
Abstract
Dirigent proteins (DIRs) are thought to play important roles in plant secondary metabolism. They lack catalytic activity but direct the outcome of bimolecular coupling reactions toward regio- and stereospecific product formation. Functionally described DIRs confer specificity to the oxidative coupling of coniferyl alcohol resulting in the preferred production of either (+)- or (−)-pinoresinol, which are the first intermediates in the enantiocomplementary pathways for lignan biosynthesis. DIRs are extracellular glycoproteins with high β-strand content and have been found in all land plants investigated so far. Their ability to capture and orientate radicals represents a unique naturally evolved concept for the control of radical dimerization reactions. Although oxidative coupling is commonly used in biological systems, its wider application in chemical synthesis is often limited by insufficient selectivity. This minireview gives an overview of functionally described DIRs and their molecular characteristics and wants to inspire further research for their use in biotechnological applications.
Similar content being viewed by others

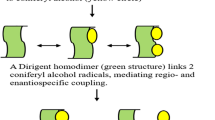
Avoid common mistakes on your manuscript.
Introduction
Chemical synthesis of natural products has been widely used to overcome their limited availability from natural sources as caused by their low abundance in source organisms or difficult isolation and purification schemes. Although a great variety of molecules are accessible by classical chemical or biomimetic approaches (Nicolaou et al. 2000, 2003; Gravel and Poupon 2008; Lindsley et al. 2011), the production procedures are often very complicated because of the complex structure and the defined absolute configuration of many natural substances (Finefield et al. 2012). Oxidative coupling is a prominent reaction type in the biosynthesis of many natural products, especially for phenolic compounds (Keseru and Nogradi 1998; Dewick 2009). Despite considerable progress in recent years in terms of selectivity (Kuhl et al. 2012; Giri et al. 2009), a wider application of this reaction type in chemical synthesis has still not caught on, since the control of regio- and stereospecificity remains challenging (Lessene and Feldman 2002; Whiting 1991; Iqbai et al. 1994; Chioccara et al. 1993; Lindsley et al. 2011). The application of oxidative coupling is nonetheless promising because of its fast reaction rate and efficiency under mild reaction conditions, alleviating the need for substrate activation or protection groups (Jasperse et al. 1991). While biotechnological approaches with enzymes as catalysts have successfully been used to solve the problem of selectivity in many cases (Koeller and Wong 2001; Bornscheuer et al. 2012), their potential in phenol coupling reactions remains largely unexplored.
Stereo- and regiospecific control of phenoxy radical coupling
Radical coupling in general is a two-step process. Firstly, radicals are generated from the substrate by the action of an oxidizing agent. In the second step, the two radicals are quenched by the formation of a covalent C–C or C–O bond. In the presence of conjugated double bond systems, delocalization of the unpaired electrons can lead to the generation of multiple products. In natural product (bio-)synthesis, where stereo- and enantiospecific features of the product are often associated with bioactivity (Mori 2011; Leffingwell 2003), the generation of side products or stereoisomers of incorrect absolute configuration can be considered as a waste of energy and resources. Therefore, the (bio-)synthesis of compounds generated by oxidative phenoxy coupling requires regio- and enantioselective control. While regioselectivity during radical coupling may be, in part at least, provided by thermodynamically favored mesomeric structures of the generated radicals, enantioselectivity is energetically indiscriminant. Nonetheless, optimization by natural selection resulted in the evolution of at least two mechanisms that confer regio- and enantiospecificity to phenoxy radical coupling. In the first one, the absolute configuration of the product is determined by the oxidizing enzyme. In Aspergillus niger, for example, the P450 enzyme KtnC was shown to control regio- and enantiospecificity of the oxidative coupling of demethyl siderin to P-(+)-orlandin, an intermediate in P-(+)-kotanin biosynthesis (Girol et al. 2012), and in Daldinia eschscholzii, a laccase catalyzes the formation of (−)-dalesconol with app. 67 % enantiomeric excess (e.e.) (Fang et al. 2012). In these examples, radical formation and subsequent coupling are achieved by the same enzyme, whereas the second mechanism requires two components: an oxidizing agent to generate radicals by one-electron abstraction and a second factor that forces the free radicals to undergo regio- and stereospecific coupling. This mechanism was discovered in 1997 by Lewis and coworkers, who identified a protein in the insoluble cell wall fraction of Forsythia intermedia that affected the free radical coupling of coniferyl alcohol in the presence of inorganic oxidants, resulting in the formation of (+)-pinoresinol with 100 % e.e. (Davin et al. 1997). The protein was named dirigent protein (DIR) from the Latin word “dirigere” (to guide or to align) (Davin et al. 1997), and its discovery led to a new concept for the control of intermolecular phenoxy radical coupling in plant secondary metabolism. In the following sections, the up-to-date knowledge of DIRs is summarized, focusing on functionally characterized DIRs and their molecular properties and evaluating their potential for biotechnological application.
Dirigent proteins in natural product biosynthesis
The few DIRs that have been characterized to date are all involved in lignan biosynthesis. Lignans are structurally defined as 8,8′-coupled phenylpropanoid dimers (Moss 2000). They are widespread in the plant kingdom (Umezawa 2003), and some of them exhibit strong antioxidative or bioactive properties (MacRae and Towers 1984; Saleem et al. 2005). The canonical pathway for lignan biosynthesis was first established in Forsythia (Lewis and Davin 1999; Suzuki and Umezawa 2007). E-coniferyl alcohol derived from the phenylpropanoid pathway serves as universal precursor for all lignans in angio- and gymnosperms (Lewis and Davin 1999). The only exception known to date is liridendrin which is synthesized from sinapyl alcohol (Fujimoto and Higuchi 1977; Katayama and Ogaki 2001) and was first described in Liridendron tulipifera, a deciduous tree from eastern North America (Dickey 1958). In the first step of the pathway, oxidation of E-coniferyl alcohol—putatively catalyzed by a laccase (Davin et al. 1997)—leads to the formation of radicals, which dimerize to yield pinoresinol (Fig. 1). The successive reduction of pinoresinol by pinoresinol lariciresinol reductases (PLRs) generates larici- and secoisolariciresinol, respectively (Dinkova-Kostova et al. 1996). Secoisolariciresinol is oxidized by secoisolariciresinol dehydrogenases (SIRD) concomitant with the formation of a lacton moiety to afford matairesinol (Xia et al. 2001; Moinuddin et al. 2006). Further modifications of matairesinol or upstream precursors account for the numerous lignan structures described (Umezawa 2003).

Overview of the coniferyl alcohol radical coupling reaction and its link to lignan biosynthesis. Coniferyl alcohol (1) is oxidized by one-electron oxidation. The resulting radicals are stabilized by resonance and couple in open solution to (±)-pinoresinol (2), (±)-dehydrodiconiferyl alcohol (3), and erythro/threo -(±)-guaiacylglycerol coniferyl alcohol ethers (4). In the presence of AtDIR5/6 (−)-2 and in the presence of FiDIR1, ScDIR1 or TpDIR5/8 (+)-2 are formed, which are converted by PLRs and SIRDs to (+)- or (−)-secoisolariciresinol (5) in the subsequent steps of the lignan biosynthetic pathway
Lignan biosynthesis is under stereochemical control. While plant-derived dibenzylbutyrolignans are optically pure in most cases, furofuran-, furan-, and dibenzyllignans exhibit at least an enantiomeric excess (Umezawa 2003). The enantiomeric configuration of lignans is determined during the first step of biosynthesis, as two chiral centers are introduced during the formation of a covalent bond between C-8 and C-8′. This once established absolute configuration is maintained throughout the downstream biosynthetic pathway (Fig. 1).
