
Abstract
Conventional method of species identification in Eimeria employs phenotypic characters of the oocysts and the site of infection in the chicken intestine, which are subjective analyses. PCR-based identification of Eimeria spp. is known to be specific and sensitive. We used internal transcribed spacer 1 (ITS-1)-based nested PCR to follow the distribution of Eimeria spp. in the field, which may be of significant value in the management of coccidiosis in chickens. In the present study, intestinal samples of chicks from commercial poultry farms, in India, suspected of having contracted Eimeria infections were analyzed using ITS-1 PCR. The PCR-amplified ITS-1 regions were also sequenced from these samples. Of 26 field samples analyzed, 19 showed the presence of multiple infections of Eimeria spp. Incidence of Eimeria tenella (80%) was found to be highest in these samples followed by Eimeria mitis (53%), Eimeria acervulina (42%), Eimeria brunetti, and Eimeria maxima (23%). Incidence of Eimeria necatrix was found to be the lowest (15%) in the samples analyzed, while none of the samples analyzed showed the presence of ITS-1 sequence from Eimeria praecox. The ITS-1 sequences amplified from Eimeria spp. in the present study showed few variations from the ITS sequences available in the GenBank database. Further studies will be required to determine whether these differences are unique to geographical locations.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Coccidiosis is one of the most economically important diseases of poultry (Ruff 1999). The disease is prevalent wherever intensive rearing of poultry birds is practiced. Coccidiosis in chicken is caused by seven species of Eimeria, viz. Eimeria acervulina, Eimeria brunetti, Eimeria maxima, Eimeria mittis, Eimeria necatrix, Eimeria tenella, and Eimeria praecox.
Identification of species in the infected birds has important implications for the disease management as well as for studying their epidemiology and population biology (Woods et al. 2000). Conventional methods of identification, based on morphological features of the sporulated oocysts and infection site on the intestine, are tedious to perform and have serious limitations due to overlapping characteristics among different species (Long and Joyner 1984).
In eukaryotic cells, multiple copies of the highly conserved ribosomal RNA (rRNA) genes are located in tandem. A single cluster contains 18S, 5.8S, and 28S rRNA genes and the rRNA sequences are interspersed by internal transcribed spacers (ITS-1 and ITS-2). The ITS-1 sequences are highly variable between species and are relatively conserved within species. ITS sequences, therefore, have provided genetic markers for the identification of species in various organisms including Eimeria (Hnida and Duszynski 1999; Mugridge et al. 2000; Stucki et al. 1993; Barta et al. 1997; Schnitzler et al. 1998). In particular, primers specific for ITS-1 have been developed for the detection of Eimeria spp. from fecal and intestinal samples using PCR assays (Lew et al. 2003; Schnitzler et al. 1998, 1999).
One of the major challenges faced by the poultry industry worldwide in controlling coccidiosis is the species-specific, at times strain-specific, nature of the immunity (Smith et al. 2002). Strain-specific protective immunity is particularly prevalent in E. maxima (Smith et al. 2002). Identification of immunoprotective antigens for organisms like Eimeria is far from complete, and the absence of a robust assay system complicates such a search (Blake et al. 2004). The lack of defined immunoprotective antigens has hindered the identification of polymorphism between the species and strains by genotypic analysis (Beck et al. 2009). Though the evolutionary relationships based on internal transcribed spacer sequence did not correlate well with the demonstrated immunological cross-reactivities among different strains of E. maxima, these strains could be differentiated based on the changes in the internal transcribed spacer nucleotide sequences (Barta et al. 1998). Thus, sequencing the ITS region of field isolates of Eimeria may be useful in assessing the strain diversity.
