
Abstract
Main conclusion
A green algal phenol oxidase was firstly purified, confirmed to be a laccase, and a hetero-oligomeric quaternary structure is suggested. The operation of a laccase-mediator system is firstly described in algae.
Laccases (EC 1.10.3.2) catalyze the oxidation of a multitude of aromatic substrates. They are well known in higher plants and fungi, while their presence in green algae appears uncertain. Extracellular laccase-like enzyme activity has previously been described in culture supernatants of the green soil alga Tetracystis aeria [Otto et al. in Arch Microbiol 192:759–768, (2010)]. As reported herein, the T. aeria enzyme was purified 120-fold by employing a combination of anion exchange and size exclusion chromatography. The purified enzyme was confirmed to be a laccase according to its substrate specificity. It oxidizes 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS), syringaldazine, and 2,6-dimethoxyphenol (pH optima of pH ≤2.5, 7.0, and 6.5; K m values of 28.8, 40.5, and 1,830 µM; respectively), but not l-tyrosine or Fe2+. ABTS is by far the most efficient substrate. Two polypeptides, A (~110 kDa) and B (71 kDa), were co-purified by the applied procedure, both being highly N-glycosylated (≥~53 and ≥27 %, respectively). As suggested by various gel electrophoretic analyses, the native enzyme (apparent molecular mass of ~220 kDa) most probably is a hetero-oligomer with the composition AB 2 , wherein A is the catalytic subunit and B forms a disulfide-linked homo-dimer B 2 . The decolorization of anthraquinone (Acid Blue 62 and Remazol Brilliant Blue R) and diazo dyes (Reactive Black 5) was studied in the presence of redox-mediating compounds (ABTS and syringaldehyde), demonstrating the operation of the laccase-mediator system in algae for the first time. Thus, laccases from green algae may participate in the biotransformation of a wide spectrum of natural and xenobiotic compounds.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Laccases (EC 1.10.3.2, p-diphenol: O2 oxidoreductases) are mostly extracellular glycoproteins belonging to the group of multicopper oxidases. They catalyze the one-electron oxidation of a wide range of substrates such as mono- and polyphenols and aromatic amines, concomitantly reducing molecular oxygen to water. Substrates are oxidized to radicals, which may undergo cross-linking or depolymerization reactions thereafter (Claus 2004). Appropriate substrates may also act as so-called redox mediators, which enable the indirect oxidation of an even wider range of compounds including lignin and, e.g., polyaromatic hydrocarbons (Morozova et al. 2007b). Laccase was firstly described by Yoshida (1883) in the Japanese lacquer tree Rhus vernicifera. The enzyme causes the wound sap of the tree to harden in contact with air, whereas other plant laccases are thought to contribute to the polymerization of monolignols and hence to the biosynthesis of lignin (Sterjiades et al. 1992). Plant laccases are further involved in a plethora of physiological processes like cytokinin homeostasis (Galuszka et al. 2005), resistance to phenolic pollutants (Wang et al. 2004), flavonoid polymerization in seed coats (Pourcel et al. 2005) and iron metabolism (Hoopes and Dean 2004). Only a few laccases from plants are characterized biochemically (Morozova et al. 2007a), but phylogenetic analysis reveals a high diversity and widespread occurrence of the enzymes in higher plants (McCaig et al. 2005). Apart from plants, laccases are known in prokaryotes and insects (Claus 2004) and by far the most laccases studied as yet are produced by higher fungi (Baldrian 2006). The fungal enzymes are assumed to participate in the breakdown of lignin, but may also fulfill several other functions related to detoxification processes, morphogenesis, pathogenicity and more (Claus 2004). Laccases are found in lichens as well, where they are almost certainly produced by the fungal rather than by the algal symbiont (Lisov et al. 2012). Laccases have attracted extensive interest due to their ecological importance as well as manifold applications, such as the bioremediation of soils and waters, usage in the food and textile industries and for chemical syntheses (Riva 2006).
While the biotransformation of aromatic compounds by fungi and bacteria has been studied intensively, the ability of algae to degrade diverse phenolic and other aromatic compounds, like synthetic dyes and polyaromatic hydrocarbons, is just increasingly recognized (Semple et al. 1999; Ghasemi et al. 2011; Subashchandrabose et al. 2013). Phenol metabolism in algae may proceed via hydroxylation and ring cleavage pathways (Semple et al. 1999). In view of the phylogenetic distribution of laccases, it could be expected that these enzymes are present not only in higher, but in more ancestral plants as well (Dean and Eriksson 1994; McCaig et al. 2005). In red algae, laccase-like enzyme activity was described in an early report by Rönnerstrand (1943). The putative presence of algal laccases has been discussed with respect to the evolution of lignin and related polymers (Dean and Eriksson 1994) and has also been proposed to contribute to the biodegradation of diverse phenolic pollutants (Subashchandrabose et al. 2013; Chiaiese et al. 2011; Kılıç et al. 2011). Recently, various green algae were found to oxidize typical laccase substrates extracellularly (La Russa et al. 2008; Otto et al. 2010; Chiaiese et al. 2011; Kılıç et al. 2011). Attempts to identify laccase-encoding genes in green algae, by screening sequenced genomes or using degenerate primers, have not been successful so far (Richter et al. 2012; McCaig et al. 2005) except for one report, where six metagenome-derived laccase-like sequence reads are mentioned to be assigned to green algae (Zakrzewski 2012). In a previous study, we have described the production and some features of a laccase-like enzyme in culture supernatants of the green soil alga Tetracystis aeria (Otto et al. 2010). However, no algal phenol oxidase has been purified and characterized to date and their occurrence still appears enigmatic.
In order to address the remaining uncertainty about the presence and nature of laccase-like phenol oxidases in green algae, we aimed to purify the enzyme from T. aeria, thus enabling the investigation of its basic structural and kinetic properties. Furthermore, the degradation of synthetic dyes by the algal enzyme in the presence of redox-mediating compounds is firstly reported herein.
Materials and methods
Cultivation of algae and enzyme production
Tetracystis aeria SAG 89.80 [Culture Collection of Algae at the University of Göttingen (SAG), Germany] was grown in liquid mineral medium, as described previously (Otto et al. 2010). For enzyme production, 400-mL algal cultures were supplemented with 20-µM CuSO4 at the beginning of cultivation (Otto et al. 2010) and cultures were harvested after 7 weeks of growth. The chlorophyll (a + b) content, used as an indicator for algal biomass, was determined after extraction with 80 % acetone according to Ziegler and Egle (1965) and increased from 3.4 (±1.1) to 12.2 (±1.2) µg mL−1 (mean ± SD, n = 4) during that period.
