
Abstract
Laccases (EC 1.10.3.2) are versatile multi-copper oxidases so far found in higher plants, fungi, insects, prokaryotes and lichens. In the present study, the production of an extracellular laccase-like enzyme by the coccoid green soil alga Tetracystis aeria was investigated and the enzyme was partly characterized, thereby providing the first description of a laccase-like enzyme in soil algae. Enzyme production in algae cultures was considerably increased by addition of the fungal laccase inducer copper sulphate. Maximal enzyme production was observed during the stationary growth phase. Peroxidase or tyrosinase activity was not detected. The native enzyme exhibits an apparent molecular mass of about 212 kDa as observed with size exclusion chromatography and about 210–260 kDa as estimated by zymograms. The enzyme efficiently oxidizes 2,2′-azino-bis(3-ethylbenzthiazoline-6-sulphonic acid) (ABTS), 2,6-dimethoxyphenol (2,6-DMP), syringaldazine (SGZ) and the anthraquinone dye Acid Blue 62, while guaiacol and Remazol Brilliant Blue R are only poorly oxidized. The apparent kinetic parameters obtained for ABTS, 2,6-DMP and SGZ oxidation are within the range reported for fungal laccases. Oxidation of the phenolic substrate 2,6-DMP displays a remarkably high pH optimum (pH 8.0–8.5), which is interesting with respect to potential biotechnological applications.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Eukaryotic soil algae can be found on nearly every kind of terrestrial surfaces as well as in the air, including a range of extreme environments (Seckbach 2007). Soil algae contribute to the formation and stability of soils and to energy and matter fluxes in soil ecosystems (Zancan et al. 2006). The soil algal biomass approximately averages some hundred kilograms fresh weight per hectare in temperate farmland soils, which is about one order of magnitude lower than the bacterial and fungal biomass (Oesterreicher 1990).Footnote 1 Shtina (1974) observed the algal biomass in fertile soil to be renewed at least three times in a month (See footnote 1).
In soil food webs, algae are primary producers and serve as food for soil fauna organisms like collembola (Scheu and Folger 2004). Algae might positively or negatively influence other soil organisms like bacteria and fungi and possibly compete for nutrients (Safonova and Reisser 2005). While adapting to different habitats, algae might have evolved new physiological capacities like desiccation tolerance, as has been shown for desert algae forming microbial crusts (Cardon et al. 2008). Viable algal cells can often be found in aphotic layers of soils, where autotrophic growth is excluded. Although heterotrophic growth of soil algae is well known from laboratory cultures, its role for natural environments remains unclear (Round 1984).
Green algae have been shown to biodegrade various xenobiotics like triphenylmethane and azo dyes, p-nitrophenol and phenanthrene (Daneshvar et al. 2007; Acuner and Dilek 2004; Lima et al. 2003; Safonova et al. 2005). Algae were effectively used for the treatment of wastewaters from the paper industries without the need for co-substrates and might turn out to offer an alternative to fungal wastewater treatment (Tarlan et al. 2002). In algae, phenolic compounds may be metabolized via hydroxylation and ring cleavage (Semple et al. 1999). However, the enzymes involved in the degradation of the aforementioned compounds by algae have not yet been characterized.
Laccases (EC 1.10.3.2, p-diphenol : O2 oxidoreductases) are multi-copper oxidases which catalyze the one-electron oxidation of a broad range of substrates. Radicals formed as primary oxidation products from substrates like mono- and polyphenols or aromatic amines may in turn react non-enzymatically, leading to the cross-linking of monomers or the decomposition of polymers (Claus 2004). Complex polymers like lignin can be oxidized indirectly by the use of low-molecular-weight compounds, called redox mediators (Baldrian 2006). Natural functions of laccases include the degradation as well as the biosynthesis of lignin. Laccases are further thought to be involved in morphogenesis and detoxification processes and may influence immunity and pathogenicity (Claus 2004). Laccases were also found to be useful for a multitude of biotechnological applications, e.g. in the food, textile, pulp and paper industries and in bioremediation processes (Riva 2006). Until now, laccases and laccase-like multicopper oxidases were found in higher plants, fungi, insects, prokaryotes and lichens (Claus 2004; Lisov et al. 2007).
As yet, indications for the occurence of extracellular laccase-like phenoloxidases in algae are limited to the demonstration of the oxidation of typical laccase substrates by cell-free culture supernatants of green microalgae (La Russa et al. 2008), whereas information about structural or catalytic properties of such enzymes is still missing. Species of the green algal genus Tetracystis inhabit soils worldwide (Ettl and Gaertner 1995) and therefore may be considered as appropriate models to study algal phenoloxidases in the context of soil environments. The coccoid green soil alga Tetracystis aeria Brown & Bold strain SAG 89.80 (Chlorophyta, Chlorococcales) was isolated from air over Pampa, Texas (USA) (Brown and Bold 1964). T. aeria has also been described for Japanese soils and for contemporary glacier moraines in West Antarctica (Nakano 1983; Massalski et al. 2001). The aim of the present study was to investigate factors affecting the production of an extracellular laccase-like enzyme by T. aeria strain SAG 89.80 and to partly characterize the enzyme, thereby also firstly providing information about some structural and catalytic properties of laccase-like phenoloxidases in eukaryotic algae.