In contrast, when E-coniferyl alcohol is oxidized in vitro by organic or inorganic oxidizing agents, or enzymatically through the action of non-specific laccases or peroxidases (Sterjiades et al. 1992; Frías et al. 1991; Davin et al. 1997; Bao et al. 1993; Chioccara et al. 1993), bimolecular coupling of the resulting radicals leads to racemic mixtures of 55.6 % (±)-dehydrodiconiferyl alcohols, 27.7 % (±)-pinoresinols, and (under the addition of water) 16.7 % erythro/threo-(±)-guaiacylglycerol coniferyl alcohol ethers (Halls et al. 2004) (Fig. 1). While the stereoselectivity of subsequent enzymes in the biosynthetic pathway could potentially explain the accumulation of selected enantiomerically pure end-products in vivo (von Heimendahl et al. 2005), such a pathway would be highly inefficient, as only a small fraction of the initial coupling products would actually end up in lignan biosynthesis. The employment of DIRs in combination with an oxidizing agent to control the coupling outcome in regio- and stereospecific terms provides a more efficient alternative.
The hallmarks of DIR-mediated couplings include (1) the dependence on an oxidizing agent, (2) altered product ratio as compared to the free coupling reaction, and (3) the loss of selectivity when DIRs are denatured, as first described for FiDIR1 from Forsythia (Davin et al. 1997). In agreement with the proposed biosynthetic pathway for lignans, coniferyl alcohol radicals are the only substrates of FiDIR1, and other monolignol coupling reactions are not affected (Davin et al. 1997). FiDIR1 directs the coupling of coniferyl alcohol radicals toward the formation of (+)-pinoresinol (Davin et al. 1997). Lignans derived from (+)-pinoresinol are widespread in phylogenetically diverse taxa of the plant kingdom including gymno- and angiosperms (Austrobaileyales and Lamiales). The presence of (+)-pinoresinol-forming DIRs related to FiDIR1 might thus be expected in all these taxa. This has been confirmed for several species including western red cedar (Thuja plicata) and Schisandra chinensis (Table 1). The heartwood of western red cedar contains large amounts of 8–8′ linked lignans, mainly plicatic acid and derivatives thereof (Yasuda et al. 1989). The absolute configuration of plicatic acid is 2R, 3S, 4R ((−)-plicatic acid) (Gardener et al. 1966; Swan et al. 1967), which is in agreement with (+)-pinoresinol being its precursor (Kim et al. 2002a). Nine DIRs were identified in T. plicata, and as expected, those that were characterized functionally (TpDIR5 and TpDIR8) were found to direct the coupling of E-coniferyl alcohol radicals toward (+)-pinoresinol (Kim et al. 2002b). S. chinensis, a medicinal plant native to northern China, produces dibenzocyclooctadiene lignans with different enantiomeric configurations, including schizandrins and gomisins (reviewed in Lu and Chen 2009). While the biosynthetic pathway of Schisandra lignans has not been fully established, DIRs are likely to be involved for enhanced efficiency. Recently, such a DIR has been characterized (ScDIR1), and similar to previously characterized DIRs from Thuja and Forsythia, it was found to mediate (+)-pinoresinol formation in vitro (Kim et al. 2012). One of the pharmaceutically and economically most important (+)-pinoresinol-derived lignans is podophyllotoxin. Its derivatives etopside and teniposide are widely used in the treatment of various cancers (reviewed in Gordaliza et al. 2004). Podophyllotoxin and derivatives have been found in several Podophyllum (mayapple) (Bedows and Hatfield 1982; Jackson and Dewick 1983) and Linum (flax) species (Berlin et al. 1986; Broomhead and Dewick 1990; Fuss 2003). Its biosynthesis occurs via the general lignan pathway including matairesinol (Xia et al. 2000; Seidel et al. 2002). With an absolute configuration of (−)-podophyllotoxin (Petcher et al. 1973), (+)-pinoresinol would be the appropriate precursor. Two DIRs have been identified in Podophyllum peltatum, but their expected specificity for (+)-pinoresinol formation has not been confirmed yet (Xia et al. 2000).
Lignans with opposite enantiomeric configuration have been isolated from various plants like (+)-secoisolariciresinol from Wikstroemia sikokiana (Okunishi et al. 2000), (−)-lariciresinol from Arabidopsis thaliana (Nakatsubo et al. 2008), (+)-matairesinol from Daphne odora and Daphne genkwa (Okunishi et al. 2001), as well as (+)-secoisolariciresinol-O-β-d-diglucopyranoside and (−)-pinoresinol-O-β-d-diglucopyranoside from flax (Linum usitatissimum) seeds (Qiu et al. 1999). Those lignans are derived from (−)-pinoresinol and, therefore, have to be synthesized by an enantiocomplementary biosynthetic pathway (Suzuki and Umezawa 2007). The existence of parallel pathways for the synthesis of lignans with opposite enantiomeric configuration is supported by the identification of PLRs that show substrate specificity for either (+)- or (−)-pinoresinol. PLR from Linum album, for example, reduces (+)-pinoresinol, while L. usitatissimum PLR acts upon the (−)-enantiomer (von Heimendahl et al. 2005). Furthermore, two PLRs with opposing enantioselectivity have been characterized in western red cedar, suggesting that enantiocomplementary pathways for lignan biosynthesis may exist even within a single species (Fujita et al. 1999). Consistent with this notion, Arctium lappa produces lignans with opposite absolute configurations in seeds and petioles, respectively (Suzuki et al. 2002).
The presence of enantiocomplementary pathways for lignan biosynthesis implies the existence of two types of DIRs, the well-characterized type for the production of (+)-pinoresinol, and a second type mediating (−)-pinoresinol formation. First indications for the existence of an enantiocomplementary pair of DIRs were obtained in A. lappa. Using enzyme preparations from petioles in phenoxy radical coupling assays, the preferred formation of (+)-pinoresinol (33 % e.e.) was observed, while (−)-pinoresinol was preferentially formed (22 % e.e.) by protein extracts from ripening seeds (Suzuki et al. 2002). However, the corresponding DIRs have not been identified yet. Indirect evidence for the existence of a (−)-pinoresinol forming DIR was obtained also in A. thaliana roots, which accumulate (−)-lariciresinol at 88 % e.e. (Nakatsubo et al. 2008). Further investigations of the lignan biosynthetic pathway in A. thaliana revealed that lariciresinol is formed by two pinoresinol reductases (AtPrR1 and AtPrR2). In double mutants lacking functional atprr1 and atprr2 genes, lariciresinol was completely absent, rather pinoresinol accumulated with 74 % e.e. in favor of the (−)-enantiomer, indicating that pinoresinol formation is under stereospecific control (Nakatsubo et al. 2008). Considering these genetic data, A. thaliana provided an ideal model system for the identification of the enantiocomplementary DIR responsible for (−)-pinoresinol formation. Among 24 homologs identified by sequence comparison, AtDIR6 and AtDIR5 showed the highest sequence similarity with the known (+)-pinoresinol-forming DIRs (Pickel et al. 2010). AtDIR6 was cloned and expressed in a plant cell culture system. The purified recombinant protein exhibited the long-sought enantiocomplementary activity, mediating the formation of (−)-pinoresinol in vitro (Pickel et al. 2010). The (−)-pinoresinol-forming activity was later confirmed for AtDIR6 and also found for AtDIR5 (Kim et al. 2012).
