
Abstract
Validation of suitable reference genes is critical in quantitative real-time polymerase chain reaction (qRT-PCR) analysis. Suitable and reliable reference genes for the normalization of gene expression data are characterized by high gene expression stability across tissues and different experimental conditions. This study evaluated the gene expression stability of ten reference genes commonly used in Arabidopsis thaliana for their suitability in qRT-PCR analysis in Tripterygium wilfordii Hook.f. The orthologous sequences of these ten candidate genes were identified from T. wilfordii transcriptomic data (Project No. SRX472292). Five algorithms including GeNorm, NormFinder, BestKeeper, ΔCt, and RefFinder were used to assess the gene expression stability of these putative reference genes in different plant tissues and different stress conditions. The results identified ACTINT7 and TBP as the most suitable reference genes across all samples. The gene expressions of TwHMGR (3-hydroxy-3-methylglutaryl coenzyme A reductase, KU246037.1) and of TwDXR (1-deoxy-D-xylulose-5-phosphate reductoisomerase, KJ174341.1) were investigated to validate the suitability of the reference genes. The validation analysis confirmed the suitability of ACTINT7 and TBP as the best reference genes for elucidating secondary metabolite biosynthesis pathway in T. wilfordii. In summary, this study identified the most suitable and reliable reference genes for future qRT-PCR- based studies in T. wilfordii.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Tripterygium wilfordii Hook.f. is a herb used in Traditional Chinese Medicine which has a diverse range of bioactivities and thus, has been used in the treatment of an array of ailments including inflammation, systemic lupus erythematosus and rheumatoid arthritis for hundreds of years [1,2,3]. About 415 secondary metabolites were isolated from T. wilfordii, mainly terpenoids and alkaloids [4]. Triptolide, a main diterpenoid epoxide, has been reported to have immunosuppressive and anticancer activities [5,6,7]. Sesquiterpene pyridine alkaloids (such as wilforgine, wilforine, wilfortrine and wilfordine) have immunosuppressive, antitumor-promoting and cytotoxic, antiviral and anti-inflammatory properties [8,9,10,11,12]. Traditionally, the secondary metabolites such as terpenoids and alkaloids are obtained by extraction from the root of T. wilfordii. However, T. wilfordii is not widely cultivated and the content of secondary metabolites in natural plants is far below demand. Therefore, it is necessary to understand the secondary metabolites biosynthetic pathway for producing the compound through synthetic biology strategies.
The selection of target genes for the genetic manipulation of plants towards the production of bioactive metabolites often relies on quantitative transcription reverse polymerase chain reaction (qRT-PCR) analysis [13]. The qRT-PCR data of target genes is normalized against that of reference genes that, in turn, must be characterized by high levels of stability of gene expression, across different developmental stage and different experimental settings. This data normalization procedure mitigates many sources of experimental errors associated with the qRT-PCR method [14, 15]. Ideal reference genes should be stable and reliable throughout the diverse experimental conditions. However, no single reference genes have been shown to be universally constant during development and diverse experimental conditions. Furthermore, many studies have shown that traditionally used popular housekeeping genes used to normalize gene expression vary considerably under different conditions [16,17,18,19,20,21].
To elucidate biosynthesis pathway of bioactive metabolites, accurate quantification of key genes expression is needed which will help us to understand gene function in secondary metabolite biosynthesis. qRT-PCR has become the preferred method due to its high sensitivity, specificity, and accuracy in detecting the target gene expression. In previous studies, many reference genes were used to study the secondary metabolites biosynthesis for T. wilfordii, including elongation factor 1-α (EF-1α) [22,23,24,25], ACTIN7 [26], β-Actin [27,28,29,30,31], polypyrimidine tract-binding protein (PTB) [32].
Until now, no systematic validation of reference genes has been performed in T. wilfordii. Therefore, we evaluated the expression stability of ten genes (PP2A, TBP, TUB, 26S, SAND, GAPDH, TIP41, eIF-4α, EF-1α, and ACTINT7) among the tissues of T. wilfordii and under a range of abiotic stresses, such as drought, salinity, and methyl jasmonate (MeJA) and salicylic acid (SA) in T. wilfordii hairy roots by qRT-PCR. To validate the reliability of the reference genes, the expression levels of the TwHMGR and TwDXR, which encode for enzymes that participate in the isoprenoid biosynthesis pathways in T. wilfordii, were used to assess the performance of the newly selected reference genes to confirm their suitability for qRT-PCR analysis in T. wilfordii.
Materials and methods
Plant materials and experimental treatments
Tripterygium wilfordii plants were grown in the greenhouse of Northwest A&F University which transferred from a natural growing in Fujian Province of China. 0.8–1 g of roots, stems and leaves were collected randomly each from six cottage plantlets with the same growth and development status, each tissue part was repeated three times. The abiotic stress treatments were performed on hairy roots (HRs) of T. wilfordii. The culture of HRs was carried out as described previously [33]. Stock solutions of MeJA and SA were prepared and added to the final media (0.1 mM) [34]. The samples were collected from plants exposed to different stress treatments after 0, 3, 6, 9, 12, and 24 h for follow-up analysis. Salinity stress was induced by the addition of NaCl to the media to the final concentration of 150 mM. Drought stress was induced by the addition of PEG 8000 (20% w/v) to the media [35]. The different plant tissues were collected, immediately frozen in liquid nitrogen, and kept at − 80 °C for further analysis. All the treatments were gathered in three biological replications.