Materials and methods
Algal strain and culture conditions
Tetracystis aeria Brown & Bold strain SAG 89.80 [Culture Collection of Algae at the University of Göttingen (SAG), Germany] was grown axenically in Erlenmeyer flasks containing varying volumes (see below) of liquid mineral medium (Bold’s Basal Medium, BBM; Bischoff and Bold 1963), which consisted of NaNO3 (2.9 mM), KH2PO4 (1.3 mM), K2HPO4 (430 μM), NaCl (430 μM), MgSO4 (300 μM), CaCl2 (170 μM), Fe-Na2EDTA (25 μM), H3BO3 (3.2 μM), MnSO4 (1.5 μM), NaMoO4 (1.2 μM), ZnSO4 (0.7 μM), Co(NO3)2 (0.7 μM) and CuSO4 (0.4 μM). Flasks were incubated at 90 rpm and 20°C, employing a light:dark cycle of 14:10 h under light intensities of 86 μmol photons m−2 s−1. For determination of extracellular enzyme activities cell-free culture supernatants were obtained after 10 days of cultivation, unless stated otherwise. Algal cells were gently removed by centrifugation for 10–15 min at 3,000×g. Particles remaining in the supernatant were removed by further centrifugation at 15,000×g for 10 min.
The potential laccase inducers o-dianisidine dihydrochloride (79 μM; purified for use with peroxidase and peroxidase-coupled reactions according to the supplier; Sigma–Aldrich, St. Louis/MO, USA), p-xylidine (12 μM; 99% purity; Aldrich, Steinheim, Germany), syringaldazine (SGZ; 0.2 μM; ≥98% purity; Sigma, Deisenhofen, Germany), veratryl alcohol (1.0 mM; 96% purity; Sigma–Aldrich), guaiacol (18.2 μM; ≥98% purity; Sigma–Aldrich) and vanillic acid (1.5 mM; 97% purity; Sigma–Aldrich) were added to culture volumes of 15 ml after 1 day of cultivation. CuSO4 was added at 10, 20 and 30 μM to T. aeria cultures at the beginning of incubation, in addition to the basal CuSO4 concentration in BBM of 0.4 μM.
To determine cell-bound enzyme activities, cells grown in culture volumes of 10 ml, which were supplemented with 20 μM CuSO4 at the beginning of cultivation, were mechanically disintegrated by vortexing for 10 min at maximum speed, using 2 ml tubes and glass beads (diameter 0.25–0.30 mm and 1.00–1.05 mm at a proportion of 3:1).
To obtain concentrated culture supernatants, cell-free culture supernatants obtained from 28- to 31-day-old cultures (culture volumes of 190–400 ml), which were supplemented with 20 μM CuSO4 at the beginning of cultivation were filtered through RC 55 filter papers (Whatman, Dassel, Germany). Thereafter, filtrates were 45-fold concentrated, using a 150 ml-stirred cell equipped with an Omega polysulfone membrane (10 kDa cut-off; Pall GmbH Life Sciences, Dreieich, Germany).
Chlorophyll determination
Total chlorophyll (a and b) concentrations were determined spectrophotometrically at 647 and 664 nm after extraction with 80% acetone, using the equations of Ziegler and Egle (1965).
Biochemical enzyme characterization
Enzyme activities were routinely determined following the oxidation of 3.0 mM 2,2′-azino-bis(3-ethylbenzthiazoline-6-sulphonic acid) (ABTS; 98% purity; Sigma) in 0.1 M citrate/0.2 M phosphate buffer (pH 4.0) (Martin et al. 2007) at 25°C, using a Specord 250 spectrophotometer (Analytik Jena, Jena, Germany) unless stated otherwise.
Where indicated in the text, H2O2 (4.4 mM), catalase (~1,000 U ml−1; from bovine liver; Sigma) and NaN3 (50 μM) were additionally included in the ABTS assay. Dialysis of cell-free culture supernatants against distilled water was performed with CelluTrans membranes (8–10 kDa Cut-off; Carl Roth, Karlsruhe, Germany) by shaking for 1 day at 20°C. For enzyme inactivation, samples were incubated for 30 min in a boiling water bath. Laccase from Rhus vernicifera (crude acetone powder; Sigma) was used as a reference.
The pH dependence of the oxidation of ABTS (2.0 mM), 2,6-dimethoxyphenol (2,6-DMP; 2.0 mM; ≥97% purity; Fluka, Buchs, Switzerland; Martin et al. 2007), SGZ [20 μM; 99% purity; Sigma–Aldrich; ε530 = 65 mM−1 cm−1 (Grassin and Dubourdieu 1989)], guaiacol [4.0 mM; 99.5% purity; Merck, Darmstadt, Germany; ε470 = 6.74 mM−1 cm−1 (Das et al. 1997)], as well as the anthraquinonic dyes Remazol Brilliant Blue R [RBBR; 150 μM; 50% purity (technical grade); Acros Organics, Geel, Belgium; ε590 = 12.7 mM−1 cm−1] and Acid Blue 62 (ABu62, 200 μM; 98% purity; Yorkshire Europe, Tertre, Belgium; ε590 = 15.2 mM−1 cm−1) by the concentrated culture supernatant was followed at 25°C and wavelengths of 420, 470, 530, 470, 590 and 590 nm, respectively, using a GENios+ microplate reader (Tecan, Crailsheim, Germany). A pH range of 2.5–8.0 was obtained using 0.1 M citrate/0.2 M phosphate buffer. For 2,6-DMP, a pH range of 7.0–12.0 was additionally applied, using 0.04 M Britton-Robinson buffer (Britton and Robinson 1931). For RBBR and ABu62, calibration curves were acquired to calculate the extinction coefficients given above.