In addition to their role in lignan biosynthesis, DIRs are likely to be involved in other phenoxy radical coupling processes as well. This includes the biosynthesis of gossypol in cotton (Gossypium hirsutum). Gossypol, a highly bioactive sesquiterpenoid dimer (Wang et al. 2008; Jiang et al. 2012), is formed by phenoxy radical coupling of two molecules of hemigossypol (Veech et al. 1976; Benedict et al. 2006) (Fig. 2a). Thereby a chiral axis is introduced leading to two possible atropisomers, (+)-S- and (−)-R-gossypol, with the (−)-atropisomer exhibiting greater biological activity (Band et al. 1989; Matlin et al. 1985). The enantiomeric composition of gossypol varies among Gossypium species and even among different varieties. Most cotton species contain (−)- and (+)-gossypol at a ratio of app. 2:3, while, e.g., G. hirsutum var. marie-galante (moco cotton) accumulates (+)-gossypol at 90 % e.e. (Cass et al. 1991). The highest levels of (−)-gossypol were detected in Gossypium barbadense (Stipanovic et al. 2009) and some wild species (Stipanovic et al. 2005), but never exceeded 68 % e.e. Strong evidence for the involvement of DIRs in the final step of gossypol biosynthesis was obtained in G. hirsutum var. marie-galante. Protein extracts from embryos and flower petals promoted the formation of (+)-gossypol with 46 to 59 % e.e. in vitro, while control reactions yielded racemic product (Benedict et al. 2006; Liu et al. 2008). Two putative DIR-like proteins have been cloned from G. barbadense (Zhu et al. 2007), and two sequences from G. hirsutum are available in GenBank (cf. Table 1), but an involvement in gossypol formation has not been confirmed.

Reaction scheme of the radical coupling of hemigossypol (6) to racemic gossypol (7) or (−)-R-gossypol (R -7) under the influence of putative DIRs and the enantioselective generation of ε-viniferin (9) from resveratrol (8) with (+)-ε-viniferin ((+)-9) accumulating in Vitaceae and (−)-ε-viniferin ((−)-9) in other taxa
In addition to the systems described, the widespread occurrence of radical coupling steps in biosynthetic pathways leading to enantiomerically pure natural products and the considerable size of DIR gene families in higher plants (Ralph et al. 2006, 2007) provide a great potential for a more general involvement of DIRs in stereochemical control of secondary metabolism. The concept of DIR-mediated coupling control may also account for the specificity observed in the biosynthesis of distilbenoids (Rivière et al. 2012), biflavanoids (Iwashina 2000), disesquiterpenoids (Zhan et al. 2011), tannins (Ascacio-Valdés et al. 2011; Khanbabaee and van Ree 2001; Bors and Michel 2002), and other natural products.
For example, resveratrol and derivatives thereof can be oxidized by fungal laccases (Pezet et al. 1991; Nicotra et al. 2004; Ponzoni et al. 2007) or horseradish peroxidase (Langcake and Pryce 1977). Dimerization or oligomerization of the resulting radicals yields di- or oligostilbenoids, respectively. Using inorganic oxidants, the distilbene (±)-ε-viniferin was obtained as a racemate (Takaya et al. 2005). Also the laccase-mediated oxidation of 3,5-dimethoxy-4′-hydroxystilbene yielded a racemic trans-resveratrol dehydrodimer as the main product (Ponzoni et al. 2007). These studies show clearly that there is no thermodynamic preference for the production of one enantiomer over the other. In planta, however, ε-viniferin biosynthesis is stereochemically controlled (Fig. 2b). The formation of (+)-ε-viniferin in Vitaceaeous plants (He et al. 2008; Rivière et al. 2012) and of (−)-ε-viniferin in Dipterocarpaceae (Ito et al. 2009) and Cyperaceae (Kurihari et al. 1990) points toward the involvement of corresponding enantiocomplementary DIR activities.
In general, the concept of DIR-mediated coupling control may apply in all cases, where opposite enantiomers of a given metabolite accumulate in vivo, maybe in different species or in different tissues, while uncontrolled coupling in vitro results in diverse or racemic products. The action of DIRs is also not necessarily limited to bimolecular radical coupling.
From a functional point of view, the term “dirigent protein” should be extended to include all proteins that guide the transformation of reactive molecules, like radicals, epoxides, and others, toward one of multiple possible products, by excluding all but one of the possible reaction channels. In that broader sense, also allene oxide cyclases (AOCs), which mediate the stereospecific cyclization of an unstable epoxide (Ziegler et al. 2000), show dirigent activity.
Molecular characteristics of dirigent proteins
Primary structure
In a detailed phylogenetic analysis, app. 150 DIR sequences have been grouped into six subfamilies (Ralph et al. 2007). The functionally characterized pinoresinol-forming DIRs cluster in subfamily a. As no functional data are available for any subfamily other than subfamily a, the members of the other subfamilies are referred to as DIR-like proteins (Ralph et al. 2006). This also includes DIR-like proteins from Gossypium that are located within subfamily b and putatively involved in gossypol formation. Although sequence similarity can fall into the arbitrary range between or within subfamilies, six conserved motives have been identified that are characteristically present in all DIRs and DIR-like proteins (Ralph et al. 2006). While some DIR-like proteins contain additional domains of varying size (Kittur et al. 2007; Ralph et al. 2006), the functionally characterized DIRs are relatively small proteins of app. 180 amino acids, with molecular weights ranging from 18 to 21 kDa (Table 1), and they are encoded by intronless genes (Kim et al. 2002a; Ralph et al. 2006, 2007).
Post-translational modifications
Characteristic for pinoresinol-forming DIRs is an N-terminal signal peptide, which targets the protein for secretion into the apoplastic space (Table 1). Cleavage of the signal peptide has been validated experimentally for FiDIR1 (Gang et al. 1999), TpDIR8 (Kim et al. 2002b), and AtDIR6 (Pickel et al. 2010) (Table 1). Depending on the expression system, small differences were observed between the predicted and the actual sites of processing. Secretion was confirmed for FiDIR1 by in situ localization within the cell wall of F. intermedia stems (Burlat et al. 2001; Gang et al. 1999; Davin and Lewis 2000). Also AtDIR6 was found to be secreted when expressed in a plant cell culture (Pickel et al. 2010). However, only a small fraction was present in the culture supernatant; the majority of AtDIR6 remained non-covalently bound to the cell wall by ionic interactions.
The experimentally determined molecular weight of purified DIRs differs considerably from that predicted on basis of the amino acid sequences (Gang et al. 1999; Kim et al. 2002a; Pickel et al. 2010). The mass difference could be attributed to extensive glycosylation, since several isoforms were observed for heterologously expressed FiDIR1 and AtDIR6 that were converted into a single smaller species by chemical or enzymatic deglycosylation (Pickel et al. 2010; Gang et al. 1999). The sequences of the pinoresinol-forming DIRs comprise two to five N-glycosylation consensus motives (NxS/T) (Marshall 1972; Gavel and Heijne 1990), whereas four to seven potential N-glycosylation sites are predicted for DIRs from Gossypium (Table 1; Fig. 3). For AtDIR6, glycosylation at the predicted sites (N59 and N123) was confirmed by mass spectrometry (Pickel et al. 2010). The glycan chain was found to be of the paucimannosidic-type (Pickel et al. 2012), which is common in plant proteins and has also been shown for the subtilisin-like proteinases P69B (Bykova et al. 2006) and SBT3 from Solanum lycopersicum (Cedzich et al. 2009). Considering that paucimnannosidic glycans are derived from bigger complex N-type glycans by the successive removal of two Lewisa epitopes (0.5 kDa) (Lerouge et al. 1998), the occurrence of the five different AtDIR6 isoforms with mass differences of app. 0.5 kDa can be explained by incomplete processing during secretion.

Sequence alignment of functionally characterized DIRs involved in the formation of (+)-pinoresinol (I, FiDIR1, ScDIR1, TpDIR5/8), (−)-pinoresinol (II, AtDIR5/6) and without detected functionality towards coniferyl alcohol radicals (II, AtDIR13) as well as Gossypium DIRs putative involved in the coupling of hemigossypol (III, GhDIR1/2 and GbDIR1/2). Predicted glycosylation sites of pinoresinol-forming DIRs are marked by a red bar, those of potentially gossypol-forming DIRs by a blue bar. Cysteine residues participating in disulfide formation as shown for AtDIR6 (Pickel et al. 2012) are highlighted in yellow. Functionally relevant phenylalanines of ScDIR1 are indicated in green (Kim et al. 2012), the corresponding residue exchanged for Met in the inactive AtDIR13 is shown in red. The region implicated in coupling control is framed in red (Kim et al. 2012); amino acids that are differentially conserved between (+)- and (−)-pinoresinol forming DIRs in this region are marked by dots
Glycosylation of secreted proteins may assist in the correct folding of proteins during passage through the secretory pathway; it may be important for the regulation of enzyme activity, enhance protein solubility, or contribute to its stability (Shental-Bechor and Levy 2008, 2009; Hanson et al. 2009). Also for AtDIR6, glycosylation was found to by functionally important. Deglycosylation of AtDIR6 over time correlated with a gradual loss of soluble protein and dirigent activity (Kazenwadel et al. 2012). The formation of inclusion bodies upon expression in Escherichia coli (personal observation) provides further indirect evidence for the necessity of glycosylation and would be consistent with a requirement for glycosylation during protein folding, for protein stability or solubility. The inability to obtain soluble protein in E. coli may also be due to the reducing conditions in the bacterial cytoplasm that do not support disulfide bridge formation. Two cysteine residues are conserved at the N- and C-termini of (+)- and (−)-forming DIRs (Figs. 3 and 4a). Their engagement in the formation of a disulfide bond was experimentally confirmed for AtDIR6 (Pickel et al. 2012). The disulfide bridge may contribute to protein stability by stabilizing the tertiary structure of DIRs.

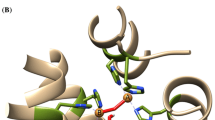
Structural model of AtDIR6. The model shows nine antiparallel β-strands (blue) and a short 310-helix (green) separated by loop regions (red, a), that form a calyx-like structure with a bottom-closed hydrophobic cavity (b: section of a with capped surface shown in beige). Surface renderings indicate positive (blue) and negative charges (red). The glycan attachment sites (N59 and N123, cyan), disulfide-forming cysteines (C40 and C186, yellow), and the functionally important F82 (orange) are highlighted (the figure was generated with UCSF Chimera 1.6.2)
Tertiary structure
As there is no crystal structure available, the tertiary structure of DIRs is unknown. However, structural modeling allowed a first insight into the three-dimensional topology of DIRs (Pickel et al. 2012). Even though there is little sequence conservation and no obvious homology with any other known protein family (Gang et al. 1999), structural and functional similarity was observed with allene oxide cyclases (Schaller and Stintzi 2009) and a structural homology search identified allene oxide cyclase 2 from A. thaliana (AtAOC2) as a likely homolog and good potential template for the modeling of AtDIR6 (Pickel et al. 2012). AOCs are involved in the stereospecific generation of cis-(+)-oxophytodienoic acid from 12,13-(S)-epoxy-octadecatrienoic acid (12,13-EOT) in the octadecanoid pathway for jasmonic acid biosynthesis (Ziegler et al. 2000; Hofmann and Pollmann 2008; Schaller and Stintzi 2009). The structural model of AtAOC2 resembles an eight-stranded antiparallel β-barrel with a central hydrophobic cavity for substrate binding (Hofmann et al. 2006). As compared to the structure of AtAOC2, the AtDIR6 model features an additional, ninth β-strand that extends the upper rim of the barrel and a short 310-helix (Pickel et al. 2012) (Fig. 4). Both features are supported by CD-spectroscopic data and secondary structure predictions (Pickel et al. 2012; Halls and Lewis 2002). The general validity of the model is confirmed by the two glycosylation sites that are located at the solvent-exposed outer surface of the barrel and by the position of the two cysteines engaged in the experimentally confirmed disulfide bridge, which is located at the bottom end of the barrel linking the N- and the C-terminus of the protein (Pickel et al. 2012) (Fig. 4a).
Hypothesized reaction mechanism of DIR-mediated coupling
Kinetic analysis of DIRs clearly support a two-step model of oxidative phenol coupling control, involving an oxidizing agent to generate the substrate radicals and DIRs to direct the coupling process in a regio- and stereospecific manner. As the presence of DIRs does not affect the overall conversion rate (Halls et al. 2004) and a high DIR to radical ratio is positively correlated to the amount and e.e. of pinoresinol that is generated (Pickel et al. 2010; Halls et al. 2004), DIR-mediated and free radical coupling have been recognized as competitive processes. While oxidation of coniferyl alcohol after binding to DIRs remains a possibility, this notion is consistent with the mode of action proposed by Halls et al. (2004), according to which the coniferyl alcohol radical is the substrate of DIRs. In the proposed model (Fig. 5), the radicals are bound by DIRs in a stoichiometry of about two per DIR homodimer, with the first radical binding reversibly followed by irreversible binding of the second, thereby initiating the formation of a C–C bond (Halls et al. 2004). Such a mode of action is also supported by the large difference in binding affinity for the alcohol and the radical. A K D of 370 μM was determined for the binding of coniferyl alcohol to FiDIR1. For the coniferyl alcohol radical, on the other hand, a K M of 10 nM was estimated from the saturation behavior of (+)-pinoresinol formation with respect to the apparent steady-state concentration of coniferyl alcohol radicals (Halls et al. 2004). The DIR homodimer provides a topology, which forces the unpaired electrons of the bound radicals to form a covalent bond in a regio- and enantiospecific manner. The resulting quinonmethide intermediate undergoes intramolecular cyclization reactions and is released. The coupling process and the release of pinoresinol were determined as the rate limiting steps (Halls et al. 2004).