RNA extraction for cDNA synthesis
RNA isolation was performed according to Miao et al. with minor modifications [36]. The extraction buffer contained the following: 2 M NaCl, 200 mM Tris (pH 8.0), 25 mM EDTA, 2% CTAB (w/v), 2% PVPP (w/v) as well as 5% β-mercaptoethanol (v/v). RNA concentration and purity were measured using an M200 Pro-multimode reader (Tecan, Switzerland). RNA integrity was further assessed by electrophoresis using 1% agarose gels. For each sample, about 300 ng of total RNA was treated with gDNA Eraser (Takara, Japan) and used to make cDNA with PrimeScript™ RT (Takara, Japan).
qRT-PCR conditions
The qRT-PCR analysis was performed in an iCycler iQ™ Real-Time PCR Detection System (BioRad, USA). Each reaction contained nuclease-free water (9.5 μL, Takara, Japan), SYBR Premix Ex Taq™ II (12.5 μL), forward primer (1 μL, 10 μM) and reverse primer (1 μL, 10 μM), and cDNA (1 μL, unquantified). The cycling condition was: 95 °C for 3 min, followed by 40 cycles of 95 °C for 10 s, 52 °C for 30 s and 72 °C for 30 s followed by 95 °Cfor 10 s. Controls for reverse transcription were included in the PCR reactions to reveal any DNA contamination. Each PCR analysis was performed with three biological replicates and three technical replicates and the mean values were used for the analysis. Amplification efficiency (E) and correlation coefficient (R2) were of primers were calculated by standard curve method, and all the samples were diluted five times. The E value is calculated by formula (% Efficiency = (10[− 1/slope] − 1) × 100%) [37].
Selection of candidate reference genes and primer design
Ten reference genes, frequently used in Arabidopsis studies, were investigated in this study: protein phosphatase 2A (PP2A), TATA-box binding protein (TBP), TUB beta-7 (TUB), ribosomal RNA 26S (26S), SAND family protein (SAND), Glyceraldehyde-3-phosphate Dehydrogenase (GAPDH), Tap42-interacting protein (TIP41), eukaryotic translation initiation factor 4a − 1 (eIF-4α), elongation factor − 1α (EF-1α) and Actin 7 (ACTINT7) study (Table 1) [38, 39]. The sequences of these Arabidopsis genes were blasted against the root transcriptome (the accession number SRX472292) of T. wilfordii. qRT-PCR primers were designed based on the identified orthologous sequences for the ten putative reference genes using Primer Premier 5 software (amplicon length: 97–148 bp, and melting temperature (Tm): 51–59 °C).
Analysis of gene expression stability
Ten selected reference genes were compared regarding the stability of their gene expression with four algorithms: GeNorm, NormFinder, BestKeeper, and the ΔCt. Finally, RefFinder (http://www.fulxie.0fees.us), a comprehensive evaluation platform which integrates the four algorithms mentioned above, was used to produce the final ranks regarding their overall stability.
GeNorm method
The GeNorm algorithm assumes that two suitable reference genes must have a constant gene expression ratio across samples, and therefore, is a measure of variation between a single candidate reference gene and each of the target gene analyzed [40]. Suitable qRT-PCR reference genes must have expression stability values of M < 1.5. This algorithm is one of the most commonly used to assess the performance of putative reference genes.
NormFinder method
The NormFinder method measures stability value (SV) of gene expression for reference genes by measuring the variance in gene expression within and between groups [41]. According to the GeNorm method, genes with the lowest SV are less variable in terms of gene expression and thus, better reference genes.
Delta-Ct (ΔCt) method
The Delta-Ct (ΔCt) method [42] is somewhat similar to the GeNorm method because it compares gene expression (ΔCt values) within a group to that of other groups. The reference gene that produces the lowest standard deviation (SD) across all samples is considered the most stable.
BestKeeper method
The BestKeeper method estimates Cq values for all candidate reference genes by measuring the coefficient of variation (CV) and the standard deviation (SD) for each reference gene. The most stable reference gene will exhibit the lowest Cq values [43].
RefFinder method
RefFinder analyzes the rankings provided by the GeNorm, NormFinder, BestKeeper and ΔCt algorithms and calculates a geometric mean for each gene. The best reference genes are the ones with the lowest geometric mean values [44].
Validation of reference gene analysis
TwHMGR (KU246037.1/GAVZ01053357.1) and TwDXR (KJ174341.1/GAVZ01038138.1) encode for enzymes that play a role in the biosynthesis of isoprenoids in T. wilfordii [26, 33, 45]. Variation in the transcript levels of these genes were used to validate the performance of the previously identified best reference genes. To evaluate the validity of the selection of reference genes, the expression levels of the TwHMGR and TwDXR genes were analyzed in different tissues and MeJA-induced abiotic stress treatments. For each experimental condition, the expression of TwHMGR and TwDXR were normalized using the two best reference genes (ACTIN7 and TBP), and a relatively unstable gene (TUB) recommended by RefFinder. RT-qPCR was performed as described above. The average Ct value was calculated from three biological and technical replicates and was used for relative expression analyses. The relative expression of the TwHMGR and TwDXR gene was subsequently calculated according the 2−∆∆CT method [46]. The expression levels of target genes were assessed using one-way ANOVA using SPSS v.17.0 with a signifcance level set at P < 0.05.
