
Abstract
In various diseases, including diabetes, extracellular vesicles (EVs) have been detected in circulation and tissues. EVs are small membrane vesicles released from various cell types under varying conditions. Recently, endothelial cell-derived EVs (EEVs) were identified as a marker of endothelial dysfunction in diabetes, but the ensuing mechanisms remain poorly understood. In this study, we dissected the ensuing pathways with respect to nitric oxide (NO) production under the condition of type 2 diabetes. Human umbilical vein endothelial cells (HUVECs) were stimulated with glucose alone and with glucose in combination with angiotensin II (Ang II) for 48 h. In supernatants from glucose + Ang II-stimulated HUVECs, release of EEVs was assessed using Western blotting with an anti-CD144 antibody. EEV release was significantly increased after stimulation of HUVECs, and high glucose + Ang II-derived EEVs impaired ACh-induced vascular relaxation responses and NO production in mice aortic rings. Furthermore, high glucose + Ang II-derived EEVs induced ERK1/2 signalling and decreased endothelial NO synthase (eNOS) protein expression in mice aortas. Furthermore, in the presence of the MEK/ERK1/2 inhibitor PD98059, high glucose plus Ang II treatment stimulated EEVs in HUVECs and those EEVs prevented the impairments of ACh-induced relaxation and NO production in mice aortas. These data strongly indicate that high glucose and Ang II directly affect endothelial cells and the production of EEVs; the resultant EEVs aggravate endothelial dysfunction by regulating eNOS protein levels and ERK1/2 signalling in mice aortas.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The prevalence of hypertension is increased in patients with type 2 diabetes mellitus [38], resulting in increased risk of cardiovascular complications [46]. This complex and multifactorial disease commonly presents with vascular dysfunction [31], and both macrovascular and microvascular complications, leading to the development of atherosclerosis [11, 29].
Endothelial dysfunction is a consistent finding in patients with diabetes [5] and refers to the inability of the endothelium to regulate vascular homeostasis. These abnormalities in endothelial function are detected early in the development of cardiovascular complications, often before symptoms are clinically evident [42]. Endothelial dysfunction is commonly assessed as alterations in endothelial-dependent vascular relaxation with concomitant changes in protein expression. Nitric oxide (NO) is synthesised by endothelial nitric oxide synthase (eNOS), and as a major mediator of endothelial-dependent vascular relaxation, it is critically involved in the regulation of other protective properties of healthy endothelium [24, 47]. Recent evidence supports a central role of the interaction between eNOS levels and extracellular signal-regulated kinase (ERK1/2) activation in endothelial cells [12].
Extracellular vesicles (EVs) are lipid bilayer structures that are shed from surfaces of various cell types, including endothelial cells, leukocytes and platelets [33]. Several classes of EVs, including exosomes, microvesicles and microparticles, are produced by different mechanisms [45] and comprise cytoplasm and cell membranes from their cells of origin [13, 53]. EVs are released under normal and pathological conditions to mediate cell-cell communication and signalling, and endothelial cell-derived EVs (EEVs) originating from activated or apoptotic endothelial cells reportedly contribute to vascular remodelling and repair [3]. Recently, vascular endothelial cadherin (CD144)-positive EVs were identified in type 2 diabetic patients with cardiovascular complications [2, 22]. Because EEVs present CD144 on their membranes, CD144-positive EVs may reflect endothelial damage [2, 14]. In accordance, Koga et al. showed increased circulating levels of CD144-positive EVs in patients with type-2 diabetes and vascular disease [22]. However, although it is unknown whether circulating EEVs cause endothelial dysfunction, diabetic conditions may contribute to the development of endothelial dysfunction via increased production of EEVs and EEVs-related molecules.
Release of EEVs is dependent on microenvironments, and is influenced by angiotensin II (Ang II) and glucose concentrations. Glucose stimulation alters exocytosis rates and release of exosomes at high glucose concentrations reflects increased rates of exocytosis, increased EV production or a combination of these factors [39, 41]. Induction of EEV release by Ang II has been demonstrated [37], and treatment with Ang II receptor type 1 blockers and lipid-lowering agents, such as simvastatin decrease plasma EV levels [23, 34]. Currently, it is unclear whether glucose and/or Ang II directly influence EV generation. Thus, in the present study we investigated direct effects of EEVs on vascular endothelial function and EEV generation in Ang II and/or glucose-stimulated cells.