Proposed reaction mechanism of DIR-mediated phenoxy radical coupling (Halls et al. 2004). Firstly, coniferyl alcohol (1, black circle) is oxidized to the corresponding radical (1•, white circle). A DIR homodimer (gray structure) successively binds two coniferyl alcohol radicals mediating regio- and enantiospecific coupling to either (+)- or (−)-pinoresinol (2, black double circle). Finally, the product is released
The proposed reaction mechanism according to which the DIR homodimer captures two coniferyl alcohol radicals and orients them in a way to favor stereospecific 8-8′ coupling is consistent with cross-linking experiments and calibrated size exclusion chromatography that confirm native FiDIR1 and AtDIR6 as homodimers (Davin et al. 1997; Halls and Lewis 2002; Pickel et al. 2012), whereas a homotetrameric structure was determined for AtDIR6 heterologously expressed in Pichia pastoris (Kazenwadel et al. 2012).
The hydrophobic binding cavity of DIRs was probed by site-directed mutagenesis. Substitution of F90, F113, and F163 to A or Y abolished the dirigent activity of ScDIR1, revealing functional importance for these conserved residues, possibly in the stabilization of coniferyl alcohol radicals by π–π interactions (Kim et al. 2012). This finding may explain the apparent lack of activity for AtDIR13 (Kim et al. 2012). In AtDIR13, the residue corresponding to F90 in ScDIR1 is replaced by methionine (M78; Fig. 2). The functional importance of F90 (or F82 in AtDIR6) is further supported by its conservation in AtAOC2 and pinoresinol-forming DIRs (Fig. 3) and by its location in the hydrophobic pocket of both classes of proteins (Pickel et al. 2012) (Fig. 4).
With the characterization of AtDIR6 (Pickel et al. 2010) and AtDIR5 (Kim et al. 2012) as being (−)-pinoresinol-forming and enantiocomplementary to previously characterized DIRs, protein regions and individual residues potentially involved in controlling the coupling mode could be identified by sequence comparisons (Pickel et al. 2010; Kim et al. 2012). Domain-swapping between ScDIR1 (N98 to P146) and AtDIR6 (K90 to L138) resulted in a conversion of enantiospecificity (Kim et al. 2012). Further studies and detailed site-directed mutagenesis will be required to resolve which of the residues that are differentially conserved in this region are responsible for the control of stereospecificity.
Evolution of DIRs
Structural homology search and subsequent modeling of AtDIR6 provided insight into the evolutionary ancestry of DIRs and revealed a distant relationship with lipocalins in the calycin superfamily (Pickel et al. 2012). Characteristic for lipocalins are their β-barrel structure with a central hydrophobic cavity for binding of small hydrophobic molecules and their function in transport processes (Flower et al. 2000). The suggested evolution of DIRs from hydrophobic ligand-binding proteins is consistent with their proposed reaction mechanism: If the bound ligand is unstable, it is only a small step for the respective binding protein to become a dirigent protein, since the chiral protein environment will impart its stereochemical influence on the reaction. Similar to the mode of action proposed for DIRs, product formation in AOCs is also directed by steric restrictions imposed by the protein environment on the enzyme-bound substrate, an unstable allylic epoxide (12,13-EOT; Hofmann et al. 2006). As members of the calycin superfamily, ancestral AOCs primarily may have been binding proteins for 12,13-EOT that were optimized as catalysts in the course of evolution (Pickel et al. 2012). Similarly, chalcone isomerase evolved as a highly efficient and stereospecific enzyme from non-enzymatic fatty acid-binding proteins (Ngaki et al. 2012). Considering the reactivity of phenoxy radicals, optimization as catalysts to lower the activation energy barrier may not be necessary in case of DIRs. We would therefore expect that proteins with dirigent activity (as opposed to proteins with enzymatic activity) are frequent among lipocalins and that the dirigent concept will be more generally applicable to binding proteins for unstable ligands (Pickel et al. 2012).
Biotechnological applications of DIRs
The functional characterization of FiDIR1 revealed a novel way of how to direct the coupling of coniferyl alcohol radicals qualitatively and quantitatively toward (+)-pinoresinol under appropriate reaction conditions (Davin et al. 1997). Although a full control of the coupling outcome has not been achieved for other DIRs under the conditions employed (Pickel et al. 2010; Kazenwadel et al. 2012; Kim et al. 2002b, 2012), further optimization of oxidative capacity-to-DIR ratio and the chemical environment may eventually allow for complete repression of side product formation. Another approach to reduce the competing undirected coupling of free radicals in favor of the directed coupling of DIR-bound radicals is to decrease the spatial distance between oxidant and DIR by cross-linking or employment of nanostructures (Ren et al. 2011; Roessl et al. 2010).
For a more general application of DIRs as tools for stereospecific radical coupling in organic synthesis, their narrow substrate and product specificity is the most serious limitation. Although various DIRs for the formation of (+)- and (−)-pinoresinol have been described, there is still no firm evidence for the involvement of DIRs in other radical coupling processes. Nevertheless, the large number of DIRs and DIR-like proteins with unknown function in plants (Ralph et al. 2007), as well as the high sequence diversity within the DIR family and the prevalence of enantiopure natural products in the plant kingdom, provides the potential for the discovery of novel DIR-mediated coupling reactions. Alternatively, the substrate and product spectra of DIRs and their scope for organic synthesis may be increased by engineering of artificial DIRs. As demonstrated by the successful reversal of the coupling mode of pinoresinol-forming DIRs (Kim et al. 2012), the engineering of dirigent activities is possible. It will be thrilling to identify the minimal DIR structure and to generate minimal DIRs tailored to direct specific coupling outcomes in the future.
A second bottleneck for the utilization of DIRs in organic synthesis is their limited availability. As purification from native sources is generally not feasible in economical terms and expression in E. coli does not yield functional DIRs due to the lack of appropriate glycosylation and secretion systems, eukaryotic expression systems are the method of choice. Heterologous expression of AtDIR6 in P. pastoris, which is routinely used as protein expression host, has recently been achieved at a level of 47 mg/L (Kazenwadel et al. 2012). The purified protein showed 90 % activity compared to the one obtained from plant cells as expression host (Pickel et al. 2010).
The application of phenoxy radical coupling in organic synthesis is often prohibited in economic and ecological terms because of limited specificity and the requirement of high concentrations of sometimes toxic oxidants. The combined use of DIRs with an appropriate oxidant offers an attractive answer to these problems. The development of appropriate DIRs may provide a biotechnological solution for the regio- and stereospecific control of phenoxy radical coupling reactions, thereby providing synthetic access to a plethora of pharmaceutically interesting compounds.
