
Abstract
Hyperinsulinemia associated with type 2 diabetes may contribute to the development of vascular diseases. Although we recently reported that enhanced contractile responses to serotonin (5-hydroxytryptamine, 5-HT) are observed in the arteries of type 2 diabetes models, the causative factors and detailed signaling pathways involved remain unclear. The purpose of this study was to investigate whether high insulin would be an amplifier of 5-HT-induced contraction in rat carotid arteries and whether the contraction involves phosphoinositide 3-kinase (PI3K)/3-phosphoinositide-dependent protein kinase 1 (PDK1) signaling, an insulin-mediated signaling pathway. In rat carotid arteries organ-cultured with insulin (for 24 h), (1) the contractile responses to 5-HT were significantly greater (vs. vehicle), (2) the insulin-induced enhancement of 5-HT-induced contractions was largely suppressed by inhibitors of the insulin receptor (IR) (GSK1838705A), PI3K (LY294002), and PDK1 (GSK2334470), and (3) the levels of phosphorylated forms of both PDK1 and myosin phosphatase target subunit 1 (MYPT1) were greater upon 5-HT stimulation. In addition, in rat carotid arteries organ-cultured with an activator of PDK1 (PS48), the 5-HT-induced contraction was greater, and this was suppressed by PDK1 inhibition but not PI3K inhibition. In addition, MYPT1 and PDK1 phosphorylation upon 5-HT stimulation was enhanced (vs. vehicle). These results suggest that high insulin levels amplify 5-HT-induced contraction. Moreover, the present results indicated the direct linkage between IR/PI3K/PDK1 activation and 5-HT-induced contraction in rat carotid arteries for the first time.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The prevalence of diabetes, particularly type 2 diabetes, has emerged as a significant problem worldwide in recent years. Although type 2 diabetes is remarkably associated with an increased incidence of vascular complications [5, 11, 13, 18, 31, 52, 53], the exact relationship between type 2 diabetes and vascular disease is not completely understood because multiple factors are involved in the development of these phenomena. Therefore, it is important to investigate and understand causal factors for altered vascular functions to prevent development of vascular complications in type 2 diabetes.
Serotonin (5-hydroxytryptamine, 5-HT) is a potent vasoactive amine that is considered to play pivotal roles in the physiological control of vascular tone and blood pressure as well as in the genesis and development of cardiovascular diseases such as atherosclerosis and hypertension [14, 22, 23, 55, 66, 67]. Several reports suggest a role of 5-HT in the pathogenesis of diabetic vascular complications [16, 21, 44, 48]. For instance, we recently found that the 5-HT-induced contractions were increased in superior mesenteric arteries of type 2 diabetic ob/ob mice [36] and in the carotid arteries of type 2 diabetic Goto-Kakizaki (GK) rats [40]. Although responsiveness to 5-HT is altered in chronic type 2 diabetes, the causal factors of alterations in 5-HT signaling remain unclear.
It is a well-established theory that one of the important causative factors related to diabetic vascular dysfunction may be associated with hyperinsulinemia and insulin resistance [4, 5, 24, 61]. In vascular cells, including endothelial cells (ECs) and smooth muscle cells (VSMCs), various reports suggest that intracellular insulin signaling is altered under hyperinsulinemia and insulin resistance [4, 5, 24, 61, 62]. Indeed, several reports suggest that insulin resistance plays a key role in the development of hypertension and impairment of endothelium-dependent relaxation observed in insulin-receptor substrate (IRS)-1- or IRS-2-deficient mice [1, 30]. A major feature of insulin resistance in vascular cells is the specific impairment of insulin-induced IRS/phosphoinositide 3-kinase (PI3K) signaling with alteration of insulin signaling through mitogen-activated protein kinase (MAPK) and other growth pathways [5, 24, 61]. Previously, we reported that the coexistence of a high insulin level and established diabetes led to (1) excessive peroxynitrite generation and resulted in impaired endothelium-dependent relaxation in aortae [29] and (2) increased endothelin-1-induced aortic contraction via enhanced ETA receptor/extracellular-signal-regulated kinase (ERK) signaling [27]. Therefore, insulin signaling could modulate arterial contractile responses to endogenous ligands; however, the mechanism by which signaling between insulin and 5-HT is integrated remains elusive.
Among kinases known to be involved in insulin signaling, there is an emerging body of evidence suggesting that 3-phosphoinositide-dependent protein kinase 1 (PDK1), which is cytoplasmic membrane-associated enzyme activated by PI3K [2, 10, 54, 64, 69], plays a pivotal regulatory role in many cellular processes and signaling pathways including cell survival, growth, proliferation, and metabolism [15, 20, 59, 69, 70]. PDK1 activates a group of protein kinases belonging to the protein kinase A (PKA)/PKG/PKC kinase family that plays important roles in mediating diverse biological processes including vascular function [3, 6, 42, 69]. Indeed, Chen et al. [9] recently reported that PDK1 regulates platelet activation and arterial thrombosis. Tawaramoto et al. [63] found that EC-specific deletion of PDK1 enhances insulin sensitivity via reducing visceral fat and suppressing angiogenesis. In VSMCs, Weber et al. [68] found that platelet-derived growth factor (PDGF)-induced migration is reactive oxygen species (ROS)-dependent and the Src/PDK1/p21-activated protein kinase1 pathway contributes to ROS-sensitive migration. However, uncertainty surrounds the mechanisms by which PDK1 might contribute to vascular contractile response.
In the present study, we hypothesized that exposure to high insulin levels would amplify 5-HT-induced contraction in the carotid arteries of rats. Furthermore, we postulated that PDK1 might be involved in such alterations. To investigate our hypothesis, we used organ culture of the entire vascular wall [25, 26, 29, 51] because in this way, it is possible to incubate the vessel with a constant concentration of insulin over a prolonged period.
Methods
Reagents
The reagent sources were as follows: insulin, 5-HT, and monoclonal β-actin antibody (Sigma Chemical Co, St. Louis, MO, USA); (3S,6R)-1-[6-(3-amino-1H-indazol-6-yl)-2-(methylamino)-4-pyrimidinyl]-N-cyclohexyl-6-methyl-3-piperidinecarboxamide (GSK2334470) and (2Z)-5-(4-chlorophenyl)-3-phenyl-2-pentenoic acid (PS48) (Tocