
Abstract
Development of an effective vaccine against leishmaniasis is possible due to the fact that individuals cured from cutaneous leishmaniasis (CL) are protected from further infection. First generation Leishmania vaccines consisting of whole killed parasites reached to phase 3 clinical trials but failed to show enough efficacies mainly due to the lack of an appropriate adjuvant. In this study, an efficient liposomal protein-based vaccine against Leishmania major infection was developed using soluble Leishmania antigens (SLA) as a first generation vaccine and cytidine phosphate guanosine oligodeoxynucleotides (CpG ODNs) as an immunostimulatory adjuvant. 1, 2-Dioleoyl-3-trimethylammonium-propane was used as a cationic lipid to prepare the liposomes due to its intrinsic adjuvanticity. BALB/c mice were immunized subcutaneously (SC), three times in 2-week intervals, with Lip-SLA-CpG, Lip-SLA, SLA + CpG, SLA, or HEPES buffer. As criteria for protection, footpad swelling at the site of challenge and spleen parasite loads were assessed, and the immune responses were evaluated by determination of IFN-γ and IL-4 levels of cultured splenocytes, and IgG subtypes. The group of mice that received Lip-SLA-CpG showed a significantly smaller footpad swelling, lower spleen parasite burden, higher IgG2a antibody, and lower IL-4 level compared to the control groups. It is concluded that cationic liposomes containing SLA and CpG ODNs are appropriate to induce Th1 type of immune response and protection against leishmaniasis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Leishmania infection causes a group of diseases ranging from a self-healing cutaneous lesion to potentially fatal visceral form of disease, known as leishmaniasis. Controls strategies are not always successful; available drugs need multiple injections and practically show a limited efficacy in some endemic areas (Croft et al. 2006; Firooz et al. 2006; Santos et al. 2008). For centuries, it is well known that long-lasting protection induces upon recovery from CL caused by natural infection or leishmanization (Khamesipour et al. 2006). Attempts to develop vaccines against leishmaniasis resulted in identifying numerous candidate antigens, but only first generation vaccines consisting of parasite fractions or whole killed Leishmania with or without adjuvant reached to phase 3 clinical trials (Khamesipour et al. 2006; Sharifi et al. 1998; Momeni et al. 1999; Khalil et al. 2000). However, the results were not conclusive in some trials, and in general they showed a limited prophylactic efficacy (Noazin et al. 2009). Meanwhile, one preparation showed to have efficacy in post-kala-azar dermal leishmaniasis (PKDL) and reduced the dose of antimonate to treat patients (Musa et al. 2008). It seems that the main reason for the low efficacy of first generation vaccines is lack of a suitable adjuvant (Khamesipour et al. 2006; Melby 2002; Modabber 1995; Keshavarz Valian et al. 2008). Furthermore, new candidate vaccines against leishmaniasis, particularly those based on recombinant proteins and DNA (Marques-da-Silva et al. 2005), seem to be less immunogenic than first generation vaccines and almost all Leishmania antigens in this category induce protection in animal model if applied with an appropriate adjuvant, particularly IL-12 (Coler and Reed 2005). Therefore, it is essential to search for an adjuvant(s) to enhance the immunogenicity of antigens and to induce protection against leishmaniasis.
Synthetic oligodeoxynucleotides, containing unmethylated CpG motifs, are efficient inducers of Th1 response and cytotoxic T lymphocyte (CTL) which are required for protection against a range of viral, bacterial, and some parasitic pathogens in animal models (Wilson et al. 2009). In fact, synthetic CpG ODNs mimic the immunostimulatory activity of bacterial DNA and act as an immuno-adjuvant to boost antigen-specific immune responses by 5–500-fold which might be due to maintaining close physical contact between the CpG ODNs and the immunogen (Klinman et al. 2004; Krieg 2002). Liposomes are used as delivery vehicles to provide a close association of CpG ODNs with antigens and to enhance the immune responses (Klinman 2004; Klinman et al. 2004). Enhancement of immune response against poorly immunogenic Ags is possible through encapsulation of Ags into liposomes, particularly cationic ones (Chikh et al. 2001; Nakanishi et al. 1999).
It is well known that liposomes direct peptide antigens into the MHC II pathways of APCs, resulting in enhancement of antibody and/or antigen-specific T-cell response based on the formulations (Rao and Alving 2000). Moreover, there are reports demonstrating that cationic liposomes induce generation of CD8+ T-cell response, which requires in turn antigen presentation in the context of the MHC I pathway (Chikh et al. 2001; Hafez et al. 2001; Nakanishi et al. 1999). Furthermore, it has been shown that cationic liposomes are superior to neutral and negatively charged liposomes in inducing antigen-specific cytotoxic T lymphocyte (CTL) responses and antibody production (Nakanishi et al. 1997, 1999) and use as delivery systems for nucleic acids in gene therapy (Karmali and Chaudhuri 2007; Wasungu and Hoekstra 2006). The ability of liposome–DNA complexes to enter the cell by endocytosis, escape from the endosome, and deliver nucleic acids into the cytoplasm has been well documented (Zabner et al. 1995; Hoekstra et al. 2007).
We have already assessed the role of CpG ODNs in enhancement of immune response against two recombinant antigens including rgp63 or rLmSTI1 when entrapped into the liposomes (Badiee et al. 2008; Jaafari et al. 2007). In the current study, SLA as a first generation vaccine, encapsulated in cationic liposomes with or without CpG ODNs, was used to immunize BALB/c mice. The extent of protection and the type of generated immune response were studied in immunized BALB/c mice and compared with the control groups which received either HEPES buffer or SLA alone.