Materials and methods
Cell culture and EEVs generation
Human umbilical vein endothelial cells (HUVECs; Kurabo, Osaka, Japan) were cultured in endothelial growth medium (Kurabo) and were maintained at 37 °C in a 5% CO2 incubator. Cells from passages 4–7 were used in all experiments and were seeded onto 12-well plates at 3 × 105 cells/well. Then the cells were treated at approximately 80% confluence. Ang II (10−7 mol/L) was diluted in a complete cell culture medium containing 5- or 22- × 10−3 mol/L glucose (low and high glucose) and was added to the cells and incubated for 48 h. Subsequently, culture medium was collected and cells and cell debris were precipitated by centrifugation at 5000 g for 10 min. Supernatants were then centrifuged at 100,000 g for 2 h to yield pellet EEVs [8, 27]. The EEVs were then washed in phosphate-buffered saline (PBS), were resuspended in 50 μL of PBS and were used immediately in experiments. EEVs from low glucose-treated HUVECs were defined as low glucose EEVs (LGEVs), those from high glucose-treated HUVECs were defined as high glucose EEVs (HGEVs), those from low glucose plus Ang II-treated HUVECs were defined as low glucose + Ang II EEVs (LG-Ang EVs) and those from high glucose plus Ang II-treated HUVECs were defined as high glucose + Ang II EEVs (HG-Ang EVs). In separate experiments, HUVECs were exposed to PD98059 (10−5 mol/L) in the presence of high glucose and Ang II for 48 h and were defined as high glucose + Ang II + PD EEVs (HG-Ang-PD EVs).
Detection of EEVs
Isolated EEVs were quantified using immunoblotting with an anti-CD144 antibody. Recombinant human CD144 was purchased from Sino Biological Inc. (Beijing, China) and was diluted and used to plot the standard curve. Identities and quantities of EEVs were analysed using SDS-PAGE and Western blotting analyses.
Animals and procedures
All animal experiments were performed in accordance with the Guidelines for the Care and Use of Laboratory Animals from the Committee for the Care and Use of Laboratory Animals of Hoshi University, which is accredited by the Ministry of Education, Culture, Sports, Science, and Technology, Japan. All animal experiments were performed with male mice (body wt, 40–50 g) from the Institute of Cancer Research (ICR).
Preparation of aortic rings and assessment of vascular relaxation
In all experiments, mice were anaesthetized with isoflurane for surgical procedures (initially with 5% and then maintenance at 2%) and euthanized by thoracotomy and exsanguination. Subsequently, aortas were carefully isolated from mice as reported previously [43, 44], were dissected from surrounding fat and connective tissue, cut into 2-mm rings and placed in Krebs-Henseleit solution (KHS) containing 118.0 × 10−3 mol/L NaCl, 4.7 × 10−3 mol/L KCl, 25.0 × 10−3 mol/L NaHCO3, 1.8 × 10−3 mol/L CaCl2, 1.2 × 10−3 mol/L NaH2PO4, 1.2 × 10−3 mol/L MgSO4 and 11.0 × 10−3 mol/L glucose. Vascular rings were then mounted between two stainless steel triangles in an organ bath containing KHS (pH 7.4) at 37 °C and gassed with 95% O2/5% CO2. Subsequently, a resting tension of 1.5 g was enforced, and changes in tension of aortic rings were analysed using a force-displacement transducer linked to a PowerLab recording system (AD Instruments, Australia). After equilibration (45 min), rings were depolarised using 80 × 10−3 mol/L KCl and maximal contractions were evaluated. Vascular preparations were then washed with KHS and contracted using prostaglandin F2α (PGF2α, 10−6–3 × 10−6 mol/L). Following stabilisation of contractile responses (1-g tension state), vascular relaxation responses to cumulative increments in ACh (10−9–10−5 mol/L) or SNP (10−10–10−5 mol/L) concentrations were examined. Aortic rings were exposed to EEVs (50 μL; as above) for 30 min before PGF2α-induced precontraction. Aortic rings were precontracted with equieffective doses of PGF2α (10−6–3 × 10−6 mol/L) and relaxation induced by ACh or SNP was expressed as a percentage of PGF2α-induced contraction.
Assessment of vascular NO production
Aortic rings (4 mm) were placed in KHS and treated with 10 μL of EEVs for 30 min. Following stimulation with ACh (10−6 mol/L) or PBS for 20 min, solutions were assayed for the stable end product NO using an NO detector/high-performance liquid chromatography system (ENO20; Eicom) as previously described [19, 43].
Western blotting
Protein expression was determined in ACh or non-stimulated aortas of mice and in EEVs isolated from HUVEC culture medium. Samples were homogenised in RIPA buffer as previously described [19, 43, 49, 50] and protein concentrations were quantified using BCA protein assay kit. Western blotting was performed using standard procedures, and membranes were incubated with the following antibodies: rabbit anti-CD144 (1:1000; Thermo Scientific, Rockford, IL, USA), rabbit anti-phospho-eNOS (1:500; Cell Signalling Technology, Danvers, MA, USA), mouse anti-eNOS (1:1000; BD Biosciences, San Jose, CA, USA), rabbit anti-phospho-ERK1/2 (1:1000; Cell Signalling Technology), mouse anti-ERK1/2 (1:1000; Cell Signalling Technology) and mouse β-actin (1:5000; Sigma Chemical CO., St. Louis, MO, USA). Membranes were then washed and incubated with anti-mouse/anti-rabbit IgG antibody (Promega, Madison, WI, USA) at a dilution of 1:10,000 for 20 min at 37 °C and were probed for immunoreactive proteins using chemiluminescence. Band intensities were quantified using densitometry.