Indian poultry industry contributes nearly $2.2 billion annually to the national economy (Mohanty and Rajendran 2003). Despite the magnitude of the problem posed by coccidiosis to the poultry industry in India, the use of PCR-based technique for species identification is not reported thus far. Sequence information of ITS-1 regions of Indian isolates is therefore lacking. To our knowledge, this is the first ever report on the use of ITS-PCR for the identification of Eimeria in India. We report the ITS-1 sequence of Indian Eimeria isolates and have compared their phylogenetic relationships with the published sequences of other exotic isolates.
Materials and methods
Samples
Birds from farms that had either suffered mortality or showed symptoms of Eimeria infections were analyzed. The intestinal contents were squeezed out or intestinal scrapings were obtained from birds showing gut lesions during postmortem. The samples were stored and transported in 2% potassium dichromate solution. Presence of the coccidial oocysts was confirmed by microscopic examination. A total of 26 samples were obtained from commercial poultry farms in various parts of southern India.
DNA extraction from oocyst samples
Intestinal contents of the suspected field samples were analyzed for the presence of Eimeria oocysts. Samples containing oocyst were processed for DNA extraction as described by Zhao et al. (2001). Briefly, the intestinal contents were passed through a sieve to remove coarse materials. Then, the filtrate was centrifuged at 2,000×g and the pellet was washed thrice using distilled water. The washed pellet was reconstituted into phosphate-buffered saline and the oocyst wall broken by vortexing in the presence of glass beads (1-mm diameter; Sigma Chemical Company, USA). The vortexed material was recovered from the glass beads and pelleted by centrifugation at 1,000×g. The pellet was resuspended in 0.5-ml lysis buffer containing 660 mM EDTA, 1.3% N-lauryl sarcosine (Sigma Chemical Company), and 2 mg/ml proteinase K (Sigma Chemical Company), and the suspension was incubated at 65°C for 45 min. Then, genomic DNA was extracted by adding an equal volume of mixture containing phenol/chloroform/isoamyl alcohol in the ratio of 25:24:1 (Invitrogen, USA). Genomic DNA was precipitated from the aqueous phase by adding an equal volume of isopropyl alcohol (Merck, India). The precipitated DNA was washed twice using 70% ethanol. Some of the intestinal samples did not contain oocysts. The epithelial lining from such intestines were scraped and genomic DNA extracted using DNazol reagent (Invitrogen) as per the manufacturer’s instruction.
Identification of Eimeria spp. using PCR
The DNA extracted from intestinal sample was used to amplify the ITS-1 region. Genus-specific primers were used for the amplification of ITS-1 region from all pathogenic Eimeria spp. of chicken. PCR was performed using conditions as described by Lew et al. (2003). Briefly, the PCR reaction mix was prepared using 50–100 ng of oocyst DNA, 50 pmol each of genus-specific primers, 1 U of Taq polymerase (Bangalore Genei, India), 200 µM each of dNTPs (Eppendorf, Germany), and PCR buffer containing 1.5 mM MgCl2 (Bangalore Genei). The thermal cycling was done with an initial denaturing step at 94°C for 3 min followed by 30 cycles of 94°C for 30 s, 55°C for 30 s, and 72°C for 1 min and 30 s and a final extension step at 72°C for 7 min. The amplified product from this PCR step (1 µl) was used as template to identify the species of Eimeria in a nested PCR. A similar PCR reaction mix as described above was used for the nested PCR also. The primer sequences and the annealing temperature used in the nested PCR are provided in Table 1.
Sequencing of ITS-1 region
The amplified products of the first PCR were cloned into pCR 2.1 TOPO TA (Invitrogen) cloning vector. At least ten colonies were picked randomly for each sample. The insert sizes of ITS-1 were checked by digesting the plasmids with EcoRI which flanks the insert. We anticipated mixed infections in the field samples. Though originating from different species, some of the ITS-1 amplicons may have very similar size. An agarose gel electrophoresis cannot resolve such small differences with sufficient clarity. Therefore, minimum of two plasmids were randomly selected for each identical insert size for sequencing with M13 forward and M13 reverse primers.