Kinetic parameters were calculated upon non-linear regression from the initial rates in oxidation of 0.5–1,000 μM ABTS, 10–5,000 μM 2,6-DMP, 1–100 μM SGZ, 1–2,000 μM guaiacol, 2.5–150 μM RBBR, and 5–500 μM ABu62 by the concentrated culture supernatant in 0.1 M citrate/0.2 M phosphate buffer (pH 2.5 for ABTS, pH 3.0 for Abu62, pH 7.0 for SGZ, and pH 8.0 for guaiacol and RBBR), or in 0.04 M Britton-Robinson buffer (pH 8.0 for 2,6-DMP) at 25°C, using the OriginPro 8G software (OriginLab, Northampton/MA, USA). Enzyme activities were corrected for blank values, which were obtained by using assay buffer instead of the samples. Enzyme activities are expressed as international units (U), where 1 U corresponds to 1 μmol of substrate oxidized per minute.
The oxidation of 0.1 mM tyrosine (purity >99%; Merck) by the concentrated culture supernatant was followed in 0.1 M citrate/0.2 M phosphate buffer at pH 3.0, 5.0 and 7.0, using the GENios+ microplate reader described before at available wavelengths of 270 and 492 nm. Additional assays were carried out with 0.1 mM tyrosine at pH 3.0, using a U-2001 two-beam spectrophotometer (Hitachi, Tokyo, Japan) at commonly used wavelengths of 280 (Niladevi et al. 2008) and 490 nm (Laufer et al. 2006).
Protein concentrations were determined according to Bradford (1976), with Bovine serum albumin serving as an external standard.
Size exclusion chromatography (SEC) of the concentrated culture supernatant on a Superdex 200 10/300 GL gel filtration column (GE Healthcare, Munich, Germany) was carried out on a BioLogic HR protein liquid chromatography system (Bio-Rad, Munich, Germany), using 10 mM sodium acetate buffer (pH 6.0) supplemented with 0.15 M NaCl as an eluent at a flow rate of 0.5 ml min−1. LMW and HMW Gel Filtration Calibration Kits (GE Healthcare) were used for calibration.
Ion exchange chromatography (IEC) of the concentrated culture supernatant on a MonoQ 5/50 GL anion exchange column (GE Healthcare) was carried out on the BioLogic HR system mentioned above at a flow rate of 1.0 ml min−1, employing a linear gradient of 0–0.5 M NaCl in 10 mM sodium acetate buffer (pH 7.0) as indicated in the text.
Fractions of 0.5 and 1.0 ml were collected during SEC and IEC separations, respectively, and assessed for ABTS oxidation activity. Fractions containing enzyme activity were pooled as indicated in the text. Proteins in the pooled fractions were precipitated with five volumes of acetone (−20°C). After centrifugation (8,000×g for 15 min at 4°C), supernatants were discarded and the precipitates were resuspended in 10 mM sodium acetate buffer (pH 6.0), thereby achieving a 20-fold concentration with concomitant removal of NaCl.
Discontinuous sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) according to Laemmli (1970) was performed in a Mini PROTEAN 3 cell (Bio-Rad) with a stacking gel of 4% (w/v) acrylamide and separating gels of 9 or 6% acrylamide, using premixed buffers (Bio-Rad). Samples were denatured by dilution in one volume of Laemmli buffer (Bio-Rad) containing 5% β-mercaptoethanol and subsequent boiling for 5 min prior to electrophoresis. For zymogram analysis, β-mercaptoethanol was excluded from the sample buffer and the boiling step was omitted. Samples containing about 1.4–13 μg of protein and 3.8–26 mU of enzyme activity (unless boiled) were applied per lane. Gels were silver stained using the Silver Stain Plus Kit (Bio-Rad) according to the instructions of the supplier. For zymogram analysis, samples were applied twice. After electrophoresis gels were cut and one of the obtained parts was silver stained, whereas the other part containing identical samples was stained for enzyme activity with 2 mM ABTS in 0.1 M citrate/0.2 M phosphate buffer (pH 4.0). Apparent molecular masses were estimated by reference to commercial molecular mass marker proteins (High Range Silver Stain SDS-PAGE standards, either from Bio-Rad or from Sigma).
Isoelectric focusing (IEF) was performed on a Multiphor II gel electrophoresis system (Pharmacia Biotech, Piscataway, USA) with Servalyt Precotes 3–10 (Serva, Heidelberg, Germany) according to the instructions of the supplier. Isoelectric points (pI) were determined by comparison with the Liquid Mix IEF Marker 3–10 (Serva). Marker proteins were stained with colloidal Coomassie G-250 according to Neuhoff et al. (1988), and enzyme bands were visualized upon activity staining with ABTS as described above.
Results and discussion
ABTS oxidation activity of cell-free culture supernatants under various conditions
Cell-free culture supernatants derived from 10-day-old T. aeria cultures were assessed for ABTS oxidation activity under various conditions (Table 1). After dialysis of the cell-free culture supernatant (8–10 kDa cut-off), about 44% of the original ABTS oxidation activity remained in the retentate, lower than a value of about 64% obtained for the R. vernicifera laccase. Boiling of the samples for 30 min led to essentially complete losses of the ABTS oxidation activities. The addition of catalase, which was intended to eliminate H2O2 potentially present in T. aeria culture supernatants, did not cause a significant change of the respective enzyme activity (though a high standard deviation of about 40% was recorded for the T. aeria samples). The decrease of the ABTS oxidation activity of T. aeria culture supernatants, as observed upon addition of H2O2, indicates that the responsible enzyme is not a peroxidase. Both plant and fungal laccases have been shown to be inhibited by H2O2 (Niemetz and Gross 2003; Mai et al. 2000). ABTS oxidation by the R. vernicifera laccase increased to about 181% when H2O2 was included, possibly indicating peroxidase impurities in this commercial enzyme preparation. NaN3 addition caused a substantial decrease in ABTS oxidation activity of T. aeria culture supernatants, as would be expected for a laccase (Wood 1980). All together, the aforementioned observations are in favour of an extracellular enzyme displaying properties of a laccase.