Materials and methods
Animals, parasites, SLA, and CpG ODNs
Female BALB/c mice 6–8 weeks old were purchased from Pasteur Institute (Tehran, Iran). The mice were maintained in animal house of Pharmaceutical Research Center and fed with tap water and laboratory pellet chow (Khorassan Javane Co., Mashhad, Iran). Animals were housed in a colony room 12:12 h light/dark cycle at 21°C with free access to water and food. Experiments were carried out according to the Ethical Committee Acts of Mashhad University of Medical Sciences.
Leishmania major strain (MRHO/IR/75/ER) used in this experiment is the one which was used to prepare experimental Leishmania vaccine, leishmanin, and leishmanization (Noazin et al. 2008, 2009).
SLA preparation was carried out using the protocol developed by Scott et al. (1987) with minor modifications. Briefly, the parasites were harvested at stationary phase and washed four times using HEPES buffer (10 mM, pH 7.5). Then the number of promastigotes was adjusted to 1.2 × 109/mL in buffer solution containing enzyme inhibitor cocktail, 50 μL/mL (Sigma, St. Louis, USA), and then the preparation was incubated in ice-water bath for 10 min and the parasites were lysed using freeze–thaw method followed by probe sonication in an ice bath. The supernatant of centrifuged lysate parasites was collected, dialyzed against buffer solution, sterilized using a 0.22-μm membrane, and stored at −70°C until use. The protein concentration of the preparation was determined using BCA protein assay method (Thermo Scientific, USA).
The CpG ODNs (Microsynth, Switzerland) used in this study was a 20-mer termed 1826 (5′-TCC ATG ACG TTC CTG ACG TT-3′) with a nuclease-resistant phosphorothioate backbone, which contains two CpG motifs with known Th1 type immunostimulatory effects on murine model (Chu et al. 1997; Weeratna et al. 2001).
Liposomes preparation and characterization
Liposomes containing SLA and CpG ODNs (Lip-SLA-CpG) were prepared using lipid film method. Briefly, the lipid phase consisting of DOTAP (4 μmol/mL; Avanti Polar lipids, USA) and cholesterol (Avanti Polar lipids) (1:1 molar ratio) was dissolved in chloroform in a sterile tube. The solvent was then removed using rotary evaporator (Hettich, Germany) resulting in deposition of a thin lipid film on the tube’s wall. The lipid film was then freeze-dried (TAITEC, Japan) overnight to ensure complete removal of the solvent. The lipid film was then hydrated and dispersed in sterile buffer containing SLA (0.5 mg/mL) at 45°C. The resulting multilamellar vesicles (MLVs) were converted to 200–400 nm vesicles using the bath sonicator (Branson 5510, USA). The CpG ODNs (200 μg/mL) was then added slowly to the liposomes containing SLA using mild shaking method. To prepare liposomes containing only SLA (i.e., Lip-SLA), the same procedure was followed except the CpG ODNs was omitted.
Particle size analyzer (Nano-ZS, Malvern, UK) was used to estimate the mean diameter and zeta potential of the liposomes. The concentration of SLA encapsulated in liposomes was determined using BCA protein assay kit (Thermo Scientific, USA).
SDS–PAGE analysis of SLA and liposomal SLA
Analytical SDS–PAGE was carried out to characterize and estimate qualitatively the concentration of SLA encapsulated in the prepared Lip-SLA. The gel was consisted of running gel (10.22%, w/v, acrylamide) and stacking gel (4.78%, w/v, acrylamide). The gel thickness was 1 mm. The electrophoresis buffer was 25 mM Tris, 192 mM glycine, and 0.1% SDS, pH 8.3. Electrophoresis was carried out at 140-V constant voltage for 45 min. After electrophoresis, the gels were stained with silver for protein detection.
Immunization of BALB/c mice
Different groups of mice (eight per group) were subcutaneously (SC) immunized in their left hind footpad three times in 2-week intervals with one of the following formulations: Lip-SLA-CpG (25 μg SLA–10 μg CpG ODNs/50 μL liposome/mouse), Lip-SLA (25 μg SLA/50 μL liposome/mouse), SLA in buffer (25 μg SLA/50 μL buffer/mouse), SLA in buffer (25 μg SLA/50 μL buffer/mouse) plus CpG ODNs in buffer (10 μg CpG ODNs/50 μL buffer/mouse), or HEPES buffer (HEPES 10 mM, sucrose 10%, pH 7.5) alone.
Challenge with L. major promastigotes
The immunized mice (five per group) were challenged SC in the left footpad with 1 × 106 L. major promastigotes harvested at stationary phase in 50 μL volume, at week 2 after the last immunization booster. Lesion development was recorded in each mouse by measurement of footpad swelling using a metric caliper (Mitutoyo Measuring Instruments, Japan). Grading of lesion size was done by subtracting the thickness of the uninfected contralateral footpad from that of the infected one.
Quantitative parasite burden in spleen
The number of viable L. major parasites in the spleen of mice was estimated by a limiting dilution assay method as described previously (Badiee et al. 2007). Briefly, the mice were sacrificed at week 9 post-challenge; the spleens were aseptically removed and homogenized in RPMI 1640 supplemented with 10% v/v heat inactivated FCS (Eurobio, France), 2 mM glutamine, 100 U/mL of penicillin, and 100 μg/mL of streptomycin sulfate (RPMI-FCS). The homogenate was diluted with the media in eight serial 10-fold dilutions and then was placed in each well of flat-bottom 96-well microtiter plates (Nunc, Denmark) containing a solid layer of rabbit blood agar in tetraplicate and incubated at 26 ± 1°C for 7–10 days. The number of viable parasite per spleen was determined by ELIDA software, a statistical method for limiting dilution assay (Taswell 1981).