Phylogenetic analysis of ITS-1 sequence
The ITS-1 sequences obtained from Indian isolates were compared with that of European, Australian, American, and Chinese isolates available in the GenBank database (Table 2). The sequence alignment was performed using ClustalW program, and phylogenetic and molecular evolutionary analyses were conducted using MEGA, version 4 (Tamura et al. 2007). A maximum parsimony tree was created using the ITS-1 sequences from Indian isolates and other published sequences. The pairwise percentage identity was calculated using GeneDoc multiple sequence alignment editor, version 2.6.002 (Nicholas and Nicholas 1997).
Results
Identification of Eimeria spp. using PCR
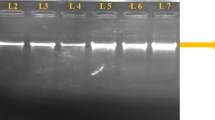
Species-specific nested PCR was used to identify Eimeria spp. All 26 intestinal samples, which were sent for postmortem analysis, were analyzed using the nested PCR. The samples originated from two major commercial poultry-rearing regions in India. Sixteen samples from Namakkal in Tamil Nadu and ten samples from various parts of Andhra Pradesh were tested for the presence of Eimeria spp. Six species out of seven tested in the nested PCR were detected from the field samples. Nineteen samples contained oocysts from multiple species of Eimeria. As many as five species could be detected from a single sample collected from a farm in Tamil Nadu. The incidence of E. tenella (80%) was the highest in those samples tested. E. necatrix was detected in relatively lesser number of farms (15%). E. praecox was not detected from any of the samples tested. The summary of the result is provided in Table 3. A representative gel picture showing the PCR amplification products from one of the field isolates is provided in Fig. 1. ITS-1 region from few of these samples was sequenced to ascertain the sequence homology between the isolates.

Nested ITS-PCR products from a representative field sample resolved on 2% agarose gel. PCR amplification was found using primers specific for E. acervulina (Ea, ∼321 bp), E. brunetti (Eb, ∼311 bp), E. mitis (Emi, ∼328 bp), E. necatrix (En, ∼383 bp), and E. tenella (Et, ∼278 bp). No amplification was observed in E. maxima (Em), E. maxima US (Em*), and E. praecox-specific primers in this particular field sample. M, 100-bp DNA ladder
Sequencing of ITS-1 region
The samples showing amplification product with universal primers were further processed for sequencing. The PCR products amplified with the universal primers were cloned into pCR 2.1 TOPO vector. Minimum of ten clones per plate were selected, and the insert size was checked using EcoRI restriction enzyme. The insert sizes varied from ∼400 to ∼700 bp. A total of 26 ITS-1 sequences could be generated from the samples analyzed. Twelve of these sequences were from E. tenella, seven from E. brunetti, four from E. acervulina, and three from E. maxima. The length of the ITS-1 region (in bp) for each sequence is provided in Table 4.
ITS-1 sequences of Indian isolates were compared with the published nested primer sequences used for species identification (Lew et al. 2003). This was done to determine the relevance of using the primers, which were essentially designed for the sequences of ITS-1 from American and Australian isolates, in the identification of Eimeria species in India. The ITS-1 sequences of all E. acervulina and E. brunetii isolates from India matched the published nested primer sequences of E. acervulina and E. brunetti, respectively. Two of the E. maxima isolates (AP_6 and TN_2) matched with the nested primer sequences of E. maxima from USA, while other E. maxima isolates (TN_9) matched with the nested primer sequences of E. maxima from Australia (Lew et al. 2003). With respect to E. tenella, six out of 12 ITS-1 sequences had a mismatch near the 5′ end of the forward primer (A→C) and one (AP_10), out of the 12 sequences had one base pair mismatch (A→G) at the 3′ end of the reverse primer. The published sequences of E. tenella nested primers and the corresponding mismatches found in the Indian isolates are underlined below.