Results
Primer specificity and PCR efficiency
The nucleotide sequences of the ten candidate genes were obtained from the Arabidopsis genome database and blasted against a T. wilfordii transcriptome database (accession number SRX472292, http://www.ncbi.nlm.nih.gov/sra/SRX472292) in order to identify their corresponding homologs to be assessed as putative reference genes for T. wilfordii. (Table 1). PCR amplification produced a single band for each primer pair (each gene) as assessed by 2% agarose gel electrophoresis (Fig. 1a). The qRT-PCR melting-curve also showed a single peak corresponding to the expected size of the amplicon (Fig. 1b). These results strongly suggest that each primer pair was able to produce specific PCR amplified products of the target genes. Table 1 summarizes the primer sequences, amplicon length, amplicon temperature, correlation coefficient, and PCR amplification efficiency values. qRT-PCR amplification efficiency varied from 91.8% for EF-1α to 107.6% for 26S, whereas the correlation coefficients varied from 0.990 for TBP to 0.998 for SAND.

Amplification products of the 10 genes. a Lanes 1–10 are PP2A, TBP, TUB, 26S, SAND, GADPH, TIP41, eIF-4α, EF-1α and ACTIN7, respectively.. M: Marker DL2000. b Melt curves of the ten reference genes
Determination of quantification cycle (Cq) values
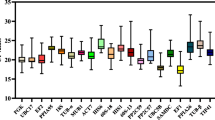
Quantification cycle values (Cq values) were obtained for the ten candidate reference genes, by performing qRT-PCR amplification reactions across the different T. wilfordii Hook.f. tissues and different abiotic stresses in root samples. The mean Cq values ranged from 16.25 to 32.98 across all samples for all the ten candidate reference genes. The 26S showed the lowest Cq (mean Cq of 18.08), suggesting that this gene had the highest expression level of all compared putative reference genes. EF-1α and ACTIN7 had slightly higher Cq values of 23.75 and 24.21 respectively, suggesting that these genes were also abundant transcripts. E1F4α, TUB, TBP, SAND, and PP2A had average Cq values between 24.25 and 28.35 and thus, were only moderately expressed. TIP41 and GADPH had the highest Cq (mean Cq of 29.71 and 30.92) suggesting that these genes transcripts were the lowest in abundance. The mean and median Cq values were similar for each gene, especially in the case of GAPDH and TBP which suggests that the Cq values were evenly distributed. The Cq values of EF-1α and TIP41 were distributed more centrally suggesting less variation across samples (Fig. 2).

Box and whisker plot displaying the Cp values for the candidate reference genes in all samples. The lower and upper edges of the box represent the 25th and the 75th percentiles, respectively; the line inside the box indicates the median value; the whiskers represent the maximum and minimum values
Determination of the optimal number of reference genes
The GeNorm algorithm was used to estimate the number of reference genes necessary for accurate data normalization using pairwise variation analysis by measuring the ratio (Vn/Vn + 1) between two sequential normalization factors (NFn and NFn + 1). Figure 3 shows pairwise variation (V) values for the reference genes in which the value is lower than 0.15 as to be optimized. The results show that a combination of two candidate reference genes efficiently normalizes the qRT-PCR data across T. wilfordii tissues and different abiotic treatments. According to the GeNorm algorithm, the best combinations were ACTINT7 + TBP across all samples, TIP41 + ACTINT7 across all tissues and across MeJA abiotic stress samples, 26S +ACTINT7 across SA abiotic stress samples, TBP + eIF-4α across drought stress samples and TBP + EF-1α across salinity stress samples.

Determination of the optimal number of reference genes for normalization by pairwise variation (V) using GeNorm
Gene expression stability analysis of candidate reference genes
The gene expression stability, in different tissue samples and in different stress treatments, was determined for all putative reference genes and analyzed using GeNorm, NormFinder, ΔCt, and BestKeeper. Finally, RefFinder summarized the results of the previously listed algorithms.
GeNorm analysis
Figure 4 summarizes the stability scores for the ten putative genes across all samples as estimated by GeNorm. ACTINT7 and TBP had the highest level of stability, whereas eIF-4α was the least stable gene across samples. Across tissues, TIP41 and ACTINT7 had the highest stability levels, while EF-1α showed the least stable expression across tissues. The most stable genes across samples submitted to the MeJA abiotic stress were TIP41 and ACTINT7, whereas TUB was the least stable. Under SA abiotic stress treatment, ACTINT7 and 26S showed the highest stability, whereas eIF-4α showed the least stable expression. TBP and eIF-4α had the highest stability in the drought stress group, whereas TBP and EF-1α showed low stability under salinity stress. TUB showed the lowest level of gene stability under both drought and salinity abiotic stresses.

Expression stability of ten candidate genes as calculated by GeNorm
NormFinder analysis
The NormFinder analysis differs from that of GeNorm as it uses ANOVA analysis to estimate intra- and inter-group variation in gene expression stability for each reference genes. According to this algorithm, ACTIN7 and TBP showed the highest level of stability in gene expression across all tissues and different abiotic stress treatments. TIP41 and ACTINT7 showed the highest stability across tissue samples. EF-1α and TIP41 were the most stably expressed genes in samples submitted to MeJA-induced abiotic stress whereas in samples submitted to SA-induced abiotic stress ACTIN7 and TBP had the highest stability. Finally, TBP and eIF-4α showed high levels of stability under drought stress, whereas TBP and EF-1α were the best reference genes under salinity stress (Table 2).