Antibody isotype assay
Blood samples were collected from mice before and at week 9 after challenge and the sera were isolated and kept frozen until being used to assess anti-SLA IgG total, IgG1, and IgG2a antibodies by ELISA method as described previously (Badiee et al. 2007). Briefly, 96-well microtiter plates (Nunc) were coated with 50 μL of 10 μg/mL of SLA in PBS buffer (0.01 M, pH 7.3) overnight at 4°C. Plates were washed and then blocked by adding 200 μL per well of 1% of bovine serum albumin in PBS–Tween and incubated for 1 h at 37°C. Serum samples were diluted to 1:200, 1:2,000, or 1:20,000 with PBS–Tween and applied to the plates. The plates were then treated with HRP–rabbit anti-mouse IgG isotype according to the manufacturer’s instructions (Zymed Laboratories Inc., USA). Optical density (OD) was determined at 450 nm using 630 nm as the reference wavelength.
In vitro spleen cells response
Three mice from each group were sacrificed at week 2 after the last booster injection (before challenge) and the spleens were aseptically removed. Mononuclear cells were isolated by Ficoll-Hypaque density centrifugation method from spleen cell suspension. The cells were washed and resuspended in RPMI 1640-FCS and seeded at 2 × 106 cells/mL in 96-well flat-bottom plates (Nunc). The cells were stimulated in vitro with either SLA (10 μg/mL), Con A (2.5 μg/mL), or medium alone, and incubated at 37°C in 5% CO2 for 72 h. The culture supernatants were collected and the levels of IL-4 and IFN-γ were checked using ELISA method according to the manufacturer’s instructions (MabTech, Sweden).
Statistical analysis
One-way ANOVA statistical test was used to assess the significance of the differences among various groups. In the case of significant F value, Tukey–Kramer multiple comparisons test was carried out as a post-test to compare the means in different groups of mice. Results with P < 0.05 were considered to be statistically significant.
Results
Liposome characterization
The mean diameter of Lip-SLA and Lip-SLA-CpG formulations was 359.2 ± 0.36 and 277.8 ± 0.174 nm (n = 3) and the zeta potential was +7.65 mV and +2.02 mV, respectively. Characterization of SLA and liposomal SLA was performed by SDS–PAGE electrophoresis (Fig. 1). SDS–PAGE analysis of SLA revealed several protein bands with different ranges of molecular weight. Also, the SDS–PAGE analysis of liposomal SLA revealed bands similar to free SLA. The calculated concentration of DOTAP, SLA, and CpG ODNs in final formulations was 0.2 μmol, 25 μg, and 10 μg per 50 μL, respectively.

SDS–PAGE analysis of SLA alone and liposomal SLA. Lane 1 low-range protein standard (Sigma, USA), 2 SLA (5 μg), 3 SLA (10 μg), 4 liposomal SLA (2.5 μg), 5 liposomal SLA (5 μg)
Challenge results
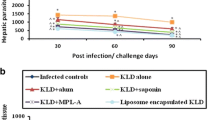
Lesion development was monitored by weekly measurement of footpad thickness (Fig. 2). The lesion size progressed at a more rapid rate in control groups which received either HEPES buffer or SLA alone than in mice immunized with SLA + CpG (P < 0.05) or Lip-SLA-CpG (P < 0.05) at week 6 after challenge. At week 8 after challenge, the lesion size in mice immunized with Lip-SLA-CpG was still significantly (P < 0.05) smaller than the control groups. The lesion size in mice immunized with SLA alone progressed continuously and no protection was observed in this group. At week 8 after challenge, progress in lesion size was slower in mice immunized with Lip-SLA compared with the mice that received SLA plus CpG, and the footpad swelling in this group of mice was significantly (P < 0.01) smaller than the group that received HEPES buffer. In control groups, the footpad swelling reached a plateau after 6 weeks (Fig. 2), but the disease progressed by metastasis to other organs and the mice lost their foot.

Footpad swelling in BALB/c mice immunized SC, three times in 2-week interval, with Lip-SLA, Lip-SLA-CpG, SLA + CpG, SLA or HEPES buffer after challenge with 106 virulent L. major promastigotes in left hind footpads. The footpad thickness was measured weekly for 8 weeks. Each point represents the average increase in footpad thickness ± SEM (n = 5)
Splenic parasite burden
The number of viable L. major was determined in the spleen of different groups of mice at week 9 after challenge (Fig. 3). Interestingly, live parasite was not detected in the group of mice immunized with Lip-SLA-CpG but the mice immunized with Lip-SLA or SLA + CpG showed live parasites in the spleen. The number of parasites in the spleen of mice immunized with Lip-SLA or SLA + CpG was significantly lower than the control groups (P < 0.01). However, there was no significant difference between these groups and the group of mice immunized with Lip-SLA-CpG.

Spleen parasite burden in BALB/c mice immunized SC, three times in 2-week interval, with Lip-SLA, Lip-SLA-CpG, SLA + CpG, SLA or HEPES buffer after challenge with L. major promastigotes. A limiting dilution analysis was performed at week 9 after the challenge on the spleen of individual mice and cultured in tetraplicate in serial of 8-fold dilutions. The number of viable parasites per spleen was determined using ELIDA software. The bar represents the average score ± SEM (n = 3)
Antibody response
In order to determine the type of generated immune response, the anti-SLA IgG antibodies and IgG1, IgG2a subclasses were titrated before (Fig. 4a–c) and after (Fig. 5a–c) challenge.