Localization and production of the enzyme in T. aeria cultures
The cell-bound ABTS oxidation activity accounted for 27 ± 7% (mean ± SD for triplicate determinations) of the total (cell-bound + extracellular) ABTS oxidation activity of T. aeria cultures, as determined after a mechanical disintegration of the algal cells. Hence, the ABTS oxidizing enzyme appears to be preferentially located extracellularly.
Among six potential organic laccase inducers tested, the addition of o-dianisidine dihydrochloride to 1-day-old T. aeria cultures caused a significant increase [P ≤ 0.05, according to one-sided Dunnett’s test (α = 5%)] in extracellular ABTS oxidation activity at culture day 10 [177 ± 46% (mean ± SD for triplicate cultures) of the corresponding control value of 0.9 U l−1]. Albeit not statistically significant, enhanced ABTS oxidation activities were also recorded for the well-known fungal laccase inducers p-xylidine (Eggert et al. 1996) and SGZ (145 ± 37% and 142 ± 35% of the control value, respectively). Veratryl alcohol and guaiacol caused no changes in ABTS oxidation activity, whereas a decrease in enzyme activity was observed upon addition of vanillic acid (105 ± 47%, 103 ± 14% and 46 ± 4% of the control value, respectively). Addition of the well-established fungal laccase inducer CuSO4 (Palmieri et al. 2000) led to a 7.0-fold increase in extracellular ABTS oxidation, as compared to cultures grown in normal BBM (Fig. 1). A considerably lower enzyme activity recorded upon addition of 30 μM CuSO4 was probably due to toxic effects of the metal. CuSO4 at 10 μM was ineffective. Normally, the total copper concentration in surface soil solutions is only 0.01–0.6 μM (Baker and Senft 1995).

Extracellular ABTS oxidation activities in 10-day-old T. aeria cultures (15 ml culture volume) supplemented with different CuSO4 concentrations at the beginning of cultivation (basal + added CuSO4 concentration is shown). Where indicated, o-dianisidine dihydrochloride (o-dian.) was additionally included at 79 µM after 1 day of cultivation. Data represent means ± standard deviations from triplicate cultures
When o-dianisidine dihydrochloride was additionally applied to cultures containing varying CuSO4 concentrations, enzyme activities essentially remained on the basal level (Fig. 1). This contrasts previously reported synergistic effects of organic inducers and CuSO4 on laccase activities (Junghanns et al. 2008) and might reflect an interference of enzyme induction and toxic effects of the inducers. Addition of o-dianisidine dihydrochloride to BBM without additional CuSO4 led to a less pronounced increase in enzyme activity in this experiment (129 ± 20% of the corresponding value without o-dianisidine dihydrochloride) than was observed in the aforementioned experiment (177 ± 46%, see above). This discrepancy may be due to a high biological variability between individual cultures, as also indicated by high SD values of the respective means and impedes to establish a rank order of efficiency of the potential organic inducers tested. Since the highest enzyme activities were observed in presence of 20 μM CuSO4 without additional organic inducers, this condition was used for enzyme production in the following experiments.
The extracellular ABTS oxidation activities of copper-induced cultures (75 ml culture volume), as well as the algal biomass (measured as the total chlorophyll concentration), were monitored over 39 days. While the chlorophyll concentration decreased after reaching a maximum of 14.8 ± 1.8 μg ml−1 (mean ± SD for triplicate cultures) at culture day 18, the enzyme activity continuously increased until a maximum of 34.2 ± 2.5 U l−1 was reached at culture day 32, which was maintained further on. Hence, maximum enzyme production is linked to the stationary growth phase of the algae as also reported for laccase production by numerous fungi (Mansur et al. 2003).
Biochemical enzyme characterization
When cell-free culture supernatant was concentrated 45-fold by ultrafiltration, the ABTS oxidation activity increased from 22 ± 1 to 995 ± 284 U l−1 (mean ± SD for triplicate determinations), corresponding to a complete recovery of the enzyme activity (Table 2).
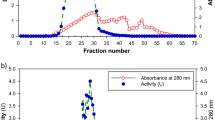
By SEC of the concentrated culture supernatant, one peak containing the major ABTS oxidation activity, which corresponded to 76% of the overall eluted enzyme activity, was eluted between 23.5 (corresponding to fraction 24) and 29.5 min (corresponding to fraction 30) (Fig. 2a). The maximum ABTS oxidation activity was detected in the eluate fraction corresponding to an apparent molecular mass of about 212 kDa. Fractions 24–30 were pooled and subjected to acetone precipitation prior to further analysis. After acetone precipitation a specific activity of 0.8 U mg−1 was achieved, corresponding to a twofold purification as based on a specific activity of 0.4 U mg−1 of the cell-free culture supernatant. The yield of enzyme activity after SEC and subsequent acetone precipitation was about 9%. Two minor ABTS oxidation peaks, which corresponded to apparent molecular masses of about 19 and below 13.7 kDa (the molecular mass of the smallest standard protein used for calibration), contained only 4 and 5% of the overall eluted enzyme activity, respectively, and were excluded from further investigations.

SEC (a) and IEC (b) of the concentrated culture supernatant of T. aeria. The ABTS oxidation activity, absorbance at 280 nm and the NaCl gradient (b) are indicated. ABTS oxidation activities represent means ± standard deviations for triplicate determinations (a), or means for duplicate determinations (b). 100% activity refers to absolute values of 46.7 ± 1.6 (a) and 44.3 U l−1 (b). 100% absorbance refers to absolute values of 0.15 (a) and 0.05 AU (b)
Concentrated culture supernatant was also subjected to IEC on a MonoQ anion exchange column for purification. One major ABTS oxidation peak, which contained 70% of the overall eluted enzyme activity and eluted between 18.5 (corresponding to fraction 19) and 21.5 min (corresponding to fraction 22), was detected (Fig. 2b). Fractions 19–22 were pooled and subjected to acetone precipitation prior to further analysis. The specific enzyme activity of the pooled fraction increased to 7.0 U mg−1, corresponding to a 19-fold enzyme purification and a yield of enzyme activity of about 33% (Table 2).