Data analysis
Vascular reactivity was analysed using 2-way repeated measures ANOVA with post hoc Bonferroni tests. Western blot and NO data were analysed using Tukey tests followed by multiple comparison tests (Graph Pad Prism 6.0; GraphPad Software, San Diego, CA, USA). Differences were considered significant when P < 0.05.
Results
Isolation and characterisation of EVs from HUVECs
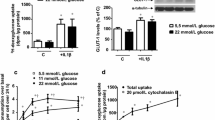
EVs were isolated from the HUVEC culture media and were identified using Western blotting for CD144 (Fig. 1). Recombinant CD144 protein EVs were expressed with a predicted size of about 120 kDa in Western blot analyses with an anti-CD144 antibody. Blots with varying concentrations of recombinant CD144 showed corresponding band intensities.

Western blotting data from EEVs; a representative Western blots of recombinant CD144 protein and EEVs from HUVECs using an anti-CD144 antibody; LGEVs, EEVs induced by low glucose (5 mmol/L); HGEVs, EEVs induced by high glucose (22 mmol/L); LG-Ang EVs, EEVs induced by low glucose + Ang II (100 nmol/L); HG-Ang EVs, EEVs induced by high glucose + Ang II. b CD144 protein expression in 20-μL aliquots of protein. Data are expressed as means ± standard errors of the mean (SE); n = 4; *P < 0.05, **P < 0.01 vs. LGEVs
Numbers of LG-Ang EVs and HG-Ang EVs were significantly higher than those of LGEVs. However, production of EEVs did not differ between LG and HG (Fig. 1b). These results show that Ang II increases EEV release from endothelial cells.
High glucose plus Ang II-derived EEVs induce endothelial dysfunction
The effects of HGEVs on ACh-induced endothelial-dependent relaxation did not differ from those observed in LGEVs (Fig. 2a). Moreover, concentration-dependent curves for the NO donor SNP showed similar relaxation effects in both groups (Fig. 2b). Additionally, relaxation due to ACh was significantly impaired in aortas treated with HG-Ang EVs compared with that in those treated with either vehicle or LG-Ang EVs (Fig. 2c). The data in Fig. 2d show no significant difference in SNP-induced relaxation in aortas treated with vehicle, LG-Ang EVs or HG-Ang EVs. These results indicate that endothelium-independent relaxation remains unchanged in aortas treated with EEVs, and that endothelium-dependent relaxation is not affected by HG-induced EEVs only. Furthermore, these results suggest that HG-Ang EVs induce endothelial dysfunction in mice aortas. Critically, numbers of EEVs were similarly increased in LG-Ang EVs and HG-Ang EVs, and LG-Ang EVs did not induce endothelial dysfunction.

High glucose plus Ang II-derived EEVs impair endothelium-dependent relaxation. Concentration-response curves following ACh (a) and SNP (b) treatments of mouse aortic rings after incubation with vehicle (PBS), LGEVs or HGEVs; Concentration-response curves following ACh (c) and SNP (d) treatments of mouse aortic rings after incubation with vehicle, LG-Ang EVs or HG-Ang EVs. Vehicle and EEVs treatments were administered in 50-μL aliquots for 30 min. Abbreviations are defined in Fig. 1. Data are expressed as means ± SE; n = 5―6; **P < 0.01 vs. Control +veh, ##P < 0.01 vs. Control +HG-Ang EVs
High glucose plus Ang II-derived EEVs decrease NO production in mice aortas
As shown in Fig. 3, EEVs did not alter NO production in aortas in comparison with aortas from vehicle-treated mice. However, although LG-Ang EVs treated aortas were not significantly altered, HG-Ang EVs reduced ACh-stimulated NO production in aortas. These data suggest that HG-Ang EVs synergistically induce rapid endothelial dysfunction, whereas the NO producing effects of EEVs, such as HG-derived EEVs and HG plus Ang II-derived EEVs, are the same as those of the endothelium-dependent relaxation responses shown in Fig. 2.

High glucose plus Ang II-derived EEVs decrease ACh-stimulated NO production. Detection of NO production in aortic rings; Aortas from mice treated with vehicle, LGEVs, HGEVs, LG-Ang EVs or HG-Ang EVs were incubated for 30 min at 37 °C and were then treated with PBS (non-stimulation; (a)) or ACh (10−6 M; (b)) for 20 min. Data are expressed as means ± SE; n = 5―6. *P < 0.05 vs. +veh
To investigate the molecular mechanisms that govern reductions of NO production following treatments with HG-Ang EVs, we determined the expression and activation of NO pathway enzymes using Western blotting. No differences in eNOS phosphorylation (Ser1177) were observed among ACh-stimulated EEVs treated aortas, whereas treatments with HG-Ang EVs significantly decreased total eNOS protein expression in non-stimulated and ACh-stimulated aortas compared those treated with HGEVs or LG-Ang EVs (Fig. 4).