-
E. tenella forward: 5′ AA(C)T TTA GTC CAT CGC AAC CCT 3′
-
E. tenella reverse: 5′ CGA GCG CTC TGC ATA CGA CA(G) 3′
The mismatch near the 5′ end of the forward primer sequence has obviously not affected the nested PCR results. However, considering the fact that many isolates had the mismatch, it is desirable to use degenerate primers incorporating both the nucleotides. In contrast, one of the 12 isolates had a mismatch with the 3′ end of the reverse primer, which is expected to affect the PCR amplification. Given the limited number of samples examined, it would be premature to conclude that this mismatch at the 3′ end reflects a general trend in Indian E. tenella isolates. Analysis of a larger sample size would help determine whether the sequence difference is a common occurrence or an isolated case.
Phylogenetic analysis of Eimeria spp. using ITS-1 sequences
A maximum parsimony tree was created using the ITS-1 sequences from Indian Eimeria spp. by comparing the available ITS-1 sequences of exotic isolates from Europe, America, Australia, and China (Fig. 2). There was a clear species-wise clustering, irrespective of the geographical location, for all the ITS-1 sequences of E. tenella, E. acervulina, and E. brunetti. The E. maxima sequences formed two separate groups. Two (AP_6 and TN_2) of the three E. maxima sequences in the present study grouped distantly compared to the third (TN_9) sequence. The sequence diversity and strain variation in E. maxima was reported earlier by other authors as well (Schnitzler et al. 1999; Lew et al. 2003; Cantacessi et al. 2008).

Maximum parsimony tree comparing the ITS-1 sequences of Indian isolates with the sequences of Australian, American, European, and Chinese isolates. Percentage of bootstrap (1,000 replicates) values is shown at each node. The tree indicates the species-wise grouping of isolates except for E. maxima
The pairwise percentage identity among the sequences was checked using GeneDoc multiple sequence alignment editor, version 2.6.002 (Table 5). The E. acervulina, E. brunetti, and E. tenella ITS-1 sequences were >96% identical when compared within the species. However, the TN_9 maxima sequence had only 30% identity with the other two E. maxima (TN_2 and AP_6) isolates. The ITS-1 sequence identity between different species of Eimeria ranged from 22% to 57%.
Discussion
Diversity in Eimeria spp. is traditionally studied by its phenotypic characters of the oocysts and life cycle stages. The published data on molecular diversity among Eimeria spp. are limited and population genetic studies have been scarce. Molecular studies of population diversity in Eimeria species remain elementary in the poultry industry. Importantly, most studies with Eimeria have been based on a few well-characterized laboratory strains, many of which were isolated more than 30 years ago and have a strong European/North American bias (Beck et al. 2009). ITS sequence information on chicken Eimeria are now beginning to emerge from the field isolates of Australia and Taiwan (Lew et al. 2003; Lien et al. 2007; Su et al. 2003). Similar sequence diversity studies for Indian isolates have not been reported. In the present study, we have determined the ITS-1 sequence of Eimeria field isolates in India. Using these sequence information, we also evaluated the utility of the available species-specific nested primers in identifying Eimeria spp. from Indian field samples.
In our study, six out of seven species tested in a nested ITS-PCR could be detected from the field samples (Table 3). Among the pathogenic species, the incidence of E. tenella was the highest followed by E. acervulina. Eimeria mitis, which is not known to cause severe infection in chicks, was also detected in 53% of the samples. E. praecox could not be detected in any of the samples tested. Since the samples were taken from birds showing intestinal lesion, it is possible that birds harboring single infection of E. praecox (which does not produce apparent lesion) were not sampled at all. Many of the samples contained multiple infections from Eimeria spp. Our results seem to agree to the observations made previously by other investigators that suggest the occurrence of mixed infection with more than one species of Eimeria being very common and that infection of E. tenella and E. acervulina are the most prevalent species worldwide (Shirley et al. 2005; McDougald et al. 1986).