Delta-Ct (ΔCt) analysis
In the ΔCt analysis, the reference gene that produces the lowest gene pair standard deviation (SD) across all samples is most stable and therefore the most suitable reference gene. The ten candidate reference genes were ranked from the highest to the lowest levels of stability of gene expression according to the ΔCt algorithm (Table 3). Overall, ACTINT7 had the highest level of stability in gene expression across all tissues, and within the SA-induced abiotic stress group. In the MeJA-induced abiotic stress group as well as across different tissues TIP41 was the most stably expressed, whereas TBP was the best reference gene under the drought and salinity stresses.
BestKeeper analysis
BestKeeper estimates expression stability by measuring the standard deviation (SD) across samples. A gene with SD > 1 is considered unacceptable for a reference gene. The SD values of all genes as estimated by BestKeeper logarithm are shown in Table 4. Across all samples, TIP41 (0.51) had the lowest SD value, followed by SAND (0.55), while EF-1α and eIF-4α had SD values were higher than 1. Across tissue samples, TIP41 was considered the most suitable reference genes (0.37), followed by ACTINT7 (0.60) and TUB (0.67). Among the MeJA treated samples, all reference genes had SD values lower than 1.0, the lowest ones obtained for the TIP41, ACTINT7 and TBP (0.31, 0.33 and 0.4, respectively). Similarly, under the SA stress treatment, all reference genes had low SD values (SD < 0.52), with the TBP (0.21), PP2A (0.3) and GADPH (0.32) showing the highest levels of stability. ACTIN7 showed the lowest SD value of 0.09 in the drought treatment group. Under salinity stress, GADPH (0.53) and TIP41 (0.67) were the two most stable genes.
RefFinder analysis
The results from the four previously described algorithms were integrated by the RefFinder algorithm and the results (Table 5). The stability rank of the ten putative reference genes was listed from the highest to the lowest as assessed by RefFinder (Fig. 5). The expression of TBP was the most stable under the drought and salinity stress. The most stable gene under MeJA-induced abiotic stress was TIP41. The most stable gene under the SA-induced stress and across different tissue samples was ACTIN7. Overall, the level of stability of the expression pattern of ACTIN7 revealed that this gene is the most reliable regarding the normalization of qRT-PCR data in T. wilfordii, across all samples.

Expression stability of ten candidate reference genes as calculated by RefFinder
Expression analysis of TwHMGR and TwDXR genes for reference gene validation
TwHMGR and TwDXR encode for enzymes directly involved in the biosynthesis of isoprenoids in T. wilfordii [26, 47]. The relative expression of TwHMGR and TwDXR were used to validate the suitability of the reference genes across different tissues as well as across root samples in the MeJA-induced abiotic stress group (Fig. 6). We used the RefFinder stability rankings to select the most stable genes (ACTIN7 and TBP) and the least stable gene (TUB) for the normalization of qRT-PCR data obtained for TwHMGR and TwDXR.

Relative quantification of TwHMGR and TwDXR expression using validated reference genes including the most or the least stable reference genes for normalization under different tissues and MeJA treatment experimental sets. aTwHMGR expression of hairy roots under MeJA treatment after 0 h, 3 h, 6 h, 9 h, 12 h and 24 h; bTwHMGR expression of different tissues; cTwDXR expression of hairy roots under MeJA treatment after 0 h, 3 h, 6 h, 9 h, 12 h and 24 h; dTwDXR expression of different tissues. Bars represent the mean ± SE of three biological replicates
For MeJA treatment, TwHMGR expression was induced significantly at 3 h and kept to increase until 6 h and then decreased at 24 h, which was normalized by the two most stable reference genes (ACTIN7 and TBP). Furthermore, the expression of TwHMGR which was normalized by the two most stable reference gene (ACTIN7 and TBP) was markedly different from the expression level which was calculated using the least suitable reference gene (TUB) (P < 0.05) (Fig. 6a). Across different tissues, transcript level of TwHMGR was the lowest in leaf while it was the highest in root that was normalized by the two most stable reference gene (ACTIN7 and TBP). However, the expression level of TwHMGR which was normalized by the two most stable reference gene (ACTIN7 and TBP) was significantly different from the expression level which was calculated using the least suitable reference gene (TUB) among different tissues (P < 0.05) (Fig. 6b).
The expression level of TwDXR began to increase at 3 h, increased until 6 h and then dropped at 24 h that was normalized by the two most stable reference genes (ACTIN7 and TBP). However, the expression level of TwDXR normalized by the two most stable reference genes (ACTIN7 and TBP) was significantly different from the expression level calculated using the least suitable reference gene (TUB) (P < 0.05) (Fig. 6c). Across different tissues, significant difference of TwDXR transcript levels was observed. The expression level of TwDXR normalized by the two most stable reference genes (ACTIN7 and TBP) was significantly different from the expression level calculated using the least suitable reference gene (TUB) (P < 0.05) (Fig. 6d).