Levels of anti-SLA IgG1 (a), IgG2a (b), and IgG (c) in pooled sera of BALB/c mice immunized SC, three times in 2-week intervals, with Lip-SLA-CpG, Lip-SLA, SLA + CpG, SLA or HEPES buffer. Blood samples were collected from the mice 2 weeks after the last booster. The anti-SLA IgG1, IgG2a, and IgG levels were assessed using ELISA method. The assays were performed in triplicate at 200-, 2,000-, or 20,000-fold dilution for each serum sample. Values are the mean ± SD

Levels of anti-SLA IgG1 (a), IgG2a (b), and IgG (c) in pooled sera of BALB/c mice immunized SC, three times in 2-week intervals, with Lip-SLA-CpG, Lip-SLA, SLA + CpG, SLA or HEPES buffer. Blood samples were collected from the mice at week 9 after challenge. The anti-SLA IgG1, IgG2a, and IgG levels were assessed using ELISA method. The assays were performed in triplicate using 200-, 2,000-, or 20,000-fold dilution for each serum sample. Values are the mean ± SD
The sera of mice immunized with Lip-SLA-CpG before challenge showed significantly (P < 0.001) higher levels of IgG2a antibodies compared to the other groups. The ratio of IgG2a/IgG1 in sera of mice immunized with Lip-SLA-CpG or SLA + CpG were higher than the other groups (Table 1). The levels of IgG1 and IgG2a in sera of mice immunized with Lip-SLA were also significantly (P < 0.001) higher than the groups that received either HEPES buffer or SLA alone. The ratio of IgG2a/IgG1 in sera of mice immunized with Lip-SLA was higher than the control groups.
Challenge with L. major promastigotes induced elevation of IgG1, IgG2a, and IgG antibody levels in nearly all the groups of mice compared with before challenge (Fig. 5a–c). The sera of mice immunized with Lip-SLA-CpG, Lip-SLA, or SLA + CpG showed significantly (P < 0.001) elevated levels of IgG2a antibodies compared with the control groups. Although the level of IgG2a Ab in sera of mice immunized with Lip-SLA was the same as the level in the mice immunized with Lip-SLA-CpG, the level of IgG1 in the sera of mice immunized with Lip-SLA was significantly (P < 0.001) higher than the mice immunized with Lip-SLA-CpG. The ratio of IgG2a/IgG1 in sera of mice immunized with Lip-SLA-CpG or SLA + CpG was higher than all the other groups (Table 2). Also, the IgG2a/IgG1 ratio in sera of mice immunized with Lip-SLA or SLA was higher than the HEPES buffer group (Table 2).
In vitro cytokine production by splenocytes
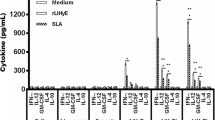
The supernatant of cultured splenocytes restimulated in vitro with SLA and analyzed at week 2 after the last booster injection to determine the level of IFN-γ and IL-4, cytokines indicative of Th1 and Th2 response, respectively. The results showed that the groups of mice immunized with Lip-SLA produced the highest amount of IFN-γ compared with the other groups. Moreover, the level of IFN-γ in mice immunized with SLA, SLA + CpG, or Lip-SLA-CpG showed no significant difference. The mice immunized with HEPES buffer produced significantly (P < 0.05) lower level of IFN-γ than all the other groups. Furthermore, the group of mice immunized with SLA alone produced significantly (P < 0.001) the highest level of IL-4 compared to all the other groups (Fig. 6a, b). The secreted IL-4 was below the detection level in the supernatant of splenic cells of mice immunized with Lip-SLA or Lip-SLA-CpG.

Cytokine levels in immunized mice at week 2 after the last booster injection. Mononuclear splenocytes were cultured in the presence of SLA (10 μg/mL) and the level of IFN-γ (a) or IL-4 (b) in the culture supernatants were detected using ELISA method. Results are shown as the mean ± SEM (n = 3)
Discussion
T cells recognize just peptides derived from cytosolic proteins bind to the major histocompatibility complex class I (MHC I) molecules or peptides derived from the lysosomal compartment bind to the MHC II molecules on the antigen presenting cells (APCs) surface, and it seems that almost any protein epitope might potentially function as an antigen, regardless of the location in the parasite. A specific polypeptide might be a strong immunogen when used as a cocktail, although individually it might be a weak immunogen or induce only partial protection (Handman 2001). Cocktail vaccines may provide a wider range of potentially protective epitopes taking advantage of these antigens that induce the required immunity (mainly CD4+ and CD8+ IFNγ-mediated responses) (Manuel et al. 2009).
In the present study, SLA was used as a first generation crude vaccine. It is shown that SLA’s components of Leishmania donovani might be a candidate vaccine for future studies if used in liposomal form (Afrin et al. 2002). SLA included most of the Leishmania soluble antigens and induced more protection than recombinant antigens such as gp63 and LAg when was used in liposomal form (Afrin et al. 2002). In the current study, immunization with SLA alone induced no protection in BALB/c mice based on footpad swelling and splenic parasite burden. The ratio of IgG2a/IgG1 (a marker for the induction of Th1-like response) in mice immunized with SLA was less than 0.8 and the high level of IL-4 production was assessed which confirmed the footpad swelling and parasite burden results. Exacerbation by SLA in the absence of any Th1-promoting adjuvant is not surprising in BALB/c mice since they have an inherent tendency to develop Th2 responses. The co-existence of Th1/Th2 responses with SLA immunization is consistent with the previous vaccine studies (Bhowmick et al. 2007; Afonso et al. 1994).