High glucose plus Ang II-derived EEVs decrease eNOS protein levels in mouse aortas. a Western blots of phospho-eNOS and total eNOS expression in aortas from mice treated with LGEVs, HGEVs, LG-Ang EVs or HG-Ang EVs for 30 min followed by treatments with ACh (10−6 mol/L) or PBS (non-stimulated) for 20 min; b–e Results are expressed as ratios of total protein to β-actin and ratios of phosphorylated protein to total protein. b Non-stimulated eNOS phosphorylation (Ser1177); c ACh-stimulated eNOS phosphorylation (Ser1177). d Non-stimulated total eNOS expression; e ACh-stimulated total eNOS expression; Data are expressed as means ± SE; n = 5. #P < 0.05, ##P < 0.01 vs. +HG-Ang EVs
Effect of EEVs on MAPK signalling
To determine the effects of EEVs on signalling via one of three major subgroups of the mitogen-activated protein kinase (MAPK) family that regulates eNOS, ERK1/2 phosphorylation levels were measured. These experiments showed significant increases in ERK1/2 phosphorylation in aortas treated with HG-Ang EVs compared with those treated with LGEVs or HGEVs (Fig. 5a, b). However, expression of total ERK1/2 did not differ among treatment groups (Fig. 5a, c).

Involvement of ERK1/2 signalling in high glucose plus Ang II-derived EEVs-induced endothelial dysfunction in mouse aortas; a Western blots of phospho-ERK1/2 and total ERK1/2 expression in aortas from mice treated with LGEVs, HGEVs, LG-Ang EVs or HG-Ang EVs (for 30 min); b, c Results are expressed as ratios of total protein to β-actin and ratios of phosphorylated protein to total protein. b ERK1/2 phosphorylation (Thr202/Tyr204); c total ERK1/2 expression; d concentration-response curves from ACh-treated aortic rings from mice treated with vehicle, HG-Ang EVs or HG, Ang II and PD98059 (10−5 mol/L)-derived EVs (HG-Ang-PD EVs); e ACh-stimulated NO production in mice aortas treated with vehicle, HG-Ang EVs or HG-Ang-PD EVs. Data are expressed as means ± SE; n = 5―6. b *P < 0.05 vs. +LGEVs; ##P < 0.01 vs. +HG-Ang EVs; (d, e) ***P < 0.001 vs. +veh; #P < 0.05 vs. +HG-Ang EVs
To investigate the role of ERK1/2 in endothelial function, experiments were performed with HG-Ang-PDEVs that were generated from HUVECs treated with high glucose plus Ang II and the selective ERK1/2 inhibitor PD98059 for 48 h. In the presence of HG-Ang EVs, vascular endothelial-dependent responses to ACh and NO production in the presence of ACh were reduced in aortas (Figs. 2c and 5d). Furthermore, ACh-induced NO production and relaxation responses were significantly increased in aortas treated with HG-Ang-PDEVs (Fig. 5d e). Hence, EEVs that are sensitive to ERK1/2 participate in ACh-induced relaxation.
Discussion
Numerous studies have demonstrated associations between numbers of circulating EEVs and endothelial function in patients with cardiovascular diseases [48, 51]. Similarly, higher numbers of EEVs in type 2 diabetic patients are correlated with impaired endothelial function [17]. Thus, EEVs may have direct effects on endothelial function, and may operate through ligand-induced endothelial signalling molecules. Previously, we reported that circulating EVs including microparticles, impair endothelial dysfunction by regulating endothelial protein expression in diabetic rats [19]. Thus, in the present, we focused on endothelium derived EVs and investigated EEV production and subsequent regulation of vascular endothelial function using EVs from HUVECs as a model of diabetic complications. Subsequently, we investigated direct effects of EEVs on vascular endothelial function in isolated mice aortas.
The present data demonstrate that (1) Ang II is a potent stimulus for EEVs generation, (2) high glucose and Ang II-costimulated HUVECs produce EEVs that affect endothelial-dependent vascular relaxation via diminished aortic NO production, (3) EEVs stimulate ERK1/2 signalling and decrease eNOS expression in aortas, and (4) ERK1/2 activating EEVs contribute to endothelial dysfunction and decreased NO production in aortas (Fig. 6). Taken together, these data suggests a positive feedforward system whereby Ang II and high glucose cotreatments promote endothelial dysfunction through EEVs. These phenomena may contribute to endothelial dysfunction in diabetes, particularly in conditions associated with hypertension.

Role of endothelial cell-derived EVs in the high glucose plus Ang II conditions. High gluose (HG) plus Ang II increased release of EEVs. HG plus Ang II-derived EEVs induced ERK1/2 activation leading to decreasing eNOS protein levels, NO production. Finally, HG plus Ang II-derived EEVs induced endothelial dysfunction in mice aortas
Elevated levels of circulating EVs were previously reported in peripheral blood from patients with diabetes or vascular complications [30, 35]. However, EVs are released from endothelial cells and various other cell types [4, 40]. Moreover, although EEVs comprise a subpopulation of EVs in human plasma, they have been associated with the pathogenesis of various cardiovascular diseases that are predominantly initiated by endothelial dysfunction [7, 16]. Furthermore, various factors have been implicated as inducers of endothelial cell vesiculation, membrane blebbing, and consequent EV release [6, 26]. We showed that Ang II directly stimulates release of EVs from endothelial cells. In these experiments, EEVs were characterised using the membrane marker CD144 in Western blotting analyses and EV release did not differ between HUVECs exposed to glucose at low and high concentrations, indicating a role of Ang II in this process. In agreement, a previous clinical study correlated plasma levels of EEVs with hypertension [17]. Furthermore, Ang II-mediated EV release mechanisms have been described previously [1, 10, 25, 52]. Herein, we collected EVs that were released from 2.7 × 104 HUVECs over 48 h and found that numbers of LG-Ang EVs and HG-Ang EVs were three times those of LGEVs (Fig. 1). The present study of circulating agents that are associated with diabetes shows novel mechanisms by which glucose and Ang II-derived EVs induce endothelial dysfunction.