Discussion
The qRT-PCR analysis is a commonly used method to quantify gene differential expression levels in gene function studies [48]. However, the accuracy of this method depends on the availability of suitable reference genes, which is used to normalize the expression of the target genes. We made an orthologue search analysis using the sequences of ten reference genes of the Arabidopsis genome against the T. wilfordii root transcriptome database (accession number SRX472292, http://www.ncbi.nlm.nih.gov/sra/SRX472292) (Table 1) to identify putative reference genes in T. wilfordii.
The images of the agarose gels of each putative reference gene PCR products as well as the dissociation curves of the qRT-PCR amplification reactions showed that the primers designed for this analysis had good specificity. At the same time, the high value of the R2 of the standard curve equation as well as the high amplification efficiency values obtained during the qRT-PCR analyses confirmed the high degree of specificity of the primers designed to amplify T. wilfordii reference genes.
qRT-PCR data was obtained across different tissues of T. wilfordii as well as across samples exposed to different abiotic stress treatments. The stability of gene expression of ten putative reference genes was assessed with five statistical algorithms (GeNorm, NormFinder, BestKeeper, ΔCt, and RefFinder). In general, GeNorm, NormFinder, ΔCt, and RefFinder were able to produce consistent stability rank for the putative reference gene (Table 5). The BestKeeper algorithm is based on a different principle as compared to the other algorithms, which might explain why the BestKeeper results differed the most from those of the other algorithms [49]. Finally, we selected the most suitable reference gene(s) as determined by GeNorm, NormFinder, ΔCt, and the RefFinder algorithms.
For validation purposes, we chose TwHMGR and TwDXR as target genes, which encode for enzymes directly involved in the biosynthesis of isoprenoids in plants. The HMGR reductase catalyzes the irreversible conversion of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) into mevalonic acid. This catalysis is a key regulatory step in isoprenoids biosynthesis leading to the biosynthesis of IPP and DMAPP [50, 51]. The (DXR) reductoisomerase catalyzes the conversion of DXP; a key protein activated upstream of the MEP pathway. Moreover, the addition of MeJA and SA to the media mimics the endogenous signals that occur during the plant response to biotic and abiotic stresses [52]. Moreover, MeJA and SA have also been used as elicitors to enhance the accumulation of secondary metabolites in adventitious roots and hairy roots of T. wilfordii Hook. f. [33, 36, 53]. Hence, we chose MeJA to induce an abiotic stress response in the T. wilfordii hairy roots in order to evaluate the stability of gene expression. Ideally, the stability of gene expression of a reference gene is not affected by tissue types and experimental conditions. As shown in Fig. 6a, c, TwHMGR and TwDXR are gradually upregulated by MeJA over the time course of the experiment until the 6 h. This pattern of gene expression was also observed when data was normalized against ACTIN7 and TBP, which had been identified as the most stable reference genes in previous studies [54,55,56]. Figure 6b and 6d show that the expression trends of TwHMGR and TwDXR in different tissues were identical when ACTIN7 and TBP were used for qRT-PCR data normalization. By contrast, large discrepancies in expression patterns appeared when the least stable reference gene (TUB) was used for data normalization.
This study is the first, to our knowledge, to evaluate and validate reference genes for the normalization of qRT-PCR data from different plant tissues and different abiotic stress conditions in T. wilfordii. The optimization of the qRT-PCR analysis depends on the identification and validation of reference genes in different tissues, different development stages, and under different experimental conditions. Our study successfully identified the most suitable reference genes for future studies based on qRT-PCR analysis of T. wilfordii.
References
Tao X, Lipsky PE (2000) The Chinese Aanti-inflammatory and immunosuppressive herbal remedy Tripterygium wilfordii Hook.f. Reuh Dis Clin N Am 26:29–50. https://doi.org/10.1016/s0889-857X(05)0118-6
Setty AR, Sigal LH (2005) Herbal medications commonly used in the practice of rheumatology: mechanisms of action, efficacy, and side effects. Semin Arthr Rheum 34:773–784. https://doi.org/10.1016/j.semarthrit.2005.01.011
Brinker AM, Ma J, Lipsky PE, Raskin I (2007) Medicinal chemistry and pharmacology of genus Tripterygium (Celastraceae). Phytochemistry 68:732–766. https://doi.org/10.1016/j.phytochem.2006.11.029