Enzyme isolation and purification
Algal cultures were sequentially passed through filter paper, 0.45-µm regenerated cellulose, and 0.2-µm cellulose acetate membrane filters (Whatman No. 1, RC 55, and OE 66, respectively; GE Healthcare; Little Chalfont, UK) to remove algal cells and undissolved material. Proteins in the obtained cell-free culture supernatants were concentrated and simultaneously exchanged to 25-mM Tris–HCl (pH 7.5) by ultrafiltration on a 30-kDa cut-off PES membrane connected to a 400-mL stirred cell (Amicon 8400; Millipore; Billerica/MA, USA). The concentrate was centrifuged (10,000×g for 20 min) and the obtained supernatant was used for chromatographic enzyme purification.
All protein chromatographic separations were carried out at room temperature on a BioLogic HR system (Bio-Rad; Munich, Germany). For ion exchange chromatography (IEC), concentrated culture supernatant (about 4–7 U per run) was loaded onto three HiTrap Q HP columns (5 mL each; GE Healthcare) connected in series, which had been pre-equilibrated with 25-mM Tris–HCl (pH 7.5). After washing with 100 mL of 30-mM NaCl, proteins were separated within a 120-mL linear gradient of 30–150 mM NaCl at a flowrate of 4 mL min−1. A bulk of brownish material was eluted subsequently upon washing with a 20-mL linear gradient of 0.15–1 M NaCl (Fig. 1a). Fractions of the major activity peak were pooled (Fig. 1a) and re-concentrated, using the stirred cell mentioned above followed by Vivaspin 15R centrifugal devices (30 kDa cut-off; Sartorius; Göttingen, Germany). For size exclusion chromatography (SEC), two Superdex 200 10/300 GL columns (GE Healthcare) were connected in series (total bed height 60–62 cm). Up to 0.75 mL of sample was injected. Elution was performed using 25-mM Tris–HCl plus 150-mM NaCl (pH 7.5) at a flowrate of 0.5 mL min−1. Fractions corresponding to the second activity peak were narrowly pooled, as indicated in Fig. 1b, and instantly re-concentrated (Amicon 0.5, 30 kDa cut-off; Millipore). Thereafter, the obtained samples were subjected to a second SEC separation under identical conditions. Active fractions were narrowly pooled as before (Fig. 1c). The SEC columns had been calibrated using LMW and HMW Gel Filtration Calibration Kits (GE Healthcare). Instantly after the second SEC, the obtained samples were diluted with 4 volumes of 25-mM Tris–HCl (pH 8.5) and loaded onto a Mono Q 5/50 GL column (GE Healthcare) for a final IEC. A 15-mL linear gradient of 30–200 mM NaCl in 25-mM Tris–HCl (pH 8.5) was employed at a flowrate of 0.5 mL min−1. Active fractions were pooled (Fig. 1d), re-concentrated (Amicon 0.5, 30 kDa cut-off; Millipore), and stored at −80 °C. The entire purification procedure took about 5–6 days.

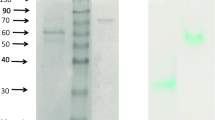
Purification of the T. aeria laccase. a–d Chromatograms. a IEC on HiTrap Q HP. b First SEC on Superdex 200. c Second SEC (re-chromatography). d IEC on Mono Q. Enzyme activities (single determinations) of pooled and non-pooled fractions, the UV absorbance at 280 nm, and the NaCl gradients (for a and d) are indicated. Activity peaks in SEC (b and c) are labeled with the corresponding apparent molecular masses. e The course of purification, as visualized by silver-stained SDS–PAGE under reducing conditions. Concentrated culture supernatant obtained by ultrafiltration (lane 1), as well as enzyme pools obtained from IEC on HiTrap Q HP (lane 2), the first SEC on Superdex 200 (lane 3), the second SEC on Superdex 200 (lane 4), and IEC on Mono Q (lane 5) were applied on a 4–15 % Tris–HCl gel. Sample amounts corresponded to 1 mU (lanes 1–2) and 4 mU (lanes 3–5) of enzyme activity. Apparent molecular masses of a protein ladder and two protein bands obtained after enzyme purification are indicated on the left and right, respectively
Protein concentrations were determined with the bicinchoninic acid method, using the Novagen BCA Protein Assay Kit (EMD Chemicals; Billerica/MA, USA) according to the instructions of the supplier. Bovine serum albumin served as a standard.
For SEC analysis of enzyme aggregation and chromatofocusing, partially purified enzyme preparations were used, which had been obtained by IEC separations on HiTrap Q and Mono Q columns. SEC was done as described above, except that only one Superdex 200 10/300 GL column was used and elution was carried out with 50 mM Na–phosphate (pH 7.0) containing 0.15-M NaCl at a flowrate of 0.75 mL min−1. For chromatofocusing, samples were loaded onto a Mono P 5/50 GL column (GE Healthcare), which had been pre-equilibrated with 25-mM Bis-Tris–HCl buffer (pH 6.5). A gradient of pH 6.5–4.0 was established using 7 % Polybuffer 74–HCl (pH 4.0) at a flowrate of 0.5 mL min−1.
Structural enzyme characterization
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) was done following the method of Laemmli (1970), using 4–15 % precast gels (10-well Tris–HCl Ready Gel; Bio-Rad) in a Mini PROTEAN 3 cell (Bio-Rad). Samples were routinely denatured by heating with 1 volume of 2× Laemmli sample buffer (Bio-Rad), which contained 5 % β-mercaptoethanol, for 5 min at 95 °C. Where indicated, samples were not heated and/or β-mercaptoethanol was omitted. Gels were stained for protein using the Silver Stain Kit (Bio-Rad). Glycoproteins were stained using the Pro-Q Emerald 300 Gel Stain Kit (Molecular Probes; Eugene/OR, USA). For enzyme activity staining, gels were soaked in 0.1 M citrate–0.2 M phosphate buffer (pH 4.0) containing 2-mM 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS; ultra pure; AppliChem; Darmstadt, Germany) for about 10–15 min. Apparent molecular masses were inferred by reference to a 10–250 kDa protein ladder (NEB; Ipswich/MA, USA).
For chemical cross-linking of protein complexes with glutaraldehyde, proteins were exchanged to 50-mM Na–phosphate buffer (pH 7.0; protein concentration ~30 µg mL−1). Glutaraldehyde was added to a final concentration of 0.01–0.1 %. After incubation at 25 °C for 5–20 min, the reaction was stopped by the addition of sample buffer prior to SDS–PAGE.
Prior to enzymatic N-deglycosylation, proteins were denatured with 0.02 % SDS and 10 mM β-mercaptoethanol at 95 °C for 10 min. Peptide-N-glycosidase F (PNGase F; Sigma–Aldrich; St. Louis/MO, USA) was added to a final concentration of 50 U L−1 and allowed to react for 2.5 h at 37 °C. The reaction was stopped by heating with sample buffer prior to SDS–PAGE.