In further experiments, elevated concentrations of EVs from endothelial cells were detected following stimulation with LG+Ang II and HG+Ang II, suggesting that Ang II stimulates EEV release. Although mechanisms for the formation of EEVs poorly understood, recent reports show that Ang II is associated with increased EV formation from endothelial cells [10]. These investigators suggest that EVs formation is partly dependent on the NADPH oxidase/Rho kinase pathway. In accordance, the present data suggest that Ang II contributes to quantitative changes in EV release from endothelial cells. However, high glucose concentrations did not promote the release of EVs from endothelial cells (Fig. 1), and release of EEVs following single exposures to high glucose or Ang II did not cause endothelial dysfunction (Fig. 2). Only HG-Ang EVs impaired ACh-induced endothelial relaxation responses and decreased NO production (Figs. 2 and 3). The results suggest that HG-Ang EVs cause qualitative changes to endothelial function, likely by activating ERK1/2 (Fig. 5). However, little is known about the effects of HG-Ang EVs on vascular mechanisms.
In further experiments, we investigated pathophysiological roles of EEVs by monitoring deleterious vascular signalling. Vascular diabetic complications are strongly associated with prolonged exposure to hyperglycemia, which is the hallmark of diabetes mellitus [36]. Thus, we compared responses to LGEVs and HGEVs in the presence of ACh and SNP and showed that NO-induced relaxation in endothelial and smooth muscle cells was not impaired by HGEVs. However, ACh-induced relaxation was only significantly attenuated in HG-Ang EVs treated aortic rings, reflecting diminished NO production despite the absence of changes in numbers of LG-Ang EVs and HG-Ang EVs. These data indicate that whereas Ang II quantitative changes EV release, HG causes qualitative changes. In addition, increased numbers of CD144-positive EVs were correlated with endothelial dysfunction, corroborating previous studies showing decreased concentrations of NO metabolites under similar conditions [9, 20].
EVs may reduce NO production from endothelial cells by negatively regulating eNOS. Therefore, to elucidate mechanisms that lead to decreased NO production, we demonstrated that EVs induce eNOS in aortic rings. In these experiments, no differences in eNOS phosphorylation were observed between mice aortas that were treated with LGEVs, HGEVs, LG-Ang EVs or HG-Ang EVs in the absence or presence of ACh. However, treatment of aortas with HG-Ang EVs led to decreased NO production and reduced eNOS expression. In conjunction with the present data, we recently reported that microparticles from diabetic rats reduced eNOS expression following acute treatments with ACh [19]. Although the ensuing mechanisms remain unknown, we speculate that EVs enhance the release of EEVs containing eNOS. Endothelial cells also release vesicles into the extracellular space under normal and stress conditions. Subsequently, EVs retain cell surface proteins from the cell of origin, along with cytosolic contents including enzymes and RNA [15]. Because endothelial eNOS is mainly located on cell surfaces, it is readily shed from endothelial cells by EVs that are induced by high glucose and Ang II. However, further research is required to characterise regulatory mechanisms for eNOS protein levels. Previous studies show that eNOS activation by ACh is mediated by increases in intracellular calcium, whereas activation by insulin or shear stress follows eNOS phosphorylation [18, 32]. However, the present EVs did not change eNOS activity by phosphorylation (Ser1177) following ACh stimulation, although loss of total eNOS expression in aortas treated with HG-Ang EVs may have decreased NO production significantly. Accordingly, treatment with LG-Ang EVs but not HG-Ang EVs maintained total eNOS expression and increased the release of NO.