The ITS-1 regions from a few samples were PCR-amplified and sequenced. Sequence information were obtained for four Eimeria spp. The ITS-1 length of E. acervulina varied from 404 to 407 bp. ITS-1 length of 398 to 404 bp is reported earlier for E. acervulina (Lew et al. 2003; Schnitzler et al. 1998, 1999). In the present study, all the E. brunetti ITS-1 sequences were 449 bp in length, whereas earlier reports suggest the length of 441 to 444 bp for E. brunetti ITS-1 sequence (Lew et al. 2003; Schnitzler et al. 1998, 1999). Similarly, the length of E. tenella ITS-1 sequences was 563 bp except in one sequence where it was 560 bp. The ITS-1 length of 562 and 563 was reported earlier for E. tenella (Lew et al. 2003; Schnitzler et al. 1998). Lew et al. (2003) reported a shorter sequence (456 bp) for ITS-1 of E. tenella. We did not encounter this short variant in the ITS sequences amplified in the present study.
For E. maxima, two different kinds of ITS-1 sequences were identified. One of the sequences (TN_9) was 446 bp in length, and this sequence was similar to the ITS-1 sequence reported by Lew et al. (2003) for the Australian isolates and a Swedish isolate reported by Schnitzler et al. (1999). The other two E. maxima ITS-1 sequences (TN_2 and AP_6) were 322 bp in length and were similar to the E. maxima sequences of American isolates (317–319 bp; Barta et al. 1998). Lew et al. (2003) and Barta et al. (1998) have published E. maxima ITS-1 sequences from Australian and American field isolates, respectively, suggesting the presence of either one of the two variant E. maxima ITS-1 sequences. On the contrary, ITS-1 sequences of E. maxima from our study showed the presence of both the sequence variants. A similar observation was made by Schnitzler et al. (1999) for the Swedish isolates. Therefore, for identification of Eimeria species in India, we suggest the use of nested primers specific for both the E. maxima variants.
Given that our work marks the first attempt involving the use of ITS-PCR for differentiating Eimeria spp. in India, it was important to ascertain the appropriateness of the species-specific primers used in the present study. Therefore, the primers used in the species-specific nested PCR were aligned with the ITS-1 sequences obtained in the present study. The primer sequences of E. acervulina and E. brunetti matched 100% with the ITS-1 sequences of the present study. The E. maxima sequences matched either with the primers designed based on US isolates or on Australian isolate. With respect to E. tenella primers, we noticed a single base pair mismatch both in the forward as well as in the reverse primers. The mismatch in the forward primer was near the 5′ end of the primer (A→C) and was present in many ITS-1 sequences of the present study. Though the mismatch near the 5′ end may not be of much significance, it may be prudent either to use a shorter primer (that terminates before the polymorphic region at the 5′ end) or a degenerate primer consisting of both the bases (A and C) because this substitution was noticed in 50% of the E. tenella ITS-1 sequences. Another single base pair mismatch was seen at the 3′ end of the reverse primer. This mismatch in the reverse primer was present in only one out of the 12 E. tenella ITS-1 sequences of the present study. It is not clear whether the mismatch is real or an error generated by Taq polymerase. Mismatches at the 3′ end of the primers are known to seriously affect the PCR amplification; therefore, it is important to sequence a larger number of E. tenella ITS-1 sequences to determine the presence or absence of this mismatch. Since the PCR product obtained using universal primer was sequenced to determine various ITS-1 sequences, we were in a position to detect the change. If this sequence mismatch at the 3′ end of the primer is real, the incidence of E. tenella may have been underreported.
The selection of clones from the universal PCR product for sequencing was completely random. Therefore, it is possible that the plasmid clones containing E. mitis and E. necatrix ITS-1 sequences were not selected for sequencing. Though the ITS-1 sequences of E. mitis and E. necatrix were not obtained in the present study, these two species were detected in the species-specific nested PCR (Fig. 1). Thus, the available primers may be used for species identification of E. mitis and E. necatrix. Since the minor variations in the primer binding sites may not be reflected in PCR amplification, our claim needs to be substantiated by sequencing the ITS-1 regions from E. mitis and E. necatrix of Indian isolates. Nevertheless, we conclude that the available nested primers for E. mitis and E. necatrix are relevant for the species identification of Indian isolates of Eimeria.