Two-dimensional (2D) SDS–PAGE was done following the approach of Xia et al. (2007). In the first dimension of SDS–PAGE, proteins were separated under zymogram conditions, i.e., samples were not heated and β-mercaptoethanol was omitted. Purified enzyme samples (0.5 µg of protein) were mixed with sample buffer and loaded to one well of a 10-well gel (4–15 %). After electrophoresis, the corresponding lane was cut from the gel for further separation in the second dimension of SDS–PAGE. Proteins were fully denatured by boiling the gel strip in sample buffer containing 5 % β-mercaptoethanol for 10 min. Thereafter, the strip was horizontally placed onto a second gel (4–15 %, preparative-well gel) and electrophoresed.
Substrate specificity and kinetic parameters
For routine determination of enzyme activity, the oxidation of ABTS (2 mM) was monitored at 420 nm (ε 420 = 36 mM−1 cm−1; Childs and Bardsley 1975) in 0.1 M citrate–0.2 M phosphate buffer (pH 4.0). One unit of enzyme activity (U) corresponds to the oxidation of 1 µmol substrate per minute.
The pH-activity profiles for the oxidation of ABTS (2 mM), syringaldazine (≥98 % purity; Sigma; St. Louis/MO, USA; ε 530 = 65 mM−1 cm−1; Harkin and Obst 1973; 20 µM in 10 % ethanol) and 2,6-dimethoxyphenol (2,6-DMP; 99 % purity; Aldrich, St. Louis/MO, USA; ε 470 = 27.5 mM−1 cm−1; Martínez et al. 1996; 2 mM) by the purified enzyme were measured at 420, 530 and 470 nm, respectively, in 0.1 M Britton and Robinson buffers (Xu 1997) of varied pH values. Kinetic parameters were obtained by measuring the oxidation of 0.5–1,000 µM ABTS (at pH 2.5), 1–50 µM syringaldazine (at pH 7.0), and 10–5,000 µM 2,6-DMP (at pH 6.5). The obtained data were fitted to the Michaelis–Menten kinetics model by non-linear regression using SigmaPlot 11.0 (Systat Software; Chicago/IL, USA).
Tyrosinase activity was measured with l-tyrosine (≥98 % purity; Sigma–Aldrich; 1 mM) as the substrate at pH 4.0 and 7.0 in 0.1 M citrate–0.2 M phosphate buffers by monitoring dopachrome formation at 475 nm (ε 475 = 3.7 mM−1 cm−1; Cabanes et al. 1987) for 15 min. Ferroxidase activity was measured with ferrous ammonium sulfate (0.2 mM; from a freshly prepared stock solution in deaerated buffer) as the substrate in 0.1 M Na–acetate buffer (pH 5.0). After 10 min of incubation at 25 °C, remaining Fe2+ was complexed by addition of ferrozine (≥97 % purity; Fluka; St. Louis/MO, USA) at 1.6 mM and the absorption at 562 nm (ε 562 = 27.9 mM−1 cm−1; Stookey 1970) was read in a Specord 250 plus spectrophotometer (Analytik Jena; Jena, Germany).
The dyes Acid Blue 62 (ABu62; >98 % purity; Yorkshire Chemicals; Tertre, Belgium), Remazol Brilliant Blue R (RBBR; pure grade; Acros Organics; Geel, Belgium) and Reactive Black 5 (RB5; 55 % purity; Sigma–Aldrich) were treated with partially purified enzyme (as obtained after the HiTrap Q followed by one SEC separation). Final concentrations of 50 µM of dye and 50 mU mL−1 of enzyme (as determined with ABTS as the substrate at pH 5.0) were added to total assay volumes of 0.2 mL in 0.1 M citrate–0.2 M phosphate buffer (pH 5.0). The potential redox mediators ABTS and syringaldehyde (98 % purity; Aldrich) were additionally included at 50 µM. Samples were incubated in sealed 96-well microplates at room temperature (~25 °C). Decolorization was monitored at wavelengths of 590 nm (ABu62 and RBBR) and 600 nm (RB5) by reference to controls where enzyme was omitted.
Except for the ferroxidase assay, all measurements were done using a GENios + microplate reader (Tecan; Crailsheim, Germany) at 25 (±1) °C. Unless stated otherwise, measurements were done in triplicates. Values were corrected for blanks obtained without enzyme.
Results
Enzyme purification
Laccase-like enzyme activity was purified from culture supernatants of T. aeria by employing a combination of IEC and SEC separations (Fig. 1a–d). The enzyme was purified 120-fold to finally yield a specific activity of 71 mU µg−1 (Table 1). Challenges of purifying and identifying the enzyme arose due to its low initial amount, activity losses during purification, soluble aggregate formation as well as certain structural features (i.e., a high degree of glycosylation and a most probably hetero-oligomeric structure; see below).
Following an initial IEC step (Fig. 1a), SEC separation gave two peaks of enzyme activity, which corresponded to apparent molecular masses of ~410 and ~220 kDa (Fig. 1b). Only fractions of the latter activity peak of ~220 kDa were used for further enzyme purification as this peak is suggested to correspond to the native state of the enzyme, in accordance with the previous estimation of about 212 kDa (Otto et al. 2010). The higher molecular mass activity peak of ~410 kDa apparently represented soluble aggregates artificially formed during initial purification steps, as was suggested by SEC separations at various stages of purification during the method development. The formation of enzyme aggregates presumably involved hydrophobic interactions among proteins, as SEC activity peaks of around 400 kDa were found to be particularly pronounced when hydrophobic interaction chromatography in the presence of the anti-chaotropic salt ammonium sulfate had been employed during earlier attempts to purify the enzyme (data not shown). Furthermore, enzyme aggregation was superficially induced by incubation of partially purified enzyme with 1-M ammonium sulfate (Fig. 2a). Increased aggregation was also observed after chromatofocusing of partially purified enzyme preparations, where a single activity peak eluted around pH 5.0 (Fig. 2b) in accordance with an isoelectric point of 4.8 previously determined by isoelectric focusing (Otto et al. 2010). The use of hydrophobic interaction chromatography and chromatofocusing for enzyme purification was finally avoided therefore. Some inevitable aggregates, formed during initial purification steps, were effectively removed by employing the two-step high resolution SEC (Fig. 1b, c). Furthermore, the SEC separation enhanced the purity of the enzyme by roughly one order of magnitude, as compared to the preceding IEC step (Table 1; Fig. 1e). Final polishing was done by IEC on a Mono Q column (Fig. 1d). SEC analysis confirmed that no further enzyme aggregates were formed during the final purification steps (data not shown). A pattern of two enzyme activity bands of ~220 and ~110 kDa was likewise obtained in zymograms of the non-purified concentrated culture supernatant (data not shown) as well as with the purified enzyme (Fig. 3a, lane 4; see below). When samples were acetone precipitated prior to zymogram analysis, the activity band of ~220 kDa tended to become predominating (data not shown). Considering this observation, the previously observed predominance of a >200 kDa zymogram activity band (Otto et al. 2010) was apparently due to protein aggregation induced by the acetone precipitation step.