Increased MAPK activation is an accepted mechanism for endothelial dysfunction in diabetes. In agreement, proinflammatory mediators were previously shown to cause endothelial apoptosis and activation, thereby increasing basal EVs’ release through MAPK dependent pathways [15]. Furthermore, endothelial-derived microparticles upregulated ERK1/2 and promoted endothelial dysfunction [37]. In the present study, HG-Ang EVs did not affect total ERK1/2 expression, but activated ERK1/2 in aortas. In contrast, HG-Ang-PD EVs normalised ACh-stimulated NO production and ACh-induced relaxation responses in aortas, suggesting effects of EV release in the presence of ERK1/2 activation. Although the mechanisms by which EEVs activate ERK1/2 require additional investigation, ERK1/2 signalling may stimulate EEVs’ release in the presence of HG plus Ang II. Accordingly, HG plus Ang II induced ERK1/2 in endothelial cells and led to cellular hypertrophy and apoptosis [21, 28], suggesting an important role of this enzyme in the release of EEVs. Alternatively, endothelial dysfunction may reflect modulation of eNOS expression via the ERK1/2 pathway. In the present study, significant increases in ACh-induced endothelial-dependent relaxation responses and NO production were observed in aortas treated with high glucose, Ang II and PD98059-derived EEVs, compared with those treated with high glucose and Ang II-derived EEVs (Fig. 5d, e). These observations indicate that (1) PD98059 inhibits the ERK1/2 activation that is induced by high glucose and Ang II in endothelia cells, (2) released EEVs do not induce ERK1/2 in aortic endothelial cells and (3) inactivated ERK1/2 is not associated with changes in eNOS protein level. These data will be helpful for further studies of cellular regulators of EEVs using the same methods. However, this observation warrants further investigations of the roles of ERK1/2 in the pathogenesis of endothelial dysfunction following direct delivery of EVs to endothelial cells.
In summary, our findings indicate that HG plus Ang II have direct stimulatory effects on EEV release and indicate involvement of a novel ERK1/2 pathway. HG plus Ang II-derived EEVs are important pathogenic factors in the development of endothelial dysfunction and may contribute to a deleterious cycle involving elevated numbers of EEVs with endothelial dysfunction and consequent aggravation of endothelial dysfunction by EEVs. Reducing numbers of circulating EEVs or blocking their effects by inhibiting ERK1/2 activation may offer effective therapeutic approaches for treating endothelial dysfunction in diabetes patients.
References
Agouni A, Andriantsitohaina R, Martinez MC (2014) Microparticles as biomarkers of vascular dysfunction in metabolic syndrome and its individual components. Curr Vasc Pharmacol 12:483–492
Amabile N, Guérin AP, Leroyer A, Mallat Z, Nguyen C, Boddaert J, London GM, Tedgui A, Boulanger CM (2005) Circulating endothelial microparticles are associated with vascular dysfunction in patients with end-stage renal failure. J Am Soc Nephrol 16:3381–3388
Arraud N, Linares R, Tan S, Gounou C, Pasquet JM, Mornet S, Brisson AR (2014) Extracellular vesicles from blood plasma: determination of their morphology, size, phenotype and concentration. J Thromb Haemost 12:614–627
Aupeix K, Hugel B, Martin T, Bischoff P, Lill H, Pasquali JL, Freyssinet JM (1997) The significance of shed membrane particles during programmed cell death in vitro, and in vivo, in HIV-1 infection. J Clin Invest 99:1546–1554
Avogaro A, Albiero M, Menegazzo L, De Kreutzenberg S, Fadini GP (2011) Endothelial dysfunction in diabetes: the role of reparatory mechanisms. Diabetes Care 34:S285–S290
Boulanger CM, Amabile N, Guérin AP, Pannier B, Leroyer AS, Mallat CN, Tedgui A, London GM (2007) In vivo shear stress determines circulating levels of endothelial microparticles in end-stage renal disease. Hypertension 49:902–908
Boulanger CM, Scoazec A, Ebrahimian T, Henry P, Mathieu E, Tedgui A, Mallat Z (2001) Circulating microparticles from patients with myocardial infarction cause endothelial dysfunction. Circulation 104:2649–2652
Brodsky SV, Malinowski K, Golightly M, Jesty J, Goligorsky MS (2002) Plasminogen activator inhibitor-1 promotes formation of endothelial microparticles with procoagulant potential. Circulation 106:2372–2378
Brodsky SV, Zhang F, Nasjletti A, Goligorsky MS (2004) Endothelium-derived microparticles impair endothelial function in vitro. Am J Physiol Heart Circ Physiol 286:H1910–H1915
Burger D, Schock S, Thompson CS, Montezano AC, Hakim AM, Touyz RM (2013) Microparticles: biomarkers and beyond. Clin Sci (Lond) 124:423–441