Lange BM, Fischedick JT, Lange MF, Srividya N, Šamec D, Poirier BC (2017) Integrative approaches for the identification and localization of specialized metabolites in Tripterygium roots. Plant Physiol 173:456–469
Chen X, Murakami T, Oppenheim JJ, Howard OM (2005) Triptolide, a constituent of immunosuppressive Chinese herbal medicine, is a potent suppressor of dendritic-cell maturation and trafficking. Blood 106:2409–2416. https://doi.org/10.1182/blood-2005-03-0854
Krakauer T, Chen X, Howard OMZ, Young HA (2005) Triptolide attenuates endotoxin- and staphylococcal exotoxin-induced T-cell proliferation and production of cytokines and chemokines. Immunopharmacol Immunotoxicol 27:53–66. https://doi.org/10.1081/IPH-51294
Liao ZH, Gong YF, Kai GY, Zu KJ, Chen M, Tan QM, Wei YM, Guo L, Tan F, Sun XF, Tang KX (2005) An intron-free methyl jasmonate inducible geranylgeranyl diphosphate- synthase gene from Taxus media and its functional identification in yeast. Mol Biol 39:11–17. https://doi.org/10.1007/s11008-005-0002-3
Jiang XH, Wong BC, Lin MC, Zhu GH, Kung HF, Jiang SH, Yang D, Lam SK (2001) Functional p53 is required for triptolide-induced apoptosis and AP-1 and nuclear factor-kappa B activation in gastric cancer cells. Oncogene 20:8009–8018. https://doi.org/10.1038/sj.onc.1204981
Antonoff MB, Chugh R, Borja-Cacho D, Dudeja V, Clawson KA, Skube SJ, Sorenson BS, Saltzman DA, Vickers SM, Saluja AK (2009) Triptolide therapy for neuroblastoma decreases cell viability in vitro and inhibits tumor growth in vivo. Surgery 146:282–290. https://doi.org/10.1159/000236859
He MF, Liu L, Ge W, Shaw PC, Jiang R, Wu LW, But PP (2009) Antiangiogenic activity of Tripterygium wilfordii and its terpenoids. J Ethnopharmacol 121:61–68. https://doi.org/10.1016/jjep.2008.09.033
Zhu WB, He SM, Li Y, Qiu PX, Shu MF, Ou YQ, Zhou YH, Leng TD, Xie J, Zheng XK, Xu D, Su XW, Yan GM (2010) Anti-angiogenic activity of triptolide in anaplastic thyroid carcinoma is mediated by targeting vascular endothelial and tumor cells. Vascul Pharmacol 52:46–54. https://doi.org/10.1016/j.vph.2009.10.006
He MF, Huang YH, Wu LW, Ge W, Shaw PC, But PP (2010) Triptolide functions as a potent angiogenesis inhibitor. Int J Cancer 126:266–278. https://doi.org/10.1002/ijc.24694
Chervoneva I, Li Y, Schulz S, Croker S, Wilson C, Waldman SA, Hyslop T (2010) Selection of optimal reference genes for normalization in quantitative RT-PCR. BMC Bioinform 11:1–15. https://doi.org/10.1186/1471-2105-11-253
Brunner AM, Yakovlev IA, Strauss SH (2004) Validating internal controls for quantitative plant gene expression studies. BMC Plant Biol 4:14. https://doi.org/10.1186/1471-2229-4-14
Jain M, Nijhawan A, Tyagi AK, Khurana JP (2006) Validation of housekeeping genes as internal control for studying gene expression in rice by quantitative real-time PCR. Biochem Biophys Res Commun 345:646–651. https://doi.org/10.1016/j.bbrc.2006.04.140
Xu ZC, Xu J, Ji AJ, Zhu YJ, Zhang X, Hu YL, Song JY, Chen SL (2015) Genome-wide selection of superior reference genes for expression studies in Ganoderma lucidum. Gene 574:352–358. https://doi.org/10.1016/j.gene.2015.08.025
He YH, Yan HL, Hua WP, Huang YY, Wang ZZ (2016) Selection and validation of reference genes for quantitative real-time PCR in Gentiana macrophylla. Front Plant Sci 7:945. https://doi.org/10.3389/fpls.2016.00945
Zhao XT, Zhang XL, Guo XB, Li SJ, Han LL, Song ZH, Wang YY, Li JH, Li MJ (2016) Identification and validation of reference genes for qRT-PCR studies of gene expression in Dioscorea opposita. Biomed Res Int 2016:3089584. https://doi.org/10.1155/2016/3089584
Huang YX, Tan HX, Yu J, Chen Y, Guo ZY, Wang GQ, Zhang QL, Chen JF, Zhang L, Diao Y (2017) Stable internal reference genes for normalizing real-time quantitative PCR in Baphicacanthus cusia under hormonal stimuli and UV irradiation, and in different plant organs. Front Plant Sci 8:668. https://doi.org/10.3389/fpls.2017.00668
Li T, Wang J, Lu M, Zhang TY, Qu XY, Wang ZZ (2017) Selection and validation of appropriate reference GENES for qRT-PCR analysis in isatis indigotica fort. Front Plant Sci 8:1139. https://doi.org/10.3389/fpls.2017.01139
Gulshan K, Anil KS (2015) Reference gene validation for qRT-PCR based gene expression studies in different developmental stages and under biotic stress in apple. Sci Hortic 197:597–606. https://doi.org/10.1016/j.scienta.2015.10.025
Miao GP, Li W, Zhang B, Zhang ZF, Ma ZQ, Feng JT, Zhang X, Zhu CS (2015) Identification of genes involved in the biosynthesis of Tripterygium wilfordii Hook.f. secondary metabolites by suppression subtractive hybridization. Plant Mol Biol Rep 33:756–769. https://doi.org/10.1007/s11105-014-0792-3