E. praecox was not detected in any of the field samples. It is premature to conclude whether the E. praecox incidence is undetectably low in India or the available nested primers are not suitable to identify the species. It is also possible that the birds with single infection of E. praecox were not sampled at all because E. praecox produce asymptomatic coccidiosis.
In the maximum parsimony tree (Fig. 2), we found a species-wise clustering for ITS-1 sequences from E. tenella, E. acervulina, and E. brunetti. The E. maxima sequences had two different lineages. Two of E. maxima sequences from India grouped along with the US sequences, and another sequence grouped with Australian sequences. These two different E. maxima sequences shared only 30% sequence similarity. Lew et al. (2003), while studying the ITS-1 sequence of Australian Eimeria isolates, found that Australian and American E. maxima grouped separately. PCR primers designed to amplify the American E. maxima did not amplify the Australian E. maxima tested, indicating that the two ITS-1 types do not coexist in the Australian E. maxima. However, in India, both the types of E. maxima seem to be prevalent. Our claim would require further validation using in vivo cross-protection studies to find out whether the two E. maxima lineages represent high strain variation within the Indian isolates of E. maxima. It is not known whether the two E. maxima lineages represent two species lineages (Lew et al. 2003). Lew et al. (2003) also proposed that these divergent ITS-1 sequences may represent rDNA pseudogenes.
In conclusion, six out of seven species of Eimeria were found in chicken intestinal samples collected in India. The available nested primer sequence can be used to identify Eimeria species in India with some minor modifications. E. maxima demonstrates apparent diversity in ITS-1 sequence. However, the immunological diversity of E. maxima isolates of India needs to be further verified because the strain variations in E. maxima may require addition of more than one strain of E. maxima in the live vaccine (Shirley et al. 2007).
References
Barta JR, Martin ED, Liberator PA, Dashkevicz M, Anderson JW, Feighner SD, Elbrecht A, Perkins-Barrow A, Jenkins MC, Danforth HD, Ruff MD, Profous-Juchelka H (1997) Phylogenetic relationships among eight Eimeria species infecting domestic fowl inferred using complete small subunit ribosomal DNA sequences. J Parasitol 83:262–271
Barta JR, Coles BA, Schito ML, Fernando MA, Martin A, Danforth HD (1998) Analysis of intra-specific variation among five strains of Eimeria maxima from North America. Int J Parasitol 28:485–492
Beck HP, Blake D, Dardé M-L, Felger I, Pedraza-Díaz S, Regidor-Cerrillo J, Gómez-Bautista M, Ortega-Mora LM, Putignani L, Shiels B, Tait A, Weir W (2009) Molecular approaches to diversity of populations of apicomplexan parasites. Int J Parasitol 39:175–189
Blake DP, Hesketh P, Archer A, Carroll F, Smith AL, Shirley MW (2004) Parasite genetics and the immune host: recombination between antigenic types of Eimeria maxima as an entree to the identification of protective antigens. Mol Biochem Parasitol 138:143–152
Cantacessi C, Riddell S, Morris GM, Doran T, Woods WG, Otranto D, Gasser RB (2008) Genetic characterization of three unique operational taxonomic units of Eimeria from chickens in Australia based on nuclear spacer ribosomal DNA. Vet Parasitol 152:226–234
Hnida J, Duszynski D (1999) Taxonomy and systematics of some Eimeria species of murid rodents as determined by the ITS-1 region of the ribosomal gene complex. Parasitol 119:349–357