Enzyme aggregation in presence of ammonium sulfate (a) and chromatofocusing (b) of partially purified T. aeria laccase. a Samples were supplemented with 1 M of ammonium sulfate (AS), incubated at room temperature and subjected to SEC after the indicated incubation times. Identical samples without AS served as controls. Enzyme activities in eluted fractions are calculated relative to the total eluted activity of the respective run. Apparent molecular masses corresponding to activity peaks are indicated as an approximation. Data represent means ± absolute deviations from duplicate determinations. b Chromatofocusing on Mono P within a gradient of pH 6.5–4.0. Enzyme activities (from single determinations) and pH values of eluted fractions are indicated

Structural characterization of T. aeria laccase-associated proteins by SDS–PAGE analyses. a SDS–PAGE under varied conditions. Samples were heated (95 °C for 5 min) with β-mercaptoethanol (β-ME) (i.e., standard conditions) (lane 1), heated without β-ME (lane 2), and non-heated without β-ME (lanes 3, 4). Gel sections were silver-stained for protein (lanes 1–3) or stained with ABTS for laccase activity (lane 4). b Chemical cross-linking of protein complexes. Untreated control (lane 1), samples treated with 0.01 % glutaraldehyde for 5 min (lane 2), with 0.1 % glutaraldehyde for 5 min (lane 3), and with 0.1 % glutaraldehyde for 20 min (lane 4) were subjected to standard SDS–PAGE. The gel was silver-stained. c 2D SDS–PAGE. Samples were applied non-heated and without β-ME for the first dimension of SDS–PAGE. The resulting lane was cut from the gel, heated in presence of β-ME, and horizontally placed onto a second gel for the second dimension of SDS–PAGE. Reference lanes of the first dimension of SDS–PAGE were either silver-stained for protein or stained for laccase activity with ABTS as indicated, and the resulting 2D gel was silver-stained. Protein spots are assigned to the corresponding polypeptides of ~110 kDa (A) and 71 kDa (B), as indicated on the right. The spots are suggested to be derived from the subunit associations AB 2 and B 2 and monomeric A, separated in the first dimension (see “Results”), as indicated below d Glycosylation analysis. Untreated sample (lanes 1, 4), sample digested with PNGase F (lanes 2, 5), and PNGase F as control (lane 3) were subjected to standard SDS–PAGE. The gel was silver-stained (lanes 1–3), after being previously stained for carbohydrate using Pro-Q Emerald 300 (lanes 4–5). Apparent molecular masses of the protein ladder (in italics) and protein bands are indicated
Two polypeptides were always co-purified by the purification procedure, which appeared as a rather diffuse band of ~110 kDa (hereafter referred to as A) and a band of 71 kDa (hereafter referred to as B) in SDS–PAGE under reducing conditions (Fig. 1e). The identity and quaternary structure of the enzyme protein hence needed to be further elucidated.
Structural enzyme characterization
In order to gather structural information about the proteins of the obtained enzyme preparation, the effect of varied treatments was analyzed by SDS–PAGE (Fig. 3a). Possible disulfide linkages among the polypeptides A and/or B were investigated using SDS–PAGE under reducing and non-reducing conditions (i.e., heating of samples to 95 °C with and without β-mercaptoethanol) (Fig. 3a, lanes 1–2). Zymograms were obtained by omitting the heating step prior to SDS–PAGE under non-reducing conditions, thus allowing a “semi-native” separation and subsequent staining for enzyme activity with the chromogenic laccase substrate ABTS (Fig. 3a, lanes 3–4). A broad protein band of ~110 kDa (polypeptide A) was consistently observed under reducing, non-reducing, as well as zymogram conditions (Fig. 3a, lanes 1–3) and also coincided with an enzyme activity band of ~110 kDa (Fig. 3a, lane 4). The polypeptide A is therefore supposed to be a catalytically active subunit of the enzyme. Another protein band of 71 kDa (polypeptide B), which appeared under reducing conditions (Fig. 3a, lane 1), diminished under non-reducing conditions and a new band of ~140 kDa appeared instead (Fig. 3a, lane 2). This observation suggests that the polypeptide B forms a disulfide-linked homo-dimer B 2 . Under zymogram conditions, the protein staining of around ~140 kDa appeared less distinct (Fig. 3a, lane 3). Additionally, both a protein and a corresponding activity band of ~220 kDa were obtained in zymograms (Fig. 3a, lanes 3–4), apparently representing the intact enzyme.
In order to further characterize the putative oligomeric protein complexes, proteins were chemically cross-linked using glutaraldehyde prior to SDS–PAGE analysis. In addition to the still remaining bands of ~110 (A) and 71 kDa (B), two new bands of 240 and 170 kDa were formed upon treatment with glutaraldehyde (Fig. 3b), thus confirming the presence of some kind of protein complexes. The values of 240 and 170 kDa most closely match molecular masses of ~250 and ~180 kDa that would approximately be expected for the putative cross-linking products AB 2 and AB, respectively. Other putative associations, namely, A 2 (expected molecular mass of ~220 kDa) or B 2 (expected molecular mass of ~140 kDa), could also not be excluded. Nevertheless, a heteromeric structure of the native enzyme with the composition AB 2 (wherein B 2 apparently was not cross-linked intramolecularly by glutaraldehyde) appeared to be more likely.
Additional information on the quaternary structure of the assumed complexes was intended to be gained by a 2D SDS–PAGE approach. Proteins were separated under zymogram conditions in the first dimension, followed by fully denaturing conditions (i.e., sample heating and β-mercaptoethanol application) in the second dimension of SDS–PAGE. The two activity bands of about 220 and 110 kDa as well as protein patches of about 220 and around 110–140 kDa, as already observed with one-dimensional SDS–PAGE under zymogram conditions (Fig. 3a, lanes 3–4), were accordingly detected in the first dimension of the 2D SDS–PAGE (Fig. 3c). Applying fully denaturing conditions for the second dimension of SDS–PAGE, both the ~110 (A) and the 71 kDa (B) polypeptides appearing after regular one-dimensional SDS–PAGE (Fig. 3a, lane 1) were retrieved at positions of about 100–140 and 70–80 kDa within the second dimension, respectively (Fig. 3c). One spot of A was found to apparently derive from the ~110 kDa activity band of the first dimension of SDS–PAGE (Fig. 3c), which accordingly represented monomeric A as the catalytically active subunit. One spot of B derived from a position of roughly 120–140 kDa within the first dimension, apparently representing the disulfide-linked homo-dimer B 2 as already observed in one-dimensional SDS–PAGE under non-reducing conditions (Fig. 3a, lane 2; see above). A second, weakly stained spot of A and a second spot of B apparently both derived from the ~220 kDa activity and protein band of the first dimension (Fig. 3c), suggesting that polypeptides A and B both are subunits of the intact protein complex. Therefore, a hetero-oligomer with the composition AB 2 finally is assumed to be the most probable quaternary structure.