After SEC as well as after IEC, the total enzyme activities of the pooled fractions increased by 86 and 248% upon acetone precipitation, respectively, as compared to the total activities of the pooled fractions before acetone precipitation. This effect most likely indicates an inhibition of enzyme activity caused by NaCl present in the SEC and IEC elution buffers, which was removed during the acetone precipitation procedure. Laccase inhibition by chloride ions is known since a long time (Naki and Varfolomeev 1981).
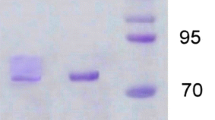
Concentrated culture supernatant and pooled enzyme fractions obtained from SEC and IEC purification of the concentrated culture supernatant were subjected to SDS-PAGE under denaturing and under zymogram conditions (Fig. 3). A single band of enzyme acticity, corresponding to an apparent molecular mass above 200 kDa, was detected for all samples in 9% polyacrylamide gel zymograms upon activity staining with ABTS (Fig. 3a). Upon application of samples to 6% polyacrylamide gels intended to obtain a better resolution of large proteins, rather diffuse patches of enzyme activity were observed, respectively (Fig. 3d). An estimation of the corresponding apparent molecular masses, as based on an extrapolation of the calibration curve provided by the concomitantly applied molecular mass markers, led to a range of about 210–260 kDa. This molecular mass range covers the apparent molecular mass of the enzyme of 212 kDa as determined by SEC (Fig. 2a).

Zymograms (a, b, d, e) and denaturing SDS-PAGE (c) of the concentrated culture supernatant of T. aeria (1) and of enzyme pools obtained from SEC (2) and IEC purification of the concentrated culture supernatant (3). Separating gels contained 9% (a–c) and 6% acrylamide (d, e). Gel pieces were either silver stained (b, c, e), or stained for enzyme activity with ABTS (a, d; arrows indicate activity bands). Numbers indicate molecular masses (kDa) of marker proteins (M) and apparent molecular masses of proteins detected in samples
Silver staining of the zymogram gels did not enable to unambiguously identify protein bands corresponding to the bands obtained upon activity staining with ABTS (Fig. 3a, b, d, e). Weak protein bands were detected at migration distances also covered by the respective enzyme activity bands for the concentrated culture supernatant and the SEC samples (Fig. 3d, e). However, no such protein bands were detected for the IEC samples, which provided the highest purity of the enzyme. By colloidal Coomassie G-250 staining according to Neuhoff et al. (1988), no protein bands corresponding to activity bands could be stained at all (data not shown). The lacking of protein staining by these techniques, as well as the appearance of rather diffuse activity spots in 6% polyacrylamide gels, may be due to a high degree of glycosylation and the presence of multiple glycoforms of the enzyme protein (Møller and Poulsen 2002; Rehm and Letzel 2010). All laccases described so far are glycoproteins (Baldrian 2006; Mayer and Staples 2002).
After denaturing SDS-PAGE, one major protein band, corresponding to an apparent molecular mass of 37 kDa, and two less intense protein bands, corresponding to apparent molecular masses of 35 and 32 kDa, were detected for all samples (Fig. 3c). No protein bands with apparent molecular masses above 116 kDa (the molecular mass of the largest protein standard applied) were detectable. The results of zymogram analysis and SDS-PAGE all together indicate that the enzyme is most likely an oligomeric protein, possibly composed of six monomers. It remains to be elucidated whether the enzyme is a heterooligomer, since the enzyme identity of the protein bands detected after SDS-PAGE (Fig. 3c) is presently unknown. Most laccases described so far are monomeric proteins, with typical molecular masses of 60–70 kDa for fungal (Baldrian 2006) and around 100 kDa for plant laccases (Sterijades et al. 1992; Nitta et al. 2002). Several oligomeric laccases have been described in fungi (see Giardina et al. 2009 for review). For instance, the ascomycete Podospora anserine produces a heterooligomeric laccase of about 390 kDa (Durrens 1981). Homodimeric fungal laccases were more often described, e.g. from basidiomycetes like Trametes villosa (subunits of 63 kDa; Yaver et al. 1996) and from the aquatic ascomycete Phoma sp. UHH 5-1-03 (subunits of 76 kDa; Junghanns et al. 2009). In lichens, homodimeric laccases (from Solorina crocea and Peltigera aphthosa) as well as a homotetrameric laccase (from Peltigera malacea) were found (subunits of 76, 97, and 85 kDa, respectively; Lisov et al. 2007; Laufer et al. 2006). Apparent molecular masses between about 140 and more than 350 kDa appear to be typical for native laccases from lichens (Laufer et al. 2009).
By IEF and subsequent enzyme activity staining, spots of enzyme activity with a partially ladder-like pattern focused for all samples in an aIcidic pH range around pH 4.5–5.2, with predominant enzyme activity spots referring to a p of about 4.8 (Fig. 4). Further, distinct but weak activity bands were detected with pI values of about 5.7, both for the concentrated culture supernatant and the SEC sample (Fig. 4). Possibly, the enzyme activity applied within the IEC sample was too low for detection of respective activity bands potentially also present in IEC samples. The pI of typical fungal laccases is around 4.0 (Baldrian 2006). Acidic pI values around 5.0 have also been found for lichen and plant laccases (Laufer et al. 2006; Sterijades et al. 1992).