Carmassi F, Morale M, Puccetti R, De Negri F, Monzani F, Navalesi R, Mariani G (1992) Coagulation and fibrinolytic system impairment in insulin dependent diabetes mellitus. Thromb Res 67:643–654
Chen C, Chai H, Wang X, Jiang J, Jamaluddin MS, Liao D, Zhang Y, Wang H, Bharadwaj U, Zhang S, Li M, Lin P, Yao Q (2008) Soluble CD40 ligand induces endothelial dysfunction in human and porcine coronary artery endothelial cells. Blood 112:3205–3216
Colombo M, Raposo G, Théry C (2014) Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol 30:255–289
Diamant M, Tushuizen ME, Sturk A, Nieuwland R (2004) Cellular microparticles: new players in the field of vascular disease? Eur J Clin Investig 34:392–401
Dignat-George F, Boulanger CM (2011) The many faces of endothelial microparticles. Arterioscler Thromb Vasc Biol 31:27–33
Faure V, Dou L, Sabatier F, Cerini C, Sampol J, Berland Y, Brunet P, Dignat-George F (2006) Elevation of circulating endothelial microparticles in patients with chronic renal failure. J Thromb Haemost 4:566–573
Feng B, Chen Y, Luo Y, Chen M, Li X, Ni Y (2010) Circulating level of microparticles and their correlation with arterial elasticity and endothelium-dependent dilation in patients with type 2 diabetes mellitus. Atherosclerosis 208:264–269
Fleming I, Bauersachs J, Fisslthaler B, Busse R (1998) Ca2+-independent activation of the endothelial nitric oxide synthase in response to tyrosine phosphatase inhibitors and fluid shear stress. Circ Res 82:686–695
Ishida K, Taguchi K, Hida M, Watanabe S, Kawano K, Matsumoto T, Hattori Y, Kobayashi T (2016) Circulating microparticles from diabetic rats impair endothelial function and regulate endothelial protein expression. Acta Physiol (Oxf) 216:211–220
Jansen F, Yang X, Franklin BS, Hoelscher M, Schmitz T, Bedorf J, Nickenig G, Werner N (2013) High glucose condition increases NADPH oxidase activity in endothelial microparticles that promote vascular inflammation. Cardiovasc Res 98:94–106
Kim MH, Kang HM, Kim CE, Han S, Kim SW (2015) Ramipril inhibits high glucose-stimulated up-regulation of adhesion molecules via the ERK1/2 MAPK signaling pathway in human umbilical vein endothelial cells. Cell Mol Biol Lett 20:937–947
Koga H, Sugiyama S, Kugiyama K, Watanabe K, Fukushima H, Tanaka T, Sakamoto T, Yoshimura M, Jinnouchi H, Ogawa H (2005) Elevated levels of VE-cadherin-positive endothelial microparticles in patients with type 2 diabetes mellitus and coronary artery disease. J Am Coll Cardiol 45:1622–1630
Labiós M, Martínez M, Gabriel F, Guiral V, Munoz A, Aznar J (2004) Effect of eprosartan on cytoplasmic free calcium mobilization, platelet activation, and microparticle formation in hypertension. Am J Hypertens 17:757–763
Landmesser U, Hornig B, Drexler H (2004) Endothelial function: a critical determinant in atherosclerosis? Circulation 109:II27–II33
Lawson C, Vicencio JM, Yellon DM, Davidson SM (2016) Microvesicles and exosomes: new players in metabolic and cardiovascular disease. J Endocrinol 228:R57–R71
Leroyer AS, Anfosso F, Lacroix R, Sabatier F, Simoncini S, Njock SM, Jourde N, Brunet P, Camoin-Jau L, Sampol J, Dignat-George F (2010) Endothelial-derived microparticles: biological conveyors at the crossroad of inflammation, thrombosis and angiogenesis. Thromb Haemost 104:456–463
Li H, Brodsky S, Kumari S, Valiunas V, Brink P, Kaide J, Nasjletti A, Goligorsky MS (2002) Paradoxical overexpression and translocation of connexin43 in homocysteine-treated endothelial cells. Am J Physiol Heart Circ Physiol 282:H2124–H2133
Liu T, Shen D, Xing S, Chen JZ, Yu Z, Wang J, Wu B, Chi H, Zhao H, Liang Z, Chen C (2013) Attenuation of exogenous angiotensin II stress-induced damage and apoptosis in human vascular endothelial cells via microRNA-155 expression. Int J Mol Med 31:188–196
Lopes-Virella MF, Virella G (1992) Immune mechanisms of atherosclerosis in diabetes mellitus. Diabetes 41:86–91
Mallat Z, Benamer H, Hugel B, Benessiano J, Steg PG, Freyssinet JM, Tedgui A (2000) Elevated levels of shed membrane microparticles with procoagulant potential in the peripheral circulating blood of patients with acute coronary syndromes. Circulation 101:841–843
Matsumoto T, Goulopoulou S, Taguchi K, Tostes RC, Kobayashi T (2015) Constrictor prostanoids and uridine adenosine tetraphosphate: vascular mediators and therapeutic targets in hypertension and diabetes. Br J Pharmacol 172:3980–4001
Michel T, Feron O (1997) Nitric oxide synthases: which, where, how, and why? J Clin Invest 100:2146–2152
Mitchell MD, Peiris HN, Kobayashi M, Koh YQ, Duncombe G, Illanes SE, Rice GE, Salomon C (2015) Placental exosomes in normal and complicated pregnancy. Am J Obstet Gynecol 213:S173–S181
Nomura S, Shouzu A, Omoto S, Nishikawa M, Fukuhara S, Iwasaka T (2006) Effect of valsartan on monocyte/endothelial cell activation markers and adiponectin in hypertensive patients with type 2 diabetes mellitus. Thromb Res 117:385–392