Lew AE, Anderson GR, Minchin CM, Jeston PJ, Jorgensen WK (2003) Inter-and intra strain variation and PCR detection of the internal transcribed spacer l (ITS-1) sequences of Australian isolates of Eimeria species from chickens. Vet Parasitol 112:33–50
Lien YY, Sheu SC, Liu HJ, Chen SC, Tsai MY, Luo SC, Wu KC, Liu SS, Su HY (2007) Cloning and nucleotide sequencing of the second internal transcribed spacer of ribosomal DNA for three species of Eimeria from chickens in Taiwan. Vet J 173:184–189
Long PL, Joyner LP (1984) Problems in the identification of species of Eimeria. J Protozool 31:535–541
McDougald LR, Fuller L, Solis J (1986) Drug-sensitivity of 99 isolates of coccidia from broiler farms. Avian Dis 30:690–694
Mohanty S, Rajendran K (2003) 2020 vision for Indian poultry industry. Int J Poult Sci 2:139–143
Mugridge NB, Morrison DA, Jakel T, Heckeroth AR, Tenter AM, Johnson AM (2000) Effects of sequence alignment and structural domains of ribosomal DNA on phylogeny reconstruction for the protozoan family Sarcocystidae. Mol Biol Evol 17:1842–1853
Nicholas KB, Nicholas HB (1997) GeneDoc: a tool for editing and annotating multiple sequence alignments. Distributed by the author
Ruff MD (1999) Important parasites in poultry production systems. Vet Parasitol 84:337–347
Schnitzler BE, Thebo PL, Mattsson JG, Tomley FM, Shirley MW (1998) Development of a diagnostic PCR assay for the detection and discrimination of four pathogenic Eimeria species of the chicken. Avian Pathol 27:490–497
Schnitzler BE, Thebo PL, Tomley FM, Uggla A, Shirley MW (1999) PCR identification of chicken Eimeria species: a simplified read-out. Avian Pathol 28:89–93
Shirley MW, Smith AL, Tomley FM (2005) The biology of avian Eimeria with an emphasis on their control by vaccination. Adv Parasitol 60:285–330
Shirley MW, Smith AL, Blake DP (2007) Challenges in successful control of the avian coccidia. Vaccine 25:5540–5547
Smith AL, Hesketh P, Archer A, Shirley MW (2002) Antigenic diversity in Eimeria maxima and the influence of host genetics and immunization schedule on cross-protective immunity. Infect Immun 70:2472–2479
Stucki U, Braun R, Roditi I (1993) Eimeria tenella: characterization of a 5S ribosomal RNA repeat unit and its use as a species-specific probe. Exp Parasitol 76:68–75
Su Y-C, Fei AC-Y, Tsai F-M (2003) Differential diagnosis of five avian Eimeria species by polymerase chain reaction using primers derived from the internal transcribed spacer 1 (ITS-1) sequence. Vet Parasitol 117:221–227
Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol 24:1596–1599
Woods WG, Richards G, Whithear KG, Anderson GR, Jorgensen WK, Gasser RB (2000) High-resolution electrophoretic procedures for the identification of five Eimeria species from chickens and detection of population variation. Electrophoresis 21:3558–3563
Zhao, Duszynski DW, Loker ES (2001) A simple method of DNA extraction for Eimeria species. J Microbiol Meth 44:131–137
Acknowledgments
The authors thank Dr. V. A. Srinivasan, Dr. D. Thiagarajan, and Mr. N. M. Ponnanna for critically reviewing the manuscript. The authors also thank Mr. N. Hanumantha Rao and Mr. P. Baji Babu for their technical help.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bhaskaran, M.S., Venkatesan, L., Aadimoolam, R. et al. Sequence diversity of internal transcribed spacer-1 (ITS-1) region of Eimeria infecting chicken and its relevance in species identification from Indian field samples. Parasitol Res 106, 513–521 (2010). https://doi.org/10.1007/s00436-009-1696-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00436-009-1696-2