IEF of the concentrated culture supernatant of T. aeria (1), enzyme pools obtained from SEC (2) and IEC purification of the concentrated culture supernatant (3), and marker proteins (M). Gel pieces were either stained for enzyme activity with ABTS (1-3) or colloidal Coomassie G-250 stained (M). Numbers indicate pI values of marker proteins and of proteins detected in samples
Kinetic properties in oxidation of selected substrates were determined with the concentrated culture supernatant. This approach seems justified, since no essential differences between ABTS-stained zymograms of the concentrated culture supernatant and those of samples from SEC and IEC separations were observed. ABTS was increasingly oxidized with decreasing pH values, while 2,6-DMP and SGZ exhibited bell-shaped pH-profiles as has often been described for laccase-catalyzed reactions (Baldrian 2006) (Fig. 5). SGZ was most efficiently oxidized in the range of pH 6.0–8.0, with maximal oxidation at pH 7.0. A broader pH range was examined for 2,6-DMP, in order to comprise the pH optimum of its oxidation. 2,6-DMP was converted over a broad pH range of 5.0–10.0, showing an alkaline optimum in oxidation at pH 8.0–8.5. Such high pH optima in substrate oxidation are only known from a few fungal (Baldrian 2006) and some bacterial laccases (Ruijssenaars and Hartmans 2004) and the laccase from the plant R. vernicifera (pH optimum of 9.0 towards SGZ; Johnson et al. 2003). The activity at high pH might be of advantage with respect to potential biotechnological applications of the enzyme. The quantification of the enzyme activities towards ABu62, RBBR and guaiacol involved considerable errors of measurement, caused by very low absorbance changes during the photometric determinations. Oxidation was maximal at pH 3.0 for ABu62 and at pH 8.0 for both RBBR and guaiacol. No oxidation of tyrosine by the concentrated culture supernatant of T. aeria was detected and hence tyrosinase activity can be excluded. Tyrosinases are not known from algae (Laufer et al. 2006).

pH dependence of the oxidation of various substrates by the concentrated culture supernatant of T. aeria in 0.1 M citrate/0.2 M phosphate (filled circles) and 0.04 M Britton-Robinson buffer (empty circles; only applied for 2,6-DMP). Data represent means ± standard deviations for triplicate determinations. 100% refer to absolute values of 212.3 (ABTS), 10.8 and 15.1 (2,6-DMP in 0.1 M citrate/0.2 M phosphate and 0.04 M Britton-Robinson buffer, respectively), 3.9 (SGZ), 5.5 (ABu62), 2.1 (RBBR) and 0.8 U l−1 (guaiacol)
The oxidation of ABTS, ABu62 and 2,6-DMP followed typical Michaelis–Menten kinetics (all correlation coefficients ≥0.96), while for SGZ oxidation the application of the Hill model yielded a better fit with the obtained data (correlation coefficient >0.99) than fitting according to Michelis-Menten kinetics. It remains to be clarified whether this observation reflects cooperative enzyme kinetics or was merely caused by an aberration of the recorded data. For both asco- and basidiomycete laccases, the oxidation of ABu62 could sufficiently be described with the Hill model in previous studies, but the reasons for such effects are still not clear (Junghanns et al. 2009). For guaiacol and RBBR, no sufficient fit with the obtained data was achieved (correlation coefficients <0.64).
Among the tested substrates, the T. aeria enzyme showed the highest affinities for SGZ and ABTS, followed by ABu62 and 2,6-DMP (Table 3). The catalytic efficiency (in terms of the V max/K M ratio) was by far highest for ABTS and followed the rank order ABTS > SGZ > ABu62 > 2,6-DMP. The observed affinities of the enzyme towards the common laccase substrates ABTS, SGZ and 2,6-DMP, as well as the rank order of the respective catalytic efficiencies resemble data typically reported for extracellular fungal laccases (Baldrian 2006). Along with the absence of tyrosinase activity, the kinetic properties of the T. aeria enzyme are in strong support of a laccase-like enzyme.
Conclusions
The ability to oxidize phenolic substrates and the anthraquinonic dye ABu62 indicates potential biotechnological applications of the enzyme concerning the treatment of effluents contaminated with phenolics and/or synthetic dyes, provided that sufficiently high amounts of the enzyme could be produced, e.g. by its expression in heterologous hosts. Concerning potential biotechnological applications of soil algae in general, especially those adapted to unfavourable environments might turn out to offer a source of biotechnologically interesting enzymes.
Regarding laccases isolated from lichens harbouring green algae, the occurence of laccase-like enzymes in green algae puts forth the possibility that both the algal as well as the fungal symbionts may potentially account for the production of such enzymes.
The biological function of the T. aeria enzyme is still not clear. Potential functions might be related to the degradation of toxic phenolic substances in soil or to pathogen defence, as postulated for laccases from other organisms (Claus 2004). Similar to fungal laccases, a role related to the degradation of lignocellulosic substrates in soils may also be considered, which could support a mixo- or heterotrophic nutrition of the alga in various ways. Brown and Bold (1964) found that T. aeria is able to extracellularly metabolize starch and to grow heterotrophically with glucose or fructose as a carbon source. Literature data also suggest that many microalgae can not only aerobically metabolize phenolic compounds but also grow on them in the light and/or in the dark (Lika and Papadakis 2009). Phenolic compounds may be released from lignocellulosic substrates through the action of laccases and other lignin-modifying enzymes. When released into the environment, the T. aeria enzyme may oxidize suitable organic substrates and thus contribute to the turnover of soil organic matter (Baldrian 2006). Although the extracellular localization of the major proportion of the enzyme activity is not in favour of a potential function of the T. aeria enzyme in cell wall formation, this possibility cannot totally be ruled out. Laccases have been implicated in lignin formation in higher plants (Mayer and Staples 2002). However, lignin-like compounds have not been described in green algae except for the Charophyceae (Delwiche et al. 1989). More detailed investigations addressing structural and catalytic properties of the T. aeria enzyme would help to clarify the question of its natural function and concomitantly contribute to a better understanding of the physiology of soil algae and their potential impacts on terrestrial ecosystems.