A possible glycosylation of the two polypeptides A and B was investigated using enzymatic N-deglycosylation and SDS–PAGE (Fig. 3d). Prior to deglycosylation, the two silver-staining protein bands of ~110 and 71 kDa (Fig. 3d, lane 1) stained for carbohydrate as well (Fig. 3d, lane 4), thus confirming their glycosylation. After deglycosylation, a protein band of 52 kDa and a doublet band of 40/38 kDa were obtained (Fig. 3d, lane 2). No staining for carbohydrate was detected after deglycosylation (Fig. 3d, lane 5), indicating complete deglycosylation. The bands of the deglycosylated polypeptides could not unambiguously be assigned to the corresponding bands of the glycosylated polypeptides. However, taking the largest deglycosylated polypeptide of 52 kDa as a basis, glycan contents of ~53 and 27 % at the minimum could be estimated for the polypeptides A and B, respectively.
Substrate specificity and kinetic parameters
The pH-dependence and kinetic parameters for the oxidation of three canonical laccase substrates, ABTS, syringaldazine, and 2,6-DMP, were determined with the purified enzyme. ABTS was increasingly oxidized with decreasing pH values in the range of pH 2.5–8.5 (Fig. 4). For syringaldazine and 2,6-DMP, bell-shaped profiles with optima around neutral pH (pH 7.0 and 6.5, respectively) were obtained (Fig. 4). The kinetic parameters for the oxidation of ABTS, syringaldazine and 2,6-DMP are given in Table 2. The oxidation of all substrates followed Michaelis–Menten kinetics. The affinity of the enzyme was high for ABTS (K m = 28.8 µM) and syringaldazine (K m = 40.5 µM) and rather weak for 2,6-DMP (K m = 1,830 µM) (Table 2). The rank order of catalytic efficiencies (V max/K m) was ABTS > syringaldazine > 2,6-DMP (Table 2). Neither l-tyrosine nor Fe2+ was oxidized by the enzyme.

pH-activity profiles for the oxidation of ABTS, syringaldazine (SGZ) and 2,6-DMP by the purified T. aeria laccase in 0.1 M Britton and Robinson buffers. Data represent mean ± SD from triplicate determinations
The decolorization of synthetic dyes in the presence of the putative redox-mediating compounds ABTS and syringaldehyde was investigated using partially purified enzyme (Fig. 5). In the absence of mediators, only ABu62 was substantially decolorized (60 % decolorization after 4 days; Fig. 5). ABu62 decolorization was initially delayed in the presence of mediators, but finally decolorization in presence of syringaldehyde exceeded that observed in its absence (72 % after 4 days; Fig. 5). Decolorization in presence of ABTS was less effective (48 % after 4 days; Fig. 5). In the ABTS-containing samples of all three dyes, absorption values increased by about 0.15 absorbance units within the first hour of incubation, which was obviously due to an interference of the absorbance of oxidized ABTS with that of the respective dye. Hence, formally negative values of decolorization were initially obtained (not shown in Fig. 5 for clarity). However, as was clearly observed later on, RBBR and RB5 were effectively decolorized in the presence of ABTS (53 and 44 % after 4 days, respectively; Fig. 5). Only slight decolorization of RBBR and RB5 was obtained in presence of syringaldehyde (8 and 6 % after 4 days, respectively; Fig. 5).

Decolorization of synthetic dyes by T. aeria laccase and putative redox mediators. Dyes (50 µM) were treated either with the partially purified enzyme alone or additionally including ABTS or syringaldehyde (SGD) in 0.1 M citrate–0.2 M phosphate buffer (pH 5.0) (see “Materials and methods” for details). Decolorization is given by reference to controls without enzyme. Apparently negative decolorization values (see “Results”) are not depicted for clarity. Data represent mean ± SD from triplicate experiments
Discussion
Within this study, we established a purification procedure for the laccase-like enzyme from T. aeria culture supernatants, which enabled the subsequent investigation of basic structural and functional characteristics of the enzyme.
High affinities of the purified enzyme towards the substrates ABTS and syringaldazine, indicated by K m values in the two-digit µM range (Table 2), and a rather low affinity towards 2,6-DMP resemble the typical kinetic properties of laccases (Baldrian 2006; Frasconi et al. 2010). Similar kinetic features were also observed with the concentrated culture supernatant of T. aeria (Otto et al. 2010). ABTS was by far the most efficient substrate in terms of the V max/K m ratio as compared to syringaldazine and 2,6-DMP (Table 2). Plant laccases often show a low or even no activity towards syringaldazine (Joel et al. 1978; Sterjiades et al. 1993; Frasconi et al. 2010). A possible oxidation of syringaldazine according to the Hill model, as assumed previously (Otto et al. 2010), is not confirmed with the pure enzyme, as all substrates are oxidized according to simple Michaelis–Menten kinetics. Along with the absence of tyrosinase activity, the oxidation of syringaldazine in the absence of hydrogen peroxide is considered to be a truly unique feature of laccases (Harkin et al. 1974; Baldrian 2006). Some laccase-like enzymes are known to act as ferroxidases (De Silva et al. 1997; Hoopes and Dean 2004). In the green alga Chlamydomonas reinhardtii, a multicopper ferroxidase was described by La Fontaine et al. (2002). However, the T. aeria enzyme shows no ferroxidase activity and hence behaves like a typical laccase.
An increasing oxidation of the non-phenolic substrate ABTS with decreasing pH values and bell-shaped pH-activity profiles toward the phenolics syringaldazine and 2,6-DMP reflect the typical behavior of laccases as well (Baldrian 2006) and confirm earlier observations obtained with concentrated culture supernatants of T. aeria (Otto et al. 2010). The oxidation of syringaldazine with a pH optimum of pH 7.0 by the purified enzyme is consistent with earlier findings (Otto et al. 2010), while the optimum of pH 6.5 toward 2,6-DMP is lower than the value of pH 8.0–8.5 previously reported for the concentrated culture supernatant (Otto et al. 2010). The pattern of zymogram activity bands remained unchanged before and after enzyme purification, indicating that the purified laccase is the sole phenol oxidase excreted by the algae. However, possibly other enzymes or, e.g., redox-mediating compounds released by the algae might have affected the oxidation of 2,6-DMP by culture supernatants at alkaline pH. Most fungal laccases have acidic pH optima (Baldrian 2006), while neutral to alkaline pH optima toward phenolic substrates are reported for the plant laccase from R. vernicifera (Omura 1961; Shiba et al. 2000).