References
Ascacio-Valdés JA, Buenrostro-Figueroa JJ, Aguilera-Carbo A, Prado-Barragán A, Rodríguez-Herrera R, Aguilar CN (2011) Ellagitannins: biosynthesis, biodegradation and biological properties. J Med Plant Res 5:4696–4703
Band V, Hoffer AP, Bands H, Rhinehardt AE, Knapp RC, Matlin SA, Anderson DJ (1989) Antiproliferative effect of gossypol and its optical isomers on human reproductive cancer cell lines. Gynecol Onc 32:273–277
Bao W, O’Malley D, Whetten R, Sederoff R (1993) A laccase associated with lignifications in loblolly pine xylem. Science 260:672–674
Bedows E, Hatfield G (1982) An investigation of the antiviral activity of Podophyllum peltatum. J Nat Prod 45:725–729
Benedict CR, Liu J, Stipanovic RD (2006) The peroxidative coupling of hemigossypol to (+)- and (+)-gossypol in cotton seed extracts. Phytochemistry 67:356–361
Berlin J, Wray V, Mollenschott C, Sasse F (1986) Formation of β-peltatin-A methyl ether and coniferin by root cultures of Linum flavum. J Nat Prod 49:435–439
Bornscheuer UT, Huismann GW, Kazlauskas RJ, Lutz S, Moore JC, Robins K (2012) Engineering the third wave of biocatalysis. Nature 485:185–194
Bors W, Michel C (2002) Chemistry of the antioxidant effect of polyphenols. Ann N Y Acad Sci 957:57–69
Broomhead AJ, Dewick PM (1990) Aryltetralin lignans from Linum flavum and Linum capitatum. Phytochemistry 29:3839–3844
Burlat V, Kwon M, Davin LB, Lewis NG (2001) Dirigent proteins and dirigent sites in lignifying tissues. Phytochemistry 57:883–897
Bykova N, Rampitsch C, Krokhin O, Standing K, Ens W (2006) Determination and characterization of site-specific N-glycosylation using MALDI-Qq-TOF tandem mass spectrometry: case study with a plant protease. Anal Chem 78:1093–1103
Cass QB, Tiritan E, Matlin SA, Freire EC (1991) Gossypol enantiomer ratios in cotton seeds. Phytochemistry 30:2655–2657
Cedzich A, Huttenlocher F, Kuhn BM, Pfannstiel J, Gabler L, Stintzi A, Schaller A (2009) The protease-associated domain and C-terminal extension are required for zymogen processing, sorting within the secretory pathway, and activity of tomato subtilase 3 (SlSBT3). J Biol Chem 284:14068–14078
Chioccara F, Poli S, Rindone B, Pilati T, Brunow G, Pietikäinen P, Stälä H (1993) Regio- and diastereoselective synthesis of dimeric lignans using oxidative coupling. Acta Chem Scan 47:610–616
Davin LB, Lewis NG (2000) Dirigent proteins and dirigent sites explain the mystery of specificity of radical precursor coupling in lignan and lignin biosynthesis. Plant Phys 123:453–461
Davin LB, Wang H, Crowell AL, Bedgar DL, Martin DM, Sarkanen S, Lewis NG (1997) Stereoselective bimolecular phenoxy radical coupling by an auxiliary (dirigent) protein without an active center. Science 275:362–367
Dewick PM (2009) Medicinal natural products: a biosynthetic approach, 3rd edn. Wiley, Chichester
Dickey EE (1958) Liriodendrin, a new lignan diglucoside from the inner bark of yellow poplar (Liriodendron tulipifera L.). J Org Chem 23:179–184
Dinkova-Kostova AT, Gang DR, Davin LB, Bedgar DL, Chu A, Lewis NG (1996) (+)-Pinoresinol/(+)-lariciresinol reductase from Forsythia intermedia. J Biol Chem 271:29473–29482
Fang W, Ji S, Jiang N, Wang W, Zhao GY, Zhang S, Ge HM, Xu Q, Zhang AH, Zhang YL, Song YC, Zhang J, Tan RX (2012) Naphthol radical couplings determine structural features and enantiomeric excess of dalesconols in Daldinia eschscholzii. Nat Comm. doi:10.1038/ncomms2031
Finefield J, Sherman DH, Kreitman M, Williams RM (2012) Enantiomeric natural products: occurrence and biogenesis. Angew Chem Int Ed 51:4802–4836
Flower D, North A, Sansom C (2000) The lipocalin protein family: structural and sequence overview. Biochim Biophys Acta 1482:9–24
Frías I, Siverio JM, González C, Trujillo JM, Pérez J (1991) Purification of a new peroxidase catalyzing the formation of lignan-type compounds. Biochem J 273:109–113
Fujimoto H, Higuchi T (1977) Biosynthesis of liriodendrin by Liriodendron tulipifera. Wood Res 62:1–10
Fujita M, Gang DR, Davin LB, Lewis NG (1999) Recombinant pinoresinol–lariciresinol reductases from western red cedar (Thuja plicata) catalyze opposite enantiospecific conversions. J Biol Chem 274:618–627
Fuss E (2003) Lignans in plant cell and organ cultures: an overview. Phytochem Rev 2:307–320
Gang DR, Costa MA, Fujita M, Dinkova-Kostova AT, Wang H, Burlat V, Martin W, Sarkanen S, Davin LB, Lewis NG (1999) Regiochemical control of monolignol radical coupling: a new paradigm for lignin and lignan biosynthesis. Chem Biol 6:143–151
Gardener JAF, Swan EP, Sutherland SA, MacLean H (1966) Polyoxyphenols of western red cedar (Thuja plicata Donn) III. Structure of plicatic acid. Can J Chem 44:52–58
Gavel Y, Heijne G (1990) Sequence differences between glycosylated and non-glycosylated Asn-X-Thr/Ser acceptor sites: implications for protein engineering. Prot Eng 3:433–442
Giri R, Shi BF, Engle KM, Maugel N, Yu JQ (2009) Transition metal catalyzed C–H activation reactions: diastereoselectivity and enantioselectivity. Chem Soc Rev 38:3242–3272
Girol CG, Fisch KM, Heinekamp T, Günther S, Hüttel W, Piel J, Brakhage AA, Müller M (2012) Regio- and stereoselective oxidative phenol coupling in Aspergillus niger. Angew Chem Int Ed 51:9788–9791
Gordaliza M, Garcia P, Corral JM, Castro M, Gomez-Zurita M (2004) Podophyllotoxin: distribution, sources, applications and new cytotoxic derivatives. Toxicon 44:441–459
Gravel E, Poupon E (2008) Biogenesis and biomimetic chemistry: can complex natural products be assembled spontaneously? Eur J Org Chem 1:27–42
Halls S, Lewis NG (2002) Secondary and quaternary structures of the (+)-pinoresinol forming dirigent protein. Biochemistry 41:9455–9461
Halls S, Davin LB, Kramer D, Lewis NG (2004) Kinetic study of coniferyl alcohol radical binding to the (+)-pinoresinol forming dirigent protein. Biochemistry 43:2587–2595
Hanson SR, Culyba EK, Hsu T-L, Wong C-H, Kelly JW, Powers ET (2009) The core trisaccharide of an N-linked glycoprotein intrinsically accelerates folding and enhances stability. Proc Natl Acad Sci U S A 106:3131–3136
He S, Wu B, Pan Y, Jiang L (2008) Stilbene oligomers from Parthenocissus laetevirens: isolation, biomimetic synthesis, absolute configuration, and implication of antioxidative defense system in the plant. J Org Chem 73:5233–5241
Hofmann E, Pollmann S (2008) Molecular mechanism of enzymatic allene oxide cyclization in plants. Plant Phys Biochem 46:302–308