Miao GP, Han J, Zhang JF, Zhu CS, Zhang X (2017) A MDR transporter contributes to the different extracellular production of sesquiterpene pyridine alkaloids between adventitious root and hairy root liquid cultures of Tripterygium wilfordii Hook.f. Plant Mol Biol 95:51–62. https://doi.org/10.1007/s11103-017-0634-4
Zhang B, Liu Y, Chen MM, Feng JT, Ma ZQ, Zhang X, Zhu CS (2018) Cloning, expression analysis and functional characterization of squalene synthase (SQS) in Tripterygium wilfordii. Molecules 23:269. https://doi.org/10.3390/molecules23020269
Su P, Guan HY, Zhang YF, Wang X, Gao LH, Zhao YJ, Hu TY, Zhou JW, Ma BW, Tu LC, Tong YR, Huang LQ, Gao W (2017) Probing the Single Key Amino Acid Responsible for the Novel Catalytic Function of ent-Kaurene Oxidase Supported by NADPH-Cytochrome P450 Reductases in Tripterygium wilfordii. Front Plant Sci 8:1756. https://doi.org/10.3389/fpls.2017.01756
Zhang J, Huo YB, Liu Y, Feng JT, Ma ZQ, Zhu CS, Zhang X (2017) Differential expressed analysis of Tripterygium wilfordii unigenes involved in terpenoid backbone biosynthesis. J Asian Nat Prod Res 19(8):823–832. https://doi.org/10.1080/10286020.2016.1232713
Zhang YF, Zhao YJ, Wang JD, Hu TY, Tong YR, Zhou JW, Song YD, Gao W, Huang LQ (2017) Overexpression and RNA interference of TwDXR regulate the accumulation of terpenoid active ingredients in Tripterygium wilfordii. Biotech Lett 40(2):1–7. https://doi.org/10.1007/s10529-017-2484-1
Tong Y, Zhang M, Su P, Zhao Y, Wang X, Zhang X (2016) Cloning and functional characterization of an isopentenyl diphosphate isomerase gene from Tripterygium wilfordii. Biotechnol Appl Biochem 63(6):863–869. https://doi.org/10.1002/bab.1427
Guan HY, Zhao YJ, Su P, Tong YR, Liu YJ, Hu TY, Zhang YF, Zhang XA, Li J, Wu XY, Huang LQ, Gao W (2017) Molecular cloning and functional identification of sterol C24-methyltransferase gene from Tripterygium wilfordii. Acta Pharmaceut Sin B 7(5):603–609. https://doi.org/10.1016/j.apsb.2017.07.001
Su P, Guan HY, Zhao YJ, Tong YR, Xu MM, Zhang YF, Hu TY, Yang J, Cheng QQ, Gao LH, Liu YJ, Zhou JW, Reuben JP, Huang LQ, Gao W (2018) Identification and functional characterization of diterpene synthases for triptolide biosynthesis from Tripterygium wilfordii. Plant J 93(1):50–65. https://doi.org/10.1111/tpj.13756
Zhao YJ, Zhang YF, Su P, Yang J, Huang LQ, Gao W (2018) Genetic transformation system for woody plant Tripterygium wilfordii and its application to product natural celastrol. Front Plant Sci 8:2221. https://doi.org/10.3389/fpls.2017.02221
Nikolaj LH, Allison MH, Britta H, Carl EO, Björn MH, Johan AR, Björn H (2017) The terpene synthase gene family in Tripterygium wilfordii harbors a labdane-type diterpene synthase among the monoterpene synthase TPS-b subfamily. Plant J 89:429–441. https://doi.org/10.1111/tpj.13410
Zhu CS, Miao GP, Guo J, Huo YB, Zhang X, Xie JH, Feng JT (2014) Establishment of Tripterygium wilfordii Hook.f. hairy root culture and optimization of its culture conditions for the production of triptolide and wilforine. J Microbiol Biotechnol 24:823–834. https://doi.org/10.4014/jmb.1402.02045
Sun HP, Li F, Ruan QM, Zhong XH (2016) Identification and validation of reference genes for quantitative real-time PCR studies in Hedera helix L. Plant Physiol Biochem 108:286–294. https://doi.org/10.1371/journal.pone.0106800
Vinayak HL, Tukaram DN, Suprasanna P (2010) Differential osmotic adjustment to iso- osmotic NaCl and PEG stress in the in vitro Cultures of Sesuvium portulacastrum (L.) L. J Crop Sci Biotechnol 13(4):251–256. https://doi.org/10.1007/s12892-010-0008-9
Miao GP, Zhu CS, Yang YQ, Feng MX, Ma ZQ, Feng JT, Zhang X (2014) Elicitation and in situ adsorption enhanced secondary metabolites production of Tripterygium wilfordii Hook. f. adventitious root fragment liquid cultures in shake flask and a modified bubble column bioreactor. Bioprocess Biosyst Eng 37:641–650. https://doi.org/10.1007/s00449-013-1033-0
Ginzinger DG (2002) Gene quantification using real-time quantitative PCR: an emerging technology hits the mainstream. Exp Hematol 30(6):503–512. https://doi.org/10.1016/S0301-472X(02)00806-8
Czechowski T, Stitt M, Altmann T, Udvardi MK, Scheible WR (2005) Genome-wide identification and testing of superior reference genes for transcript normalization in Arabidopsis. Plant Physiol 139:5–17. https://doi.org/10.1104/pp.105.063743
Julia H, Florian MWG (2007) Identification of reference genes for qRT-PCR studies of gene expression in giant cells and syncytia induced in Arabidopsis thaliana by Meloidogyne incognita and Heterodera schachtii. Nematology 9:317–323. https://doi.org/10.1163/156854107781352034