The potential of the enzyme to attack recalcitrant organic compounds, either directly or with the involvement of redox-mediating compounds, was exemplarily studied by assessing the decolorization of two anthraquinone dyes, ABu62 and RBBR, and the diazo dye RB5. ABu62 is already substantially decolorized by the laccase alone, as also recorded earlier when concentrated culture supernatant of the algae was employed (Otto et al. 2010). An initial delay of decolorization in the presence of mediators, ABTS and syringaldehyde (Fig. 5), is possibly due to competitive inhibition during the concurrent oxidation of dye and mediator. However, finally the decolorization is enhanced in the presence of syringaldehyde. A less effective decolorization in presence of ABTS might be caused by the interference of oxidized ABTS in the absorbance measurement. Like many fungal laccases (Soares et al. 2001; Zille et al. 2004; Pereira et al. 2009; Camarero et al. 2005), the T. aeria laccase is unable to effectively decolorize the highly recalcitrant dyes RBBR and RB5 in the absence of redox mediators, but substantial decolorization of these dyes is achieved in the presence of ABTS (Fig. 5) (Moilanen et al. 2010; Tavares et al. 2008). Some algae can degrade several azo dyes by the enzyme azo reductase (Jinqi and Houtian 1992). Kılıç et al. (2011) already suggested a possible role of a laccase in the removal of RBBR by cultures of the green alga Gonium sp., where dye removal coincided with extracellular ABTS oxidation activity. Our results confirm the potential of algal laccase in the decolorization of synthetic dyes and furthermore demonstrate the operation of the laccase-mediator system in algae for the first time.
An apparent molecular mass of ~220 kDa for the native enzyme, as indicated by SEC and zymogram analyses, is basically in accordance with a previous estimation of about 212 kDa (Otto et al. 2010). Two polypeptides A (~110 kDa) and B (71 kDa), as co-purified by our purification procedure, most probably constitute a heteromeric quaternary structure of the native enzyme with the subunit composition AB 2 , wherein B forms a disulfide-linked homo-dimer B 2 of ~140 kDa. The catalytically active polypeptide A resembles the molecular masses of 90–130 kDa reported for most (monomeric) laccases from plant origin (Morozova et al. 2007a). It should be noted that, especially for glycoproteins, apparent molecular masses obtained by SEC may differ considerably from their actual molecular masses (Beeley 1985). Furthermore, values may be underestimated by zymograms, due to a rather compact shape of not fully denatured proteins (Thurston 1994). The actual molecular mass of the intact enzyme might therefore rather be around 240–250 kDa, as would be expected by summing the apparent molecular masses of the comprised polypeptides (~250 kDa for AB 2 ) and as indicated by a cross-linking protein band of 240 kDa.
Though most laccases are monomeric enzymes, several homo- and some hetero-oligomeric laccases are known in fungi (Giardina et al. 2010) and bacteria (Diamantidis et al. 2000). The most intensively studied heteromeric laccases are POXA3a and POXA3b from the white-rot fungus Pleurotus ostreatus (Palmieri et al. 2003; Giardina et al. 2007; Ferraroni et al. 2014), which are hetero-dimers comprising a large (61 kDa) and a small (16 or 18 kDa) subunit. While the large subunit is homologous to laccases, the function of the small subunits still remains uncertain and may be related to the stabilization and activation of the heteromer (Faraco et al. 2008) and furthermore appears to exert a physiological role during fructification (Pezzella et al. 2013). Enzyme stabilizing or regulatory roles conferred by quaternary structure have likewise been suggested for other oligomeric fungal and bacterial laccases (Xu et al. 1996; Diamantidis et al. 2000). There is no indication for the function of the assumed subunit B 2 of the T. aeria enzyme so far. A role in enzyme stabilization or regulation may also apply here. Conceivably, B 2 might also act as a dirigent protein (Pickel and Schaller 2013). Dirigent proteins are homodimeric extracellular glycoproteins known to direct the regio- and stereospecific product formation of phenoxy radical coupling reactions and have been implicated with laccase-catalyzed reactions in lignan synthesis in higher plants (Pickel and Schaller 2013).
Both polypeptides A and B are highly N-glycosylated. Glycan contents of ~53 and 27 % are estimated for A and B, respectively, at the minimum. Protein microheterogeneity due to a high degree of glycosylation explains the appearance of the polypeptide A as a diffuse smear rather than a sharp band in SDS–PAGE. High glycosylation degrees of around 45 % are often observed in plant laccases, while fungal laccases usually show lower glycan contents of 10–20 % (Morozova et al. 2007a).
Conclusions
The extracellular phenol oxidase from T. aeria is confirmed to be a laccase according to the substrate specificity of the purified enzyme. It is suggested to be a hetero-oligomeric complex with the composition AB 2 . Regarding the molecular mass and glycan content of the catalytic subunit (A), the neutral pH optima toward phenolic substrates and the rather low activity toward syringaldazine, the enzyme resembles typical features of laccases from higher plants.
Putative natural functions of algal laccases may comprise the detoxification of phenolic compounds present in terrestrial and aquatic environments, a role related to the synthesis of complex compounds such as cell wall-associated polymers (Dean and Eriksson 1994) and UV-absorbing compounds (Pichrtová et al. 2013), or possibly the acquisition of nutrients through the transformation of lignocellulosic substrates, as discussed previously (Otto et al. 2010; Blifernez-Klassen et al. 2012). Apart from that, in the presence of suitable redox-mediating compounds, algal laccases may significantly contribute to the biotransformation of a wide spectrum of natural and xenobiotic aromatic compounds.
Abbreviations
- 2,6-DMP:
-
2,6-Dimethoxyphenol
- (2D) SDS–PAGE:
-
(Two-dimensional) sodium dodecyl sulfate–polyacrylamide gel electrophoresis
- ABTS:
-
2,2′-Azino-bis(3-ethylbenzothiazoline-6-sulfonic acid)
- ABu62:
-
Acid Blue 62
- IEC:
-
Ion exchange chromatography
- PNGase F:
-
Peptide-N-glycosidase F
- RB5:
-
Reactive Black 5
- RBBR:
-