References
Acuner E, Dilek FB (2004) Treatment of tectilon yellow 2G by Chlorella vulgaris. Process Biochem 39:623–631
Baker DE, Senft JP (1995) Copper. In: Alloway BJ (ed) Heavy metals in soils, 2nd edn. Blackie Academic & Professional Press, London, pp 179–205
Baldrian P (2006) Fungal laccases—occurrence and properties. FEMS Microbiol Rev 30:215–242
Bischoff H, Bold HC (1963) Some soil algae from enchanted rock and related algal species. Phycological Studies IV. The University of Texas publication 6318, Texas
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Britton HTS, Robinson RA (1931) Universal buffer solutions and the dissociation constant of veronal. J Chem Soc 1931:1456–1462
Brown RM Jr, Bold HC (1964) Comparative studies of the algal genera Tetracystis and Chlorococcum. Phycological studies V. The University of Texas publication 6417, Texas
Cardon ZG, Gray DW, Lewis LA (2008) The green algal underground: evolutionary secrets of desert cells. Bioscience 58:114–122
Claus H (2004) Laccases: structure, reactions, distribution. Micron 35:93–96
Daneshvar N, Ayazloo M, Khataee AR, Pourhassan M (2007) Biological decolourization of dye solution containing Malachite Green by microalgae Cosmarium sp. Bioresour Technol 98:1176–1182
Das N, Sengupta S, Mukherjee M (1997) Importance of laccase in vegetative growth of Pleurotus florida. Appl Environ Microbiol 63:4120–4122
Delwiche CF, Graham LE, Thomson N (1989) Lignin-like compounds and sporopollenin in Coleochaete, an algal model for land plant ancestry. Science 245:399–401
Durrens P (1981) The phenoloxidases of the ascomycete Podospora anserina: the three forms of the major laccase activity. Arch Microbiol 130:121–124
Eggert C, Temp U, Eriksson K-EL (1996) The ligninolytic system of the white rot fungus Pycnoporus cinnabarinus: purification and characterization of the laccase. Appl Environ Microbiol 62:1151–1158
Ettl H, Gaertner G (1995) Syllabus der Boden-, Luft- und Flechtenalgen. Gustav-Fischer Verlag, Stuttgart
Giardina P, Faraco V, Pezzella C, Piscitelli A, Vanhulle S, Sannia G (2009) Laccases: a never-ending story. Cell Mol Life Sci. doi:10.1007/s00018-009-0169-1
Grassin C, Dubourdieu D (1989) Quantitative determination of Botrytis Laccase in musts and wines by the syringaldazine test. J Sci Food Agric 48:316–369
Johnson DL, Thompson JL, Brinkmann SM, Schuller KA, Martin LL (2003) Electrochemical characterization of purified Rhus vernicifera Laccase: voltammetric evidence for a sequential four-electron transfer. Biochemistry 42:10229–10237
Junghanns C, Parra R, Keshavarz T, Schlosser D (2008) Towards higher laccase activities produced by aquatic ascomycetous fungi through combination of elicitors and an alternative substrate. Eng Life Sci 8:277–285
Junghanns C, Pecyna MJ, Böhm D, Jehmlich N, Martin C, von Bergen M, Schauer F, Hofrichter M, Schlosser D (2009) Biochemical and molecular genetic characterisation of a novel laccase produced by the aquatic ascomycete Phoma sp. UHH 5-1-03. Appl Microbiol Biotechnol 84:1095–1105
La Russa M, De Biasi MG, Chiaiese P, Palomba F, Pollio A, Pinto G, Filippone E (2008) Screening of green microalgae species for extracellular phenoloxidase activity useful for wastewater phycoremediation. In: Proceedings of the 4th European Bioremediation Conference, Chania, Greece (September 03–06, 2008)
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Laufer Z, Beckett RP, Minibayeva FV (2006) Co-occurence of the multicopper oxidases tyrosinase and laccase in lichens in sub-order Peltigerineae. Ann Bot 98:1035–1042
Laufer Z, Beckett RP, Minibayeva FV, Lüthje S, Böttger M (2009) Diversity of laccases from lichens in suborder Peltigerineae. Bryologist 112:418–426
Lika K, Papadakis IA (2009) Modeling the biodegradation of phenolic compounds by microalgae. J Sea Res 62:135–146
Lima SAC, Castro PML, Morais R (2003) Biodegradation of p-nitrophenol by microalgae. J Appl Phycol 15:137–142
Lisov AV, Zavarzina AG, Zavarzin AA, Leontievsky AA (2007) Laccases produced by lichens of the order Peltigerales. FEMS Microbiol Lett 275:46–52
Mai C, Majcherczyk A, Hüttermann A (2000) Chemo-enzymatic synthesis and characterization of graft copolymers from lignin and acrylic compounds. Enzyme Microb Technol 27:167–175
Mansur M, Arias ME, Copa-Patiño JL, Flärdh M, González AE (2003) The white-rot fungus Pleurotus ostreatus secretes laccase isozymes with different substrate specificities. Mycologia 95:1013–1020
Martin C, Pecyna M, Kellner H, Jehmlich N, Junghanns C, Benndorf D, von Bergen M, Schlosser D (2007) Purification and biochemical characterization of a laccase from the aquatic fungus Myrioconium sp. UHH 1–13-18–4 and molecular analysis of the laccase-encoding gene. Appl Microbiol Biotechnol 77:613–624