In this study, DOTAP, which is a cationic lipid, was used in liposome formulation based on two main purposes: (a) to prepare positively charged liposomes which interact more efficiently with the negatively charged molecules on the APCs’ cell surface and target antigens for endocytosis, and (b) to use the intrinsic adjuvanticity of this synthetic quaternary ammonium compound as shown previously (Yan et al. 2007). Cationic lipids with a quaternary ammonium group are more immunopotent than those with a tertiary ammonium group (Vangasseri et al. 2006). The mechanism of adjuvant activity of liposomes consisting DOTAP was shown before, so that DOTAP is an active stimulator of DC resulting in extracellular-signal-regulated kinase (ERK) activation and CC chemokine (or β-chemokine) induction (Yan et al. 2007).
In the current study, we used DOTAP at 200 nmol per mouse because of the published work of Chen et al. (2008). DOTAP at 15 or 30 nmol exhibited an enhanced efficacy, while with 75 or 150 nmol lipids showed the most significant tumor regression. Interestingly, mice given high doses of DOTAP (300 or 600 nmol) did not show significant tumor inhibition (Chen et al. 2008).
DOTAP has a low transition temperature (~0°C) due to two unsaturated bonds in its fatty acid chains which makes the liposomes unstable in vivo. Hence, cholesterol was used to increase in vivo stability of the prepared vesicles to become more rigid. Moreover, the presence of cholesterol in the vesicles facilitates cytoplasmic release of the antigens and avoids lysosomal degradation (Hafez et al. 2001). Increased in vivo stability of the vesicles may also further promote stimulation of CD8+ T-cell response (Ignatius et al. 2000).
Due to the presence of cationic lipid (i.e., DOTAP), the net surface charge of empty liposomes will be highly positive. However, liposomes containing SLA had a zeta potential of +7.65 mV. This might mean that the proteins of SLA have a net negative charge at pH 7.5, and then they interact electrostatically with cationic lipids in liposome bilayers and decrease the zeta potential of the liposomes. This could be the reason of high encapsulation efficiency of these positively charged liposomes for SLA.
To assess the protection rate, footpad swelling induced by L. major challenge and spleen parasite burden were checked in immunized mice. The results showed that the size of lesion in mice immunized with Lip-SLA-CpG at week 8 post-challenge was significantly (P < 0.05) smaller than the lesion size in groups of mice that received either HEPES buffer or SLA alone. Interestingly, the mice immunized with Lip-SLA or SLA + CpG showed no significant difference in footpad swelling during 7 weeks after infection. It has also been shown that CpG ODNs in free form induce short-term protection; that is why the footpad swelling in group of mice that received SLA + CpG ODNs progressed compared with those immunized with Lip-SLA, particularly after week 6. It could be concluded that the liposomal form of CpG ODNs induce more protection and for a longer period of time, as it is shown by Klinman et al. (2004).
The current results could explain the role of lipid compositions in liposome formulation on shifting of generated immune response particularly compared with Mazumder’s study (Mazumder et al. 2007). Mazumder et al. prepared positively charged liposomes using egg lecithin, cholesterol, and stearylamine, and evaluated the efficacy of pDNA and SLA to protect against challenge with L. donovani infection. They demonstrated that using cationic liposomes as a vehicle for the antigen, with pDNA either complexed or entrapped within, significantly increased the potentiating effect of pDNA (Mazumder et al. 2007), but the liposomal SLA formulation without pDNA did not have any protective efficacy. However, in the present study, Lip-SLA induced the same protection efficacy as SLA + CpG which could be due to the presence of DOTAP in the formulation. As we mentioned before, DOTAP itself has an immunostimulatory effect and can induce cellular immunity (Chen et al. 2008).
The results of cytokine assay surprisingly showed that the level of IFN-γ in the supernatant of cultured splenocytes of mice immunized with SLA alone before challenge was as high as the other immunized groups. However, the IL-4 level was high in SLA group but it was very low (under detection limit) in groups immunized with Lip-SLA or Lip-SLA-CpG. At APCs level, CpG ODNs through TLR9 augment both activation and maturation of DC as well as the induction of proinflammatory cytokines (Hemmi et al. 2000). Thus, the endogenous production of IL-12, IL-18, and other soluble mediators from activated DC induced by CpG ODNs are likely to result in a more physiologic cognate interaction between DC and T cell, which results in qualitatively and quantitatively different types of CD4+ and CD8+ T-cell response (Rhee et al. 2002).
In terms of IgG subclasses, as it is shown by others (Klinman et al. 2004; Chu et al. 1997; Krieg 2000; Tafaghodi et al. 2010), a significant shift in IgG subclass response was achieved by co-administration of SLA with CpG ODNs or encapsulation of SLA into the liposomes. Addition of CpG ODNs resulted in a significant increase in production of IgG2a antibodies and IgG2a/IgG1 ratio (~2-fold; Table 2). The enhanced Th1 type of response is an essential step against intracellular pathogens (Dumont 2002). The results of IgG subclasses showed that CpG ODNs alone is an effective adjuvant to induce Th1 response and partial protection against leishmaniasis in BALB/c mice, but stronger IgG subclasses response (particularly IgG2a) was induced when liposomes were used to deliver CpG ODNs. Improvement of CpG ODNs adjuvanticity was shown by encapsulation of CpG ODNs in liposomes (Jiao et al. 2004; Li et al. 2003; Suzuki et al. 2004).