Hofmann E, Zerbe P, Schaller F (2006) The crystal structure of Arabidopsis thaliana allene oxide cyclase: insights into the oxylipin cyclization reaction. Plant Cell 18:3201–3217
Iqbai J, Bhatia B, Nayyar N (1994) Transition metal-promoted free-radical reactions in organic synthesis: the formation of carbon–carbon bonds. Chem Rev 94:519–564
Ito T, Abe N, Oyama M, Iinuma M (2009) Absolute structures of C-glucosides of resveratrol oligomers from Shorea uliginosa. Tetrahed Lett 50:2516–2520
Iwashina T (2000) The structure and distribution of the flavonoids in plants. J Plant Res 113:287–299
Jackson DE, Dewick PM (1983) Aryltetralin lignans from Podophyllum hexandrum and Podophyllum peltatum. Phytochemistry 23:1147–1152
Jasperse CP, Curran DP, Fevig TL (1991) Radical reactions in natural product synthesis. Chem Rev 91:1237–1286
Jiang J, Slivova V, Jedinak A, Sliva D (2012) Gossypol inhibits growth, invasiveness, and angiogenesis in human prostate cancer cells by modulating NFκb/ap-1 dependent- and independent-signaling. Clin Exp Metastasis 29:165–178
Katayama T, Ogaki A (2001) Biosynthesis of (+)-syringaresinol in Liriodendron tulipifera I: feeding experiments with L-[U-14C]phenylalanine and [8-14C]sinapyl alcohol. J Wood Sci 47:41–47
Kazenwadel C, Klebensberger J, Richter S, Pfannstiel J, Gerken U, Pickel B, Schaller A, Hauer B (2012) Optimized expression of the dirigent protein AtDIR6 in Pichia pastoris and impact of glycosylation on protein structure and function. Appl Microbiol Biotechnol. doi:10.1007/s00253-012-4579-x
Keseru GM, Nogradi M (1998) Natural products by oxidative phenolic coupling: phytochemistry, biosynthesis and synthesis. In: Atta-ur-Rahman (ed) Studies in natural products chemistry, vol 20. Elsevier Science, New York, pp 322–263
Khanbabaee K, van Ree T (2001) Tannins: classification and definition. Nat Prod Rep 18:641–649
Kim MK, Jeon JH, Davin LB, Lewis NG (2002a) Monolignol radical–radical coupling networks in western red cedar and Arabidopsis and their evolutionary implications. Phytochemistry 61:311–322
Kim MK, Jeon JH, Fujita M, Davin LB, Lewis NG (2002b) The western red cedar (Thuja plicata) 8-8′ DIRIGENT family displays diverse expression pattern and conserved monolignol coupling specificity. Plant Mol Biol 49:199–214
Kim KW, Moinuddin SGA, Atwell KM, Costa MA, Davin LB, Lewis NG (2012) Opposite stereoselectivities of dirigent proteins in Arabidopsis and Schizandra species. J Biol Chem 287:33957–33972
Kittur FS, Lalgondar M, Yu HY, Bevan DR, Esen A (2007) Maize β-glucosidase-aggregating factor is a polyspecific jacalin-related chimeric lectin, and its lectin domain is responsible for β-glucosidase aggregation. J Biol Chem 282:7299–7311
Koeller KM, Wong CH (2001) Enzymes for chemical synthesis. Nature 409:232–240
Kuhl N, Hopkinson MN, Wencel-Delord J, Glorius F (2012) Beyond directing groups: transition-metal-catalyzed C–H activation of simple arenes. Angew Chem Int Ed 51:10236–10254
Kurihari H, Kawabata J, Ichikawa S, Mizutani J (1990) (−)-ε-Viniferin and related oligostilbenes from Carex pumila Thumb. (Cyperaceae. Agric Biol Chem 54:1097–1099
Langcake P, Pryce RJ (1977) Oxidative dimerisation of 4-hydroxystilbenes in vitro: production of a grapevine phytoalexin mimic. J C S Chem Comm 208–210
Leffingwell JC (2003) Chirality and bioactivity I.: pharmacology. Leffingwell Rep 3:1–27
Lerouge P, Cabanes-Macheteau M, Rayon C, Fischette-Laine AC, Gomord V, Faye L (1998) N-Glycoprotein biosynthesis in plants: recent developments and future trends. Plant Mol Biol 38:31–48
Lessene G, Feldman KS (2002) Oxidative aryl-coupling in synthesis. In: Astruc D (ed) Modern arene chemistry. Wiley-VCH, Weinheim, pp 479–538
Lewis NG, Davin LB (1999) Lignans: biosynthesis and function. In: Barton DHR, Nakanishi K, Meth-Cohn O (eds) Comprehensive natural products chemistry 1. Elsevier, Oxford, pp 639–712
Lindsley CW, Hopkins CR, Sulikowski GA (2011) Biomimetic synthesis of lignans. In: Poupon E, Nay B (eds) Biomimetic organic synthesis. Wiley-VCH, Weinheim, pp 677–693
Liu J, Stipanovic RD, Bell AA, Puckhaber LS, Magill CW (2008) Stereoselective coupling of hemigossypol to form (+)-gossypol in moco cotton is mediated by a dirigent protein. Phytochemistry 69:3038–3042
Lu Y, Chen DF (2009) Analysis of Schisandra chinensis and Schisandra sphenanthera. J Chromatogr A 1216:1980–1990
MacRae W, Towers G (1984) Biological activities of lignans. Phytochemistry 23:1207–1220
Marshall R (1972) Glycoproteins. Annu Rev Biochem 41:673–702
Matlin SA, Zhou R, Bialy G, Blye RB, Naqvi RH, Lindberg MC, Matlin SA (1985) (−)-Gossypol: an active male antifertility agent. Contraception 31:141–149
Moinuddin SGA, Youn B, Bedgar DL, Costa MA, Helms GL, Kang CH, Davin LB, Lewis NG (2006) Secoisolariciresinol dehydrogenase: mode of catalysis and stereospecificity of hydride transfer in Podophyllum peltatum. Org Biomol Chem 4:808–816
Mori K (2011) Bioactive natural products and chirality. Chirality 23:449–462
Moss GP (2000) Nomenclature of lignans and neolignans. Pure Appl Chem 72:1493–1523
Nakatsubo T, Mizutani M, Suzuki S, Hattori T, Umezawa T (2008) Characterization of Arabidopsis thaliana pinoresinol reductase, a new type of enzyme involved in lignan biosynthesis. J Biol Chem 283:15550–15557
Ngaki MN, Louie GV, Philippe RN, Manning G, Pojer F, Bowmann ME, Li L, Larsen E, Wurtele ES, Noel JP (2012) Evolution of the chalcone-isomerase fold from fatty-acid binding to stereospecific catalysis. Nature 485:530–536
Nicolaou KC, Vourloumis D, Winssinger N, Baran PS (2000) The art and science of total synthesis at the dawn of the twenty-first century. Angew Chem Int Ed 39:45–122
Nicolaou KC, Montagnon T, Snyder SA (2003) Tandem reactions, cascade sequences, and biomimetic strategies in total synthesis. Chem Comm 7:551–564
Nicotra S, Cramarossa MR, Mucci A, Pagnoni UM, Riva S, Forti L (2004) Biotransformation of resveratrol: synthesis of trans-dehydrodimers catalyzed by laccases from Myceliophtora thermophyla and from Trametes pubescens. Tetrahedron 60:595–600
Okunishi T, Umezawa T, Shimada M (2000) Enantiomeric compositions and biosynthesis of Wikstroemia sikokiana lignans. J Wood Sci 46:234–242
Okunishi T, Umezawa T, Shimada M (2001) Isolation and enzymatic formation of lignans of Daphne genkwa and Daphne odora. J Wood Sci 47:383–388