Massalski A, Mroziñska T, Olech M (2001) Ultrastructural observations on five pioneer soil algae from ice denuded areas (King George Island, West Antarctica). Polar Biosci 14:61–70
Mayer AM, Staples RC (2002) Laccase: new functions for an old enzyme. Phytochem 60:551–565
Møller HJ, Poulsen JH (2002) Staining of glycoproteins/proteoglycans in SDS-Gels. In: Walker JM (ed) The protein protocols handbook, 2nd edn. Humana Press, Totowa, pp 773–777
Nakano T (1983) Taxonomical studies on the genus Tetracystis (Chlorosarcinales, Chlorophyta) from Japanese soils. J Sci Hiroshima Univ Series B, Div 2 (Botany) 18:115–172
Naki A, Varfolomeev SD (1981) Inhibition mechanism of Polyporus versicolor laccase by halide ions. Biokhimiia 46:1694–1702
Neuhoff V, Arold N, Taube D, Ehrhardt W (1988) Improved staining of proteins in polyacrylamide gels including isoelectric focusing gels with clear background at nanogram sensitivity using Coomassie Brilliant Blue G-250 and R-250. Electrophoresis 9:255–262
Niemetz R, Gross GG (2003) Ellagitannin biosynthesis: laccase-catalyzed dimerization of tellimagrandin II to cornusiin E in Tellima grandiflora. Phytochemistry 64:1197–1201
Niladevi KN, Jacob N, Prema P (2008) Evidence for a halotolerant-alkaline laccase in Streptomyces psammoticus: purification and characterization. Process Biochem 43:654–660
Nitta K, Kataoka K, Sakurai T (2002) Primary structure of a Japanese lacquer tree laccase as a prototype enzyme of multicopper oxidases. J Inorg Biochem 91:125–131
Oesterreicher W (1990) Ökologische Bedeutung der Algen im Boden. Nachrichtenbl Deut Pflanzenschutzd 42:122–126
Palmieri G, Giardina P, Bianco C, Fontanella B, Sannia G (2000) Copper Induction of Laccase Isoenzymes in the Ligninolytic Fungus Pleurotus ostreatus. Appl Environ Microbiol 66:920–924
Rehm H, Letzel T (2010) Der Experimentator: Proteinbiochemie/Proteomics, 6th edn. Spektrum Akademischer Verlag, Heidelberg, pp 339–361
Riva S (2006) Laccases: blue enzymes for green chemistry. Trends Biotechnol 24:219–226
Round FE (1984) The ecology of algae. Cambridge University Press Archive, Cambridge
Ruijssenaars HJ, Hartmans S (2004) A cloned Bacillus halodurans multicopper oxidase exhibiting alkaline laccase activity. Appl Microbiol Biotechnol 65:177–182
Safonova E, Reisser W (2005) Growth promoting and inhibiting effects of extracellular substances of soil microalgae and cyanobacteria on Escherichia coli and Micrococcus luteus. Phycol Res 53:189–193
Safonova E, Kvitko K, Kuschk P, Möder M, Reisser W (2005) Biodegradation of Phenanthrene by the Green Alga Scenedesmus obliquus ES-55. Eng Life Sci 5:234–239
Scheu S, Folger M (2004) Single and mixed diets in Collembola: effects on reproduction and stable isotope fractionation. Funct Ecol 18:94–102
Seckbach J (ed) (2007) Algae and cyanobacteria in extreme environments. Springer, Secaucus
Semple KT, Cain RB, Schmidt S (1999) Biodegradation of aromatic compounds by microalgae. FEMS Microbiol Lett 170:291–300
Shtina EA (1974) The principal directions of experimental investigations in soil algology with emphasis on the USSR. Geoderma 12:151–156
Sterijades R, Dean JFD, Eriksson K-EL (1992) Laccase from sycamore maple (Acer pseudoplatanus) polymerizes monolignols. Plant Physiol 99:1162–1168
Tarlan E, Dilek FB, Yetis U (2002) Effectiveness of algae in the treatment of a wood-based pulp and paper industry wastewater. Bioresour Technol 84:1–5
Wood DA (1980) Production, purification and properties of extracellular laccase of Agaricus bisporus. J Gen Microbiol 117:327–338
Yaver DS, Xu F, Golightly EJ, Brown KM, Brown SH, Rey MW, Schneider P, Halkier T, Mondorf K, Dalboge H (1996) Purification, characterization, molecular cloning, and expression of two laccase genes from the white rot basidiomycete Trametes villosa. Appl Environ Microbiol 62:834–841
Zancan S, Trevisan R, Paoletti MG (2006) Soil algae composition under different agro-ecosystems in North-Eastern Italy. Agric Ecosyst Environ 112:1–12
Ziegler R, Egle K (1965) Zur quantitativen Analyse der Chloroplastenpigmente. Beitr Biol Pflanzen 41:11–37
Acknowledgments
We are thankful to François Buscot (Halle) and Susanne Theuerl (Halle) for helpful advice and discussions. The programme topic CITE (Chemicals in the Environment) of the Helmholtz Association of German Research Centres provided resources for this research.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Erko Stackebrandt.
Rights and permissions
About this article
Cite this article
Otto, B., Schlosser, D. & Reisser, W. First description of a laccase-like enzyme in soil algae. Arch Microbiol 192, 759–768 (2010). https://doi.org/10.1007/s00203-010-0603-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00203-010-0603-7