Despite backbone stabilization of CpG ODNs 1826, PS-modified ODNs are still susceptible to nuclease degradation, albeit at a lower rate (Levin 1999). Furthermore, the necessity for intracellular uptake into endosomal compartments represents a significant hurdle since charged macromolecules such as a free DNA have difficulty to cross hydrophobic membranes (Wilson et al. 2009). In the current study, liposomes were used as a delivery system to enhance the activity of CpG ODNs. Liposomes may mediate this activity enhancement in a number of ways: for example, liposomes protect CpG ODNs from nuclease activity and hamper the distribution of CpG ODNs into tissues, consequently increasing CpG ODNs half-life (De Oliveira et al. 2000); liposomes also facilitate the intracellular delivery of CpG ODNs into the cytoplasm based on the formulations (Zhang et al. 2001). Moreover, improved adjuvant effect of CpG ODNs when encapsulated into liposome might be due to the fact that TLR9, the receptor for CpG motifs, localizes to endosomal/vacuolar/vesicular compartments but not to the cell surface (Krieg 2002).
In conclusion, the current data point out that SLA in association with cationic liposomes consisting DOTAP and CpG ODNs elicit impressive protective responses against murine model of leishmaniasis. Interestingly, liposomal SLA showed no significant differences with SLA + CpG ODNs in terms of protective efficacy. The current data generated in murine model of L. major infection showed a promising role of CpG ODNs and DOTAP cationic liposomes as an adjuvant to enhance stronger immune response against SLA which is a crude Leishmania antigen.
References
Afonso LC, Scharton TM, Vieira LQ, Wysocka M, Trinchieri G, Scott P (1994) The adjuvant effect of interleukin-12 in a vaccine against Leishmania major. Science 263(5144):235–237
Afrin F, Rajesh R, Anam K, Gopinath M, Pal S, Ali N (2002) Characterization of Leishmania donovani antigens encapsulated in liposomes that induce protective immunity in BALB/c mice. Infect Immun 70(12):6697–6706
Badiee A, Jaafari MR, Khamesipour A (2007) Leishmania major: immune response in BALB/c mice immunized with stress-inducible protein 1 encapsulated in liposomes. Exp Parasitol 115(2):127–134
Badiee A, Jaafari MR, Samiei A, Soroush D, Khamesipour A (2008) Coencapsulation of CpG oligodeoxynucleotides with recombinant Leishmania major stress-inducible protein 1 in liposome enhances immune response and protection against leishmaniasis in immunized BALB/c mice. Clin Vaccine Immunol 15(4):668–674
Bhowmick S, Ravindran R, Ali N (2007) Leishmanial antigens in liposomes promote protective immunity and provide immunotherapy against visceral leishmaniasis via polarized Th1 response. Vaccine 25(35):6544–6556
Chen W, Yan W, Huang L (2008) A simple but effective cancer vaccine consisting of an antigen and a cationic lipid. Cancer Immunol Immun 57(4):517–530
Chikh GG, Kong S, Bally MB, Meunier JC, Schutze-Redelmeier MP (2001) Efficient delivery of Antennapedia homeodomain fused to CTL epitope with liposomes into dendritic cells results in the activation of CD8+ T cells. J Immunol 167(11):6462–6470
Chu RS, Targoni OS, Krieg AM, Lehmann PV, Harding CV (1997) CpG oligodeoxynucleotides act as adjuvants that switch on T helper 1 (Th1) immunity. J Exp Med 186(10):1623–1631
Coler RN, Reed SG (2005) Second-generation vaccines against leishmaniasis. Trends Parasitol 21(5):244–249
Croft SL, Sundar S, Fairlamb AH (2006) Drug resistance in leishmaniasis. Clin Microbiol Rev 19(1):111–126
De Oliveira MC, Boutet V, Fattal E, Boquet D, Grognet J-M, Couvreur P, Deverre J-R (2000) Improvement of in vivo stability of phosphodiester oligonucleotide using anionic liposomes in mice. Life Sci 67(13):1625–1637
Dumont FJ (2002) Modulation of Th1 and Th2 responses for immunotherapy. Expert Opin Ther Pat 12(3):341–367
Firooz A, Khamesipour A, Ghoorchi MH, Nassiri-Kashani M, Eskandari SE, Khatami A, Hooshmand B, Gorouhi F, Rashighi-Firoozabadi M, Dowlati Y (2006) Imiquimod in combination with meglumine antimoniate for cutaneous leishmaniasis: a randomized assessor-blind controlled trial. Arch Dermatol 142(12):1575–1579
Hafez IM, Maurer N, Cullis PR (2001) On the mechanism whereby cationic lipids promote intracellular delivery of polynucleic acids. Gene Ther 8(15):1188–1196
Handman E (2001) Leishmaniasis: current status of vaccine development. Clin Microbiol Rev 14(2):229–243
Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, Akira S (2000) A Toll-like receptor recognizes bacterial DNA. Nature 408(6813):740–745
Hoekstra D, Rejman J, Wasungu L, Shi F, Zuhorn I (2007) Gene delivery by cationic lipids: in and out of an endosome. Biochem Soc T 035(1):68–71
