integrates an exon of 22 nucleotides into the nad5 mRNA in higher plant mitochondria. EMBO J 10:3483–3493
Kubo T, Nishizawa S, Sugawara A, Itchoda N, Estiati A, Mikami T (2000) The complete nucleotide sequence of the mitochondrial genome of sugar beet (Beta vulgaris L.) reveals a novel gene for tRNA(Cys)(GCA). Nucleic Acids Res 28:2571–2576
Kudla J, Bock R (1999) RNA editing in an untranslated region of the Ginkgo chloroplast genome. Gene 234:81–86
Kurek I, Ezra D, Begu D, Erel N, Litvak S, Breiman A (1997) Studies on the effects of nuclear background and tissue specificity on RNA editing of the mitochondrial ATP synthase subunits α, 6 and 9 in fertile and cytoplasmic malesterile (CMS) wheat. Theor Appl Genet 95:1305–1311
Lamattina L, Weil JH, Grienenberger JM (1989) RNA editing at a splicing site of NADH dehydrogenase subunit IV gene transcript in wheat mitochondria. FEBS Lett 258:79–83
Liu YG, Chen Y (2007) High-efficiency thermal asymmetric interlaced PCR for amplification of unknown flanking sequences. BioTech 43:649–656
Liu YG, Whittier RF (1995) Thermal asymmetric interlaced PCR: automatable amplification and sequencing of insert end fragments from P1 and YAC clones for chromosome walking. Genomics 25:674–681
Liu YG, Mitsukawa N, Oosumi T, Whittier RF (1995) Efficient isolation and mapping of Arabidopsis thaliana T-DNA insert junctions by thermal asymmetric interlaced PCR. Plant J 8:457–463
Mazers GR, Moyret C, Jeanteur P, Theillet C-G (1991) Direct sequencing by thermal asymmetric PCR. Nucleic Acids Res 19:4783
Mower JP, Palmer JD (2006) Patterns of partial RNA editing in mitochondrial genes of Beta vulgaris. Mol Gen Genomics 276:285–293
Okuda K, Hammani K, Tanz SK, Peng L, Fukao Y, Myouga F, Motohashi R, Shinozaki K, Small I, Shikanai T (2010) The pentatricopeptide repeat protein OTP82 is required for RNA editing of plastid ndhB and ndhG transcripts. Plant J 61:339–349
Pang M, Stewart JMcD, Zhang J (2011) A mini-scale hot borate method for the isolation of total RNA from a large number of cotton tissue samples. Afr J Biotech 10:15430–15437
Picardi E, Horner DS, Chiara M, Schiavon R, Valle G, Pesole G (2010) Large-scale detection and analysis of RNA editing in grape mtDNA by RNA deep-sequencing. Nucl Acids Res 38:4755–4767
Salazar RA, Pring DR, Kempken F (1991) Editing of mitochondrial atp9 transcripts from two sorghum lines. Curr Genet 20:483–486
Schnable PS, Wise RP (1998) The molecular basis of cytoplasmic male sterility and fertility restoration. Trends Plant Sci 3:175–180
Schuster W, Unseld M, Wissinger B, Brennicke A (1990) Ribosomal protein S14 transcripts are edited in Oenothera mitochondria. Nucleic Acids Res 18:229–233
Schuster W, Ternes R, Knoop V, Hiesel R, Wissinger B, Brennicke A (1991) Distribution of RNA editing sites in Oenothera mitochondrial mRNAs and rRNAs. Curr Genet 20:397–404
Sosso D, Mbelo S, Vernoud V, Gendrot G, Dedieu A, Chambrier P, Dauzat M, Heurtevin L, Guyon V, Takenaka M, Rogowsky PM (2012) PPR2263, a DYW-subgroup pentatricopeptide repeat protein, is required for mitochondrial nad5 and cob transcript editing, mitochondrion biogenesis, and maize growth. Plant Cell 24:676–691
Suzuki H, Yu J, Wang F, Zhang J (2013) Identification of mitochondrial DNA sequence variation and development of single nucleotide polymorphic markers for CMS-D8 in cotton. Theor Appl Genet. doi:10.1007/s00122-013-2070-4
Tan HQ, Singh J (2011) High-efficiency thermal asymmetric interlaced (HE-TAIL) PCR for amplification of Ds transposon insertion sites in Barley. J Plant Mol Biol Biotechnol 2:9–14
Terauchi R, Kahl G (2000) Rapid isolation of promoter sequences by TAIL-PCR: the 5′-flanking regions of Pal and Pgi genes from yams (Dioscorea). Mol Gen Genet 263:554–560
Unseld M, Marienfeld JR, Brandt P, Brennicke A (1997) The mitochondrial genome of Arabidopsis thaliana contains 57 genes in 366,924. Nat Genet 15:57–61
Wan CY, Wilkins TA (1994) A modified hot borate method significantly enhances the yield of high-quality RNA from cotton (Gossypium hirsutum L.). Anal Biochem 223:7–12
Wang F, Feng CD, O’Connell MA, Stewart JMcD, Stewart JF (2010) RFLP analysis of mitochondrial DNA in two cytoplasmic male sterility systems (CMS-D2 and CMS-D8) of cotton. Euphytica 172:93–99
Wei L, Yan ZX, Ding Y (2008) Mitochondrial RNA editing of F0-ATPase subunit 9 gene (atp9) transcripts of Yunnan purple rice cytoplasmic male sterile line and its maintainer line. Acta Physiol Plant 30:657–662
Wilkins TA, Smart LB (1996) Isolation of RNA from plant tissue. In: Krieg PA (ed) A laboratory guide to RNA: isolation, analysis, and synthesis. Wiley-Liss Inc., New York, pp 21–42
Yura K, Go M (2008) Correlation between amino acid residues converted by RNA editing and functional residues in protein three-dimensional structures in plant organelles. BMC Plant Biol 8:79
Zhang J, Stewart JMcD (2000) Economical and rapid method for extracting cotton genomic DNA. J Cott Sci 4: 193-201
Zhang J, Stewart J McD (2001) Inheritance and genetic relationships of the D8 and D2-2 restorer genes for cotton cytoplasmic male sterility. Crop Sci 41:289–294
Zhang JF, Turley RB, Stewart JMcD (2008) Comparative analysis of gene expression between CMS-D8 restored plants and normal non-restoring fertile plants in cotton by differential display. Plant Cell Rep 27:553–561
Acknowledgments
The research was supported in part by New Mexico Agricultural Experiment Station. The authors thank Anthony Aragon and Jeremy Edwards for help with the Ion Torrent sequencing. Some of the experiments used the Keck-UNM Genomics Resource in the University of New Mexico Cancer Center. Parts of this work were supported by USPHS/NIH grant 1R01CA170250-01 (to SAN).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by S. Hohmann.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Suzuki, H., Yu, J., Ness, S.A. et al. RNA editing events in mitochondrial genes by ultra-deep sequencing methods: a comparison of cytoplasmic male sterile, fertile and restored genotypes in cotton. Mol Genet Genomics 288, 445–457 (2013). https://doi.org/10.1007/s00438-013-0764-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-013-0764-6