Vandesompele J, Preter KD, Pattyn F, Poppe B, Roy NV, Paepe AD, Speleman F (2002) Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 3(7):0034.1–0034.11. https://doi.org/10.1186/gb-2002-3-7-research0034
Andersen CL, Jensen JL, Orntoft TF (2004) Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Can Res 64:5245–5250. https://doi.org/10.1158/0008-5472.can-04-0496
Silver N, Best S, Jiang J, Thein SL (2006) Selection of housekeeping genes for gene expression studies in human reticulocytes using real-time PCR. BMC Mol Biol 7:33. https://doi.org/10.1186/1471-2199-7-33
Pfaffl MW, Tichopad A, Prgomet C, Neuvians TP (2004) Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: Bestkeeper - excel-based tool using pair-wise correlations. Biotech Lett 26:509–515. https://doi.org/10.1023/B:BILE.0000019559.84305.47
Xie F, Peng X, Chen D, Xu L, Zhang B (2012) miRDeepFinder: a miRNA analysis tool for deep sequencing of plant small RNAs. Plant Mol Biol 80:75–84. https://doi.org/10.1007/s11103-012-9885-2
Herz S, Wungsintaweekul J, Schulir CA, Hecht S, Luttgen H, Sagner S, Fellermeier M, Eisenreich W, Zenk MH, Bacher A, Rohdich F (2000) Biosynthesis of terpenoids: YgbB protein converts 4-diphosphocytidyl-2C-methyl-d-erythritol 2,4-cyclodiphosphate. Proc Natl Acad Sci USA 97:2486–2490. https://doi.org/10.1073/pnas.040554697
Livak KJ, Schmittgen TD (2001) Analysis of relative gene xpression data using real-time quantitative PCR and the 2−∆∆CT method. Methods 25:402–408. https://doi.org/10.1006/meth.2001.1262
Hemmerlin A, Harwood JL, Bach TJ (2012) A raison d’être for two distinct pathways in the early steps of plant isoprenoid biosynthesis? Prog Lipid Res 51:95–148. https://doi.org/10.1016/j.plipres.2011.12.001
Hong SY, Seo PJ, Yang M, Xiang F, Park CM (2008) Exploring valid reference genes for gene expression studies in Brachypodium distachyon by real-time PCR. BMC Plant Biol 8:112. https://doi.org/10.1186/1471-2229-8-112
Guo J, Hui L, Wu Q, Xu L, Que Y (2014) The choice of reference genes for assessing gene expression in sugarcane under salinity and drought stresses. Sci Rep 4:7042. https://doi.org/10.1038/srep07042
Enjuto M, Balcells L, Campos N, Caelles C, Arró M, Boronat A (1994) Arabidopsis thaliana contains two differentially expressed 3-hydroxy-3-methylglutaryl-CoA reductase genes, which encode microsomal forms of the enzyme. Proc Natl Acad Sci USA 91:927–931. https://doi.org/10.1073/pnas.91.3.927
Campos N, Boronat A (1995) Targeting and topology in the membrane of plant 3-hydroxy-3- methylglutaryl coenzyme A reductase. Plant Cell 7:2163–2174. https://doi.org/10.1105/tpc.7.12.2163
Pauwels L, Inzé D, Goossens A (2009) Jasmonate-inducible gene: what does it mean? Trends Plant Sci 14:87–91. https://doi.org/10.1016/j.tplants.2008.11.005
Miao GP, Zhu CS, Feng JT, Han J, Song XW, Zhang X (2012) Aggregate cell suspension cultures of Tripterygium wilfordii Hook. f. for triptolide, wilforgine, and wilforine production. Plant Cell, Tissue Organ Cult 112:109–116. https://doi.org/10.1007/s11240-012-0211-0
Jiang Q, Wang F, Li MY, Ma J, Tan GF, Xiong AS (2014) Selection of suitable reference genes for Qpcr normalization under abiotic stresses in Oenanthe javanica (BI.) DC. PLoS ONE 9(3):e92262. https://doi.org/10.1371/journal.pone.0092262
Won-Jae L, Ryoung-Hoon J, Si-Jung J, Ji-Sung P, Seung-Chan L, Raghavendra BS et al (2015) Selection of reference genes for quantitative gene expression in porcine mesenchymal stem cells derived from various sources along with differentiation into multilineages. Stem Cells Int 2015:1–14. https://doi.org/10.1155/2015/235192
Hu Y, Chen H, Luo C, Dong L, Zhang SW, He XH, Huang GX (2014) Selection of reference genes for real-time quantitative pcr studies of kumquat in various tissues and under abiotic stress. Sci Hortic 174:207–216. https://doi.org/10.1016/j.scienta.2013.12.003
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 31272110) and the Natural Science Foundation of Shaanxi Province (Grant No. 2016JM3036).
Author information
Authors and Affiliations
Contributions
CZ and JZ conceived and designed the study. JZ, YH and BZ performed the experiments. CZ and JZ wrote the paper. CZ, JF, ZM and XZ reviewed and edited the manuscript. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declared that they have no conflicts of interest to this work. We declare that we do not have any commercial or associative interest that represents a conflict of interest in connection with the work submitted.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Zhang, J., Zhu, Cs., Huo, Yb. et al. Validation of suitable reference genes for quantitative gene expression analysis in Tripterygium wilfordii. Mol Biol Rep 46, 4161–4174 (2019). https://doi.org/10.1007/s11033-019-04867-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-019-04867-8