Remazol Brilliant Blue R
- SEC:
-
Size exclusion chromatography
References
Baldrian P (2006) Fungal laccases—occurrence and properties. FEMS Microbiol Rev 30:215–242. doi:10.1111/j.1574-4976.2005.00010.x
Beeley JG (1985) Glycoprotein and proteoglycan techniques. Laboratory techniques in biochemistry and molecular biology, vol 16. Elsevier, Amsterdam
Blifernez-Klassen O, Klassen V, Doebbe A, Kersting K, Grimm P, Wobbe L, Kruse O (2012) Cellulose degradation and assimilation by the unicellular phototrophic eukaryote Chlamydomonas reinhardtii. Nat Commun 3:1214. doi:10.1038/ncomms2210
Cabanes J, García-Cánovas F, Lozano JA, García-Carmona F (1987) A kinetic study of the melanization pathway between l-tyrosine and dopachrome. Biochim Biophys Acta 923:187–195. doi:10.1016/0304-4165(87)90003-1
Camarero S, Ibarra D, Martínez MJ, Martínez ÁT (2005) Lignin-derived compounds as efficient laccase mediators for decolorization of different types of recalcitrant dyes. Appl Environ Microb 71:1775–1784. doi:10.1128/AEM.71.4.1775-1784.2005
Chiaiese P, Palomba F, Tatino F, Lanzillo C, Pinto G, Pollio A, Filippone E (2011) Engineered tobacco and microalgae secreting the fungal laccase POXA1b reduce phenol content in olive oil mill wastewater. Enzyme Microb Tech 49:540–546. doi:10.1016/j.enzmictec.2011.06.002
Childs RE, Bardsley WG (1975) The steady-state kinetics of peroxidase with 2,2′-azino-di-(3-ethyl-benzthiazoline-6-sulphonic acid) as chromogen. Biochem J 145:93–103
Claus H (2004) Laccases: structure, reactions, distribution. Micron 35:93–96. doi:10.1016/j.micron.2003.10.029
De Silva D, Davis-Kaplan S, Fergestad J, Kaplan J (1997) Purification and characterization of Fet3 protein, a yeast homologue of ceruloplasmin. J Biol Chem 272:14208–14213. doi:10.1074/jbc.272.22.14208
Dean JF, Eriksson KEL (1994) Laccase and the deposition of lignin in vascular plants. Holzforschung 48:21–33. doi:10.1515/hfsg.1994.48.s1.21
Diamantidis G, Effosse A, Potier P, Bally R (2000) Purification and characterization of the first bacterial laccase in the rhizospheric bacterium Azospirillum lipoferum. Soil Biol Biochem 32:919–927. doi:10.1016/S0038-0717(99)00221-7
Faraco V, Ercole C, Festa G, Giardina P, Piscitelli A, Sannia G (2008) Heterologous expression of heterodimeric laccase from Pleurotus ostreatus in Kluyveromyces lactis. Appl Microbiol Biotechnol 77:1329–1335. doi:10.1007/s00253-007-1265-5
Ferraroni M, Scozzafava A, Ullah S, Tron T, Piscitelli A, Sannia G (2014) Crystallization and preliminary X-ray crystallographic analysis of the small subunit of the heterodimeric laccase POXA3b from Pleurotus ostreatus. Acta Cryst F 70:76–79. doi:10.1107/S2053230X13032810
Frasconi M, Favero G, Boer H, Koivula A, Mazzei F (2010) Kinetic and biochemical properties of high and low redox potential laccases from fungal and plant origin. BBA Proteins Proteom 1804:899–908. doi:10.1016/j.bbapap.2009.12.018
Galuszka P, Frébortová J, Luhová L, Bilyeu KD, English JT, Frébort I (2005) Tissue localization of cytokinin dehydrogenase in maize: possible involvement of quinone species generated from plant phenolics by other enzymatic systems in the catalytic reaction. Plant Cell Physiol 46:716–728. doi:10.1093/pcp/pci074
Ghasemi Y, Rasoul-Amini S, Fotooh-Abadi E (2011) The biotransformation, biodegradation, and bioremediation of organic compounds by microalgae. J Phycol 47:969–980. doi:10.1111/j.1529-8817.2011.01051.x
Giardina P, Autore F, Faraco V, Festa G, Palmieri G, Piscitelli A, Sannia G (2007) Structural characterization of heterodimeric laccases from Pleurotus ostreatus. Appl Microbiol Biotechnol 75:1293–1300. doi:10.1007/s00253-007-0954-4
Giardina P, Faraco V, Pezzella C, Piscitelli A, Vanhulle S, Sannia G (2010) Laccases: a never-ending story. Cell Mol Life Sci 67:369–385. doi:10.1007/s00018-009-0169-1
Harkin JM, Obst JR (1973) Syringaldazine, an effective reagent for detecting laccase and peroxidase in fungi. Experientia 29:381–387. doi:10.1007/BF01926734
Harkin JM, Larsen MJ, Obst JR (1974) Use of syringaldazine for detection of laccase in sporophores of wood rotting fungi. Mycologia 66:469–476
Hoopes JT, Dean JF (2004) Ferroxidase activity in a laccase-like multicopper oxidase from Liriodendron tulipifera. Plant Physiol Bioch 42:27–33. doi:10.1016/j.plaphy.2003.10.011
Jinqi L, Houtian L (1992) Degradation of azo dyes by algae. Environ Pollut 75:273–278. doi:10.1016/0269-7491(92)90127-V
Joel DM, Marbach I, Mayer AM (1978) Laccase in Anacardiaceae. Phytochemistry 17:796–797. doi:10.1016/S0031-9422(00)94231-6
Kılıç NK, Karatay SE, Duygu E, Dönmez G (2011) Potential of Gonium spp. in synthetic reactive dye removal, possible role of laccases and stimulation by triacontanol hormone. Water Air Soil Pollut 222:297–303. doi:10.1007/s11270-011-0824-7
La Fontaine S, Quinn JM, Nakamoto SS, Page MD, Göhre V, Moseley JL, Kropat J, Merchant S (2002) Copper-dependent iron assimilation pathway in the model photosynthetic eukaryote Chlamydomonas reinhardtii. Eukaryot Cell 1:736–757. doi:10.1128/EC.1.5.736-757.2002
La Russa M, De Biasi MG, Chiaiese P, Palomba F, Pollio A, Pinto G, Filippone E (2008) Screening of green microalgae species for extracellular phenoloxidase activity useful for wastewater phycoremediation. In: Proceedings of the 4th European Bioremediation Conference, Chania, Greece, September 03–06, 2008
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685. doi:10.1038/227680a0
Lisov A, Zavarzina A, Zavarzin A, Demin V, Leontievsky A (2012) Dimeric and monomeric laccases of soil-stabilizing lichen Solorina crocea: purification, properties and reactions with humic acids. Soil Biol Biochem 45:161–167. doi:10.1016/j.soilbio.2011.11.004
Martínez MJ, Ruiz-Dueñas FJ, Guillén F, Martínez AT (1996) Purification and catalytic properties of two manganese-peroxidase isoenzymes from Pleurotus eryngii. Eur J Biochem 237:424–432. doi:10.1111/j.1432-1033.1996.0424k.x
McCaig BC, Meagher RB, Dean JF (2005) Gene structure and molecular analysis of the laccase-like multicopper oxidase (LMCO) gene family in Arabidopsis thaliana. Planta 221:619–636. doi:10.1007/s00425-004-1472-6
Moilanen U, Osma JF, Winquist E, Leisola M, Couto SR (2010) Decolorization of simulated textile dye baths by crude laccases from Trametes hirsuta and Cerrena unicolor. Eng Life Sci 10:242–247. doi:10.1002/elsc.200900095
Morozova OV, Shumakovich GP, Gorbacheva MA, Shleev SV, Yaropolov AI (2007a) “Blue” laccases. Biochemistry (Moscow) 72:1136–1150. doi:10.1134/S0006297907100112