Nomura S, Suzuki M, Katsura K, Xie GL, Miyazaki Y, Miyake T, Kido H, Kagawa H, Fukuhara S (1995) Platelet-derived microparticles may influence the development of atherosclerosis in diabetes mellitus. Atherosclerosis 116:235–240
Paneni F, Beckman JA, Creager MA, Cosentino F (2013) Diabetes and vascular disease: pathophysiology, clinical consequences, and medical therapy: part I. Eur Heart J 34:2436–2443
Paudel KR, Panth N, Kim DW (2016) Circulating endothelial microparticles: a key hallmark of atherosclerosis progression. Scientifica (Cairo) 2016:8514056
Pugh JA, Medina RA, Cornell JC, Basu S (1995) NIDDM is the major cause of diabetic end-stage renal disease: more evidence from a tri-ethnic community. Diabetes 44:1375–1380
Rice GE, Scholz-Romero K, Sweeney E, Peiris H, Kobayashi M, Duncombe G, Mitchell MD, Salomon C (2015) The effect of glucose on the release and bioactivity of exosomes from first trimester trophoblast cells. J Clin Endocrinol Metab 100:E1280–E1288
Sabatier F, Darmon P, Hugel B, Combes V, Sanmarco M, Velut JG, Arnoux D, Charpiot P, Freyssinet JM, Oliver C, Sampol J, Dignat-George F (2002) Type 1 and type 2 diabetic patients display different patterns of cellular microparticles. Diabetes 51:2840–2845
Savina A, Furlán M, Vidal M, Colombo MI (2003) Exosome release is regulated by a calcium-dependent mechanism in K562 cells. J Biol Chem 278:20083–20090
Tabit CE, Chung WB, Hamburg NM, Vita JA (2010) Endothelial dysfunction in diabetes mellitus: molecular mechanisms and clinical implications. Rev Endocr Metab Disord 11:61–74
Taguchi K, Matsumoto T, Kamata K, Kobayashi T (2012) G protein-coupled receptor kinase 2, with β-arrestin 2, impairs insulin-induced Akt/endothelial nitric oxide synthase signaling in ob/ob mouse aorta. Diabetes 61:1978–1985
Taguchi K, Morishige A, Matsumoto T, Kamata K, Kobayashi T (2012) Enhanced estradiol-induced vasorelaxation in aortas from type 2 diabetic mice may reflect a compensatory role of p38 MAPK-mediated eNOS activation. Pflugers Arch 464:205–215
Théry C, Ostrowski M, Segura E (2009) Membrane vesicles as conveyors of immune responses. Nat Rev Immunol 9:581–593
Uusitupa MI, Niskanen LK, Siitonen O, Voutilainen E, Pyörälä K (1993) Ten-year cardiovascular mortality in relation to risk factors and abnormalities in lipoprotein composition in type 2 (non-insulin-dependent) diabetic and non-diabetic subjects. Diabetologia 36:1175–1184
Von der Leyen HE, Gibbons GH, Morishita R, Lewis NP, Zhang L, Nakajima M, Kaneda Y, Cooke JP, Dzau VJ (1995) Gene therapy inhibiting neointimal vascular lesion: in vivo transfer of endothelial cell nitric oxide synthase gene. Proc Natl Acad Sci U S A 92:1137–1141
Wang Y, Chen LM, Liu ML (2014) Microvesicles and diabetic complications—novel mediators, potential biomarkers and therapeutic targets. Acta Pharmacol Sin 35:433–443
Watanabe S, Matsumoto T, Ando M, Adachi T, Kobayashi S, Iguchi M, Takeuchi M, Taguchi K, Kobayashi T (2016) Multiple activation mechanisms of serotonin-mediated contraction in the carotid arteries obtained from spontaneously hypertensive rats. Pflugers Arch 468:1271–1282
Watanabe S, Matsumoto T, Oda M, Yamada K, Takagi J, Taguchi K, Kobayashi T (2016) Insulin augments serotonin-induced contraction via activation of the IR/PI3K/PDK1 pathway in the rat carotid artery. Pflugers Arch 468:667–677
Werner N, Wassmann S, Ahlers P, Kosiol S, Nickenig G (2006) Circulating CD31+/annexin V+ apoptotic microparticles correlate with coronary endothelial function in patients with coronary artery disease. Arterioscler Thromb Vasc Biol 26:112–116
Yin M, Loyer X, Boulanger CM (2015) Extracellular vesicles as new pharmacological targets to treat atherosclerosis. Eur J Pharmacol 763:90–103
Yuana Y, Sturk A, Nieuwland R (2013) Extracellular vesicles in physiological and pathological conditions. Blood Rev 27:31–39
Acknowledgments
This work was partly supported by JSPS KAKENHI Grant Nos. JP15K07975 (T.K.), JP15K21419 (K.T.) and JP26460107 (T. M). The authors would like to thank Enago (www.enago.jp) for the English language review.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
All experimental protocols were approved by the local ethics committee (the Hoshi University Animal Care and Use Committee).
Conflict of interest
None.
Rights and permissions
About this article

Cite this article
Taguchi, K., Hida, M., Narimatsu, H. et al. Glucose and angiotensin II-derived endothelial extracellular vesicles regulate endothelial dysfunction via ERK1/2 activation. Pflugers Arch - Eur J Physiol 469, 293–302 (2017). https://doi.org/10.1007/s00424-016-1926-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-016-1926-2