Petcher TJ, Weber HP, Kuhn M, Von Wartburg A (1973) Crystal structure and absolute configuration of 2′-bromopodophyllotoxin–0.5 ethyl acetate. J Chem Soc Perkin Trans 2:288–292
Pezet R, Pont V, Hoang-Van K (1991) Evidence for oxidative detoxication of pterostilbene and resveratrol by a laccase-like stilbene oxidase produced by Botrytis cinerea. Physiol Mol Plant Path 39:441–450
Pickel B, Constantin MA, Pfannstiel J, Conrad J, Beifuss U, Schaller A (2010) An enantiocomplementary dirigent protein for the enantioselective laccase-catalyzed oxidative coupling of phenols. Angew Chem Int Ed 49:202–204
Pickel B, Pfannstiel J, Steudle A, Lehmann A, Gerken U, Pleiss J, Schaller A (2012) A model of dirigent proteins derived from structural and functional similarities with allene oxide cyclase and lipocalins. FEBS J 279:1980–1993
Ponzoni C, Beneventi E, Cramarossa MR, Raimondi S, Trevisi G, Pagnoni UM, Riva S, Forti L (2007) Laccase-catalyzed dimerization of hydroxystilbenes. Adv Synth Catal 349:1497–1506
Qiu SX, Lu ZZ, Luyengi L, Lee SK, Pezzuto JM, Farnsworth NR, Thompson LU, Fong HHS (1999) Isolation and characterization of flaxseed (Linum usitatissimum) constituents. Pharm Biol 37:1–7
Ralph S, Park JY, Bohlmann J, Mansfield SD (2006) Dirigent proteins in conifer defense: gene discovery, phylogeny, and differential wound- and insect-induced expression of a family of DIR and DIR-like genes in spruce (Picea spp.). Plant Mol Biol 60:21–40
Ralph SG, Jancsik S, Bohlmann J (2007) Dirigent proteins in conifer defense II: extended gene discovery, phylogeny, and constitutive and stress-induced gene expression in spruce (Picea spp.). Phytochemistry 68:1975–1991
Ren Y, Rivera JG, He L, Kulkami H, Lee DK, Messersmith PB (2011) Facile, high efficiency immobilization of lipase enzyme on magnetic iron oxide nanoparticles via a biomimetic coating. BMC Biotechnol 11:1–8. doi:10.1186/1472-6750-11-63
Rivière C, Pawlus AD, Mérillon JM (2012) Natural stilbenoids: distribution in the plant kingdom and chemotaxonomic interest in Vitaceae. Nat Prod Rep 29:1317–1333
Roessl U, Nahálka NB, Nidetzky B (2010) Carrier-free immobilized enzymes for biocatalysis. Biotechnol Lett 32:341–350
Saleem M, Kim H, Ali M, Lee Y (2005) An update on bioactive plant lignans. Nat Prod Rep 22:696–716
Schaller A, Stintzi A (2009) Enzymes in jasmonate biosynthesis—structure, function, regulation. Phytochemistry 70:1532–1538
Seidel V, Windhövel J, Eaton G, Alfermann AW, Arroo RRJ, Medarde M, Petersen M, Woolley JG (2002) Biosynthesis of podophyllotoxin in Linum album cell cultures. Planta 215:1031–1039
Shental-Bechor D, Levy Y (2008) Effect of glycosylation on protein folding: a close look at thermodynamic stabilization. Proc Natl Acad Sci U S A 105:8256–8261
Shental-Bechor D, Levy Y (2009) Folding of glycoproteins: toward understanding the biophysics of the glycosylation code. Curr Opin Struc Biol 19:524–533
Sterjiades R, Dean J, Eriksson K (1992) Laccase from sycamore maple (Acer pseudoplatanus) polymerizes monolignols. Plant Physiol 99:1162–1168
Stipanovic RD, Puckhaber LS, Bell AA, Percival AE, Jacobs J (2005) Occurrence of (+)- and (−)-gossypol in wild species of cotton and in Gossypium hirsutum var. marie-galante (Watt) Hutchinson. J Agri Food Chem 53:6266–6271
Stipanovic RD, Puckhaber LS, Liu J, Bell AA (2009) Total and percent atropisomers of gossypol and gossypol-6-methylether in seeds from pima cottons and accessions of Gossypium barbadense L. J Agri Food Chem 57:566–571
Suzuki S, Umezawa T (2007) Biosynthesis of lignans and norlignans. J Wood Sci 53:273–284
Suzuki S, Umezawa T, Shimada M (2002) Stereochemical diversity in lignan biosynthesis of Arctium lappa L. Biosci Biotechnol Biochem 66:1262–1269
Swan RJ, Klyne W, MacLean H (1967) Optical rotatory dispersion studies. XLI. The absolute configuration of plicatic acid. Can J Chem 45:319–324
Takaya Y, Terashima K, Ito J, He YH, Tateoka M, Yamaguchi N, Niwa M (2005) Biomimic transformation of resveratrol. Tetrahedron 61:10285–10290
Umezawa T (2003) Diversity in lignan biosynthesis. Phytochem Rev 2:371–390
Veech JA, Stipanovic RD, Bell AA (1976) Peroxidative conversion of hemigossypol to gossypol. A revised structure for isohemigossypol. J Chem Soc Chem Commun 144–145
von Heimendahl CBI, Schäfer KM, Eklund P, Sjöholm R, Schmidt TJ, Fuss E (2005) Pinoresinol–lariciresinol reductases with different stereospecificity from Linum album and Linum usitatissimum. Phytochemistry 66:1254–1263
Wang X, Beckham TH, Morris JC, Chen F, Gangemi DJ (2008) Bioactivities of gossypol, 6-methoxygossypol, and 6,6′-dimethoxygossypol. J Agri Food Chem 56:4393–4398
Whiting DA (1991) Selectivity, strategy and efficiency in modern organic chemistry. In: Trost BM, Fleming I (eds) Comprehensive organic synthesis. Pergamon, Oxford, pp 659–703
Xia ZQ, Costa MA, Proctor J, Davin LB, Lewis NG (2000) Dirigent-mediated podophyllotoxin biosynthesis in Linum flavum and Podophyllum peltatum. Phytochemistry 55:537–549
Xia ZQ, Costa MA, Pélissier HC, Davin LB, Lewis NG (2001) Secoisolariciresinol dehydrogenase: purification, cloning, and functional expression. J Biol Chem 276:12614–12623
Yasuda S, Hirano J, Tange J, Nadadomi W, Tachi M (1989) Manufacture of wood cement boards III: cement-hardening inhibitory components of western red cedar heartwood. J Wood Chem Technol 9:123–133
Zhan ZJ, Ying YM, Ma LF, Shan WG (2011) Natural disesquiterpenoids. Nat Prod Rep 28:594–629
Zhu L, Zhang X, Tu L, Zeng F, Nie Y, Guo X (2007) Isolation and characterization of two novel dirigent-like genes highly induced in cotton (Gossypium barbadense and G. hirsutum) after infection by Verticillium dahliae. J Plant Path 89:41–45
Ziegler J, Stenzel I, Hause B, Maucher H, Hamberg M, Grimm R, Ganal M, Wasternack C (2000) Molecular cloning of allene oxide cyclase. The enzyme establishing the stereochemistry of octadecanoids and jasmonates. J Biol Chem 275:19132–19138
Acknowledgments
We gratefully acknowledge grant support of our work by the German Research Foundation (DFG, SFB 706).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Pickel, B., Schaller, A. Dirigent proteins: molecular characteristics and potential biotechnological applications. Appl Microbiol Biotechnol 97, 8427–8438 (2013). https://doi.org/10.1007/s00253-013-5167-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-013-5167-4