Morozova OV, Shumakovich GP, Shleev SV, Yaropolov YI (2007b) Laccase-mediator systems and their applications: a review. Appl Biochem Micro 43:523–535. doi:10.1134/S0003683807050055
Omura T (1961) Studies on laccases of lacquer trees I. comparison of laccases obtained from Rhus vernicifera and Rhus succedanea. J Biochem 50:264–272
Otto B, Schlosser D, Reisser W (2010) First description of a laccase-like enzyme in soil algae. Arch Microbiol 192:759–768. doi:10.1007/s00203-010-0603-7
Palmieri G, Cennamo G, Faraco V, Amoresano A, Sannia G, Giardina P (2003) Atypical laccase isoenzymes from copper supplemented Pleurotus ostreatus cultures. Enzyme Microb Tech 33:220–230. doi:10.1016/S0141-0229(03)00117-0
Pereira L, Coelho AV, Viegas CA, Santos MM, Robalo MP, Martins LO (2009) Enzymatic biotransformation of the azo dye Sudan Orange G with bacterial CotA-laccase. J Biotechnol 139:68–77. doi:10.1016/j.jbiotec.2008.09.001
Pezzella C, Lettera V, Piscitelli A, Giardina P, Sannia G (2013) Transcriptional analysis of Pleurotus ostreatus laccase genes. Appl Microbiol Biotechnol 97:705–717. doi:10.1007/s00253-012-3980-9
Pichrtová M, Remias D, Lewis LA, Holzinger A (2013) Changes in phenolic compounds and cellular ultrastructure of Arctic and Antarctic strains of Zygnema (Zygnematophyceae, Streptophyta) after exposure to experimentally enhanced UV to PAR ratio. Microb Ecol 65:68–83. doi:10.1007/s00248-012-0096-9
Pickel B, Schaller A (2013) Dirigent proteins: molecular characteristics and potential biotechnological applications. Appl Microbiol Biotechnol 97:8427–8438. doi:10.1007/s00253-013-5167-4
Pourcel L, Routaboul JM, Kerhoas L, Caboche M, Lepiniec L, Debeaujon I (2005) TRANSPARENT TESTA10 encodes a laccase-like enzyme involved in oxidative polymerization of flavonoids in Arabidopsis seed coat. Plant Cell 17:2966–2980. doi:10.1105/tpc.105.035154
Richter H, Lieberei R, Strnad M, Novák O, Gruz J, Rensing SA, von Schwartzenberg K (2012) Polyphenol oxidases in Physcomitrella: functional PPO1 knockout modulates cytokinin-dependent development in the moss Physcomitrella patens. J Exp Bot 63:5121–5135. doi:10.1093/jxb/ers169
Riva S (2006) Laccases: blue enzymes for green chemistry. Trends Biotechnol 24:219–226. doi:10.1016/j.tibtech.2006.03.006
Rönnerstrand S (1943) Untersuchungen über Oxydase, Peroxydase und Ascorbinsäure in einigen Meeresalgen. Dissertation, University of Lund; C. W. Lindström, Lund
Semple KT, Cain RB, Schmidt S (1999) Biodegradation of aromatic compounds by microalgae. FEMS Microbiol Lett 170:291–300. doi:10.1111/j.1574-6968.1999.tb13386.x
Shiba T, Xiao L, Miyakoshi T, Chen CL (2000) Oxidation of isoeugenol and coniferyl alcohol catalyzed by laccases isolated from Rhus vernicifera Stokes and Pycnoporus coccineus. J Mol Catal B Enzym 10:605–615. doi:10.1016/S1381-1177(00)00184-3
Soares G, De Amorim MT, Costa-Ferreira M (2001) Use of laccase together with redox mediators to decolourize Remazol Brilliant Blue R. J Biotechnol 89:123–129. doi:10.1016/S0168-1656(01)00302-9
Sterjiades R, Dean JF, Eriksson KEL (1992) Laccase from sycamore maple (Acer pseudoplatanus) polymerizes monolignols. Plant Physiol 99:1162–1168. doi:10.1104/pp.99.3.1162
Sterjiades R, Dean JF, Gamble G, Himmelsbach DS, Eriksson KEL (1993) Extracellular laccases and peroxidases from sycamore maple (Acer pseudoplatanus) cell-suspension cultures. Planta 190:75–87. doi:10.1007/BF00195678
Stookey LL (1970) Ferrozine—a new spectrophotometric reagent for iron. Anal Chem 42:779–781. doi:10.1021/ac60289a016
Subashchandrabose SR, Ramakrishnan B, Megharaj M, Venkateswarlu K, Naidu R (2013) Mixotrophic cyanobacteria and microalgae as distinctive biological agents for organic pollutant degradation. Environ Int 51:59–72. doi:10.1016/j.envint.2012.10.007
Tavares AP, Cristóvão RO, Loureiro JM, Boaventura RA, Macedo EA (2008) Optimisation of reactive textile dyes degradation by laccase–mediator system. J Chem Technol Biotechnol 83:1609–1615. doi:10.1002/jctb.1952
Thurston CF (1994) The structure and function of fungal laccases. Microbiology 140:19–26. doi:10.1099/13500872-140-1-19
Wang GD, Li QJ, Luo B, Chen XY (2004) Ex planta phytoremediation of trichlorophenol and phenolic allelochemicals via an engineered secretory laccase. Nat Biotechnol 22:893–897. doi:10.1038/nbt982
Xia K, Manning M, Hesham H, Lin Q, Bystroff C, Colón W (2007) Identifying the subproteome of kinetically stable proteins via diagonal 2D SDS/PAGE. P Natl Acad Sci USA 104:17329–17334. doi:10.1073/pnas.0705417104
Xu F (1997) Effects of redox potential and hydroxide inhibition on the pH activity profile of fungal laccases. J Biol Chem 272:924–928. doi:10.1074/jbc.272.2.924
Xu F, Shin W, Brown SH, Wahleithner JA, Sundaram UM, Solomon EI (1996) A study of a series of recombinant fungal laccases and bilirubin oxidase that exhibit significant differences in redox potential, substrate specificity, and stability. BBA Protein Struct M 1292:303–311. doi:10.1016/0167-4838(95)00210-3
Yoshida H (1883) LXIII.—chemistry of lacquer (Urushi). Part I. communication from the chemical society of Tokio. J Chem Soc Trans 43:472–486. doi:10.1039/CT8834300472
Zakrzewski M (2012) Development of computational methods for the analysis of metagenome and metatranscriptome data. Dissertation, University of Bielefeld
Ziegler R, Egle K (1965) Zur quantitativen Analyse der Chloroplastenpigmente. Beitr Biol Pflanzen 41:11–37
Zille A, Ramalho P, Tzanov T, Millward R, Aires V, Cardoso MH, Ramalho MT, Gübitz GM, Cavaco-Paulo A (2004) Predicting dye biodegradation from redox potentials. Biotechnol Progr 20:1588–1592. doi:10.1021/bp049963i
Acknowledgments
We are grateful to Prof. Werner Reisser and Prof. Hauke Harms for their support. This study was sponsored by the Scholarship Programme of the German Federal Environmental Foundation (DBU; Grant no. 20010/078). The research topic Chemicals in the Environment (CITE) of the Helmholtz Centre for Environmental Research—UFZ provided additional resources for this research.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Otto, B., Schlosser, D. First laccase in green algae: purification and characterization of an extracellular phenol oxidase from Tetracystis aeria . Planta 240, 1225–1236 (2014). https://doi.org/10.1007/s00425-014-2144-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00425-014-2144-9