Ignatius R, Mahnke K, Rivera M, Hong K, Isdell F, Steinman RM, Pope M, Stamatatos L (2000) Presentation of proteins encapsulated in sterically stabilized liposomes by dendritic cells initiates CD8+ T-cell responses in vivo. Blood 96(10):3505–3513
Jaafari MR, Badiee A, Khamesipour A, Samiei A, Soroush D, Kheiri MT, Barkhordari F, McMaster WR, Mahboudi F (2007) The role of CpG ODN in enhancement of immune response and protection in BALB/c mice immunized with recombinant major surface glycoprotein of Leishmania (rgp63) encapsulated in cationic liposome. Vaccine 25(32):6107–6117
Jiao X, Wang RY-H, Qiu Q, Alter HJ, Shih JW-K (2004) Enhanced hepatitis C virus NS3 specific Th1 immune responses induced by co-delivery of protein antigen and CpG with cationic liposomes. J Gen Virol 85(6):1545–1553
Karmali PP, Chaudhuri A (2007) Cationic liposomes as non-viral carriers of gene medicines: resolved issues, open questions, and future promises. Med Res Rev 27(5):696–722
Keshavarz Valian H, Khoshabe Abdollah Kenedy L, Nateghi Rostami M, Miramin Mohammadi A, Khamesipour A (2008) Role of Mycobacterium vaccae in the protection induced by first generation Leishmania vaccine against murine model of leishmaniasis. Parasitol Res 103(1):21–28
Khalil EA, El Hassan AM, Zijlstra EE, Mukhtar MM, Ghalib HW, Musa B, Ibrahim ME, Kamil AA, Elsheikh M, Babiker A, Modabber F (2000) Autoclaved Leishmania major vaccine for prevention of visceral leishmaniasis: a randomised, double-blind, BCG-controlled trial in Sudan. Lancet 356(9241):1565–1569
Khamesipour A, Rafati S, Davoudi N, Maboudi F, Modabber F (2006) Leishmaniasis vaccine candidates for development: a global overview. Indian J Med Res 123(3):423–438
Klinman DM (2004) Use of CpG oligodeoxynucleotides as immunoprotective agents. Expert Opin Biol Ther 4(6):937–946
Klinman DM, Currie D, Gursel I, Verthelyi D (2004) Use of CpG oligodeoxynucleotides as immune adjuvants. Immunol Rev 199(1):201–216
Krieg AM (2000) The role of CpG motifs in innate immunity. Curr Opin Immunol 12(1):35–43
Krieg AM (2002) CpG motifs in bacterial DNA and their immune effects. Annu Rev Immunol 20(1):709–760
Levin AA (1999) A review of the issues in the pharmacokinetics and toxicology of phosphorothioate antisense oligonucleotides. Biochim Biophys Acta 1489(1):69–84
Li WM, Dragowska WH, Bally MB, Schutze-Redelmeier M-P (2003) Effective induction of CD8+ T-cell response using CpG oligodeoxynucleotides and HER-2/neu-derived peptide co-encapsulated in liposomes. Vaccine 21(23):3319–3329
Manuel S, Ramirez L, Pineda MA, Gonzalez V, Entringer PF, de Oliveira CI, Nascimento IP, Souza AP, Corvo L, Alonso C, Bonay P, Brodskyn C, Barral A, Barral-Netto M, Iborra S (2009) Searching genes encoding Leishmania antigens for diagnosis and protection. Scholarly Res Exchange 2009
Marques-da-Silva EA, Coelho EA, Gomes DC, Vilela MC, Masioli CZ, Tavares CA, Fernandes AP, Afonso LC, Rezende SA (2005) Intramuscular immunization with p36(LACK) DNA vaccine induces IFN-gamma production but does not protect BALB/c mice against Leishmania chagasi intravenous challenge. Parasitol Res 98(1):67–74
Mazumder S, Ravindran R, Banerjee A, Ali N (2007) Non-coding pDNA bearing immunostimulatory sequences co-entrapped with leishmanial antigens in cationic liposomes elicits almost complete protection against experimental visceral leishmaniasis in BALB/c mice. Vaccine 25(52):8771–8781
Melby PC (2002) Vaccination against cutaneous leishmaniasis: current status. Am J Clin Dermatol 3(8):557–570
Modabber F (1995) Vaccines against leishmaniasis. Ann Trop Med Parasitol 89(Suppl 1):83–88
Momeni AZ, Jalayer T, Emamjomeh M, Khamesipour A, Zicker F, Ghassemi RL, Dowlati Y, Sharifi I, Aminjavaheri M, Shafiei A, Alimohammadian MH, Hashemi-Fesharki R, Nasseri K, Godal T, Smith PG, Modabber F (1999) A randomised, double-blind, controlled trial of a killed L. major vaccine plus BCG against zoonotic cutaneous leishmaniasis in Iran. Vaccine 17(5):466–472
Musa AM, Khalil EA, Mahgoub FA, Elgawi SH, Modabber F, Elkadaru AE, Aboud MH, Noazin S, Ghalib HW, El-Hassan AM (2008) Immunochemotherapy of persistent post-kala-azar dermal leishmaniasis: a novel approach to treatment. Trans R Soc Trop Med Hyg 102(1):58–63
Nakanishi T, Kunisawa J, Hayashi A, Tsutsumi Y, Kubo K, Nakagawa S, Fujiwara H, Hamaoka T, Mayumi T (1997) Positively charged liposome functions as an efficient immunoadjuvant in inducing immune responses to soluble proteins. Biochem Biophys Res Co 240(3):793–797
Nakanishi T, Kunisawa J, Hayashi A, Tsutsumi Y, Kubo K, Nakagawa S, Nakanishi M, Tanaka K, Mayumi T (1999) Positively charged liposome functions as an efficient immunoadjuvant in inducing cell-mediated immune response to soluble proteins. J Control Release 61(1–2):233–240
Noazin S, Modabber F, Khamesipour A, Smith PG, Moulton LH, Nasseri K, Sharifi I, Khalil EA, Bernal ID, Antunes CM, Kieny MP, Tanner M (2008) First generation leishmaniasis vaccines: a review of field efficacy trials. Vaccine 26(52):6759–6767
Noazin S, Khamesipour A, Moulton LH, Tanner M, Nasseri K, Modabber F, Sharifi I, Khalil EAG, Bernal IDV, Antunes CMF, Smith PG (2009) Efficacy of killed whole-parasite vaccines in the prevention of leishmaniasis—a meta-analysis. Vaccine 27(35):4747–4753
Rao M, Alving CR (2000) Delivery of lipids and liposomal proteins to the cytoplasm and Golgi of antigen-presenting cells. Adv Drug Deliver Rev 41(2):171–188
Rhee EG, Mendez S, Shah JA, C-Y Wu, Kirman JR, Turon TN, Davey DF, Davis H, Klinman DM, Coler RN, Sacks DL, Seder RA (2002) Vaccination with heat-killed Leishmania antigen or recombinant leishmanial protein and CpG oligodeoxynucleotides induces long-term memory CD4+ and CD8+ T cell responses and protection against Leishmania major infection. J Exp Med 195(12):1565–1573
Santos DO, Coutinho CE, Madeira MF, Bottino CG, Vieira RT, Nascimento SB, Bernardino A, Bourguignon SC, Corte-Real S, Pinho RT, Rodrigues CR, Castro HC (2008) Leishmaniasis treatment—a challenge that remains: a review. Parasitol Res 103(1):1–10
Scott P, Pearce E, Natovitz P, Sher A (1987) Vaccination against cutaneous leishmaniasis in a murine model. I. Induction of protective immunity with a soluble extract of promastigotes. J Immunol 139(1):221–227
Sharifi I, FeKri AR, Aflatonian MR, Khamesipour A, Nadim A, Mousavi MR, Momeni AZ, Dowlati Y, Godal T, Zicker F, Smith PG, Modabber F (1998) Randomised vaccine trial of single dose of killed Leishmania major plus BCG against anthroponotic cutaneous leishmaniasis in Bam, Iran. Lancet 351(9115):1540–1543
Suzuki Y, Wakita D, Chamoto K, Narita Y, Tsuji T, Takeshima T, Gyobu H, Kawarada Y, Kondo S, Akira S, Katoh H, Ikeda H, Nishimura T (2004) Liposome-encapsulated CpG oligodeoxynucleotides as a potent adjuvant for inducing type 1 innate immunity. Cancer Res 64(23):8754–8760
Tafaghodi M, Khamesipour A, Jaafari MR (2010) Immunization against leishmaniasis by PLGA nanospheres encapsulated with autoclaved Leishmania major (ALM) and CpG-ODN. Parasitol Res 108(5):1265–1273
Taswell C (1981) Limiting dilution assays for the determination of immunocompetent cell frequencies. I. Data analysis. J Immunol 126(4):1614–1619
Vangasseri DP, Cui Z, Chen W, Hokey DA, Falo LD, Huang L (2006) Immunostimulation of dendritic cells by cationic liposomes. Mol Membr Biol 23(5):385–395
Wasungu L, Hoekstra D (2006) Cationic lipids, lipoplexes and intracellular delivery of genes. J Control Release 116(2):255–264
Weeratna RD, Brazolot Millan CL, McCluskie MJ, Davis HL (2001) CpG ODN can re-direct the Th bias of established Th2 immune responses in adult and young mice. FEMS Immunol Med Microbiol 32(1):65–71
Wilson KD, de Jong SD, Tam YK (2009) Lipid-based delivery of CpG oligonucleotides enhances immunotherapeutic efficacy. Adv Drug Deliver Rev 61(3):233–242
Yan W, Chen W, Huang L (2007) Mechanism of adjuvant activity of cationic liposome: phosphorylation of a MAP kinase, ERK and induction of chemokines. Mol Immunol 44(15):3672–3681
Zabner J, Fasbender AJ, Moninger T, Poellinger KA, Welsh MJ (1995) Cellular and molecular barriers to gene transfer by a cationic lipid. J Biol Chem 270(32):18997–19007
Zhang YM, Rusckowski M, Liu N, Liu C, Hnatowich DJ (2001) Cationic liposomes enhance cellular/nuclear localization of 99mTc-antisense oligonucleotides in target tumor cells. Cancer Biother Radiopharm 16(5):411–419
Acknowledgments
The financial support of the Nanotechnology Research Center and Biotechnology Research Center, Mashhad University of Medical Sciences (MUMS), and Center for Research and Training in Skin Diseases and Leprosy, Tehran University of Medical Sciences (TUMS) are gratefully acknowledged. This study was part of Pharm. D. dissertation of VHS that was done in Nanotechnology Research Center, MUMS, Iran.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Ali Badiee and Mahmoud Reza Jaafari equally designed this research.
Rights and permissions
About this article
Cite this article
Heravi Shargh, V., Jaafari, M.R., Khamesipour, A. et al. Cationic liposomes containing soluble Leishmania antigens (SLA) plus CpG ODNs induce protection against murine model of leishmaniasis. Parasitol Res 111, 105–114 (2012). https://doi.org/10.1007/s00436-011-2806-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00436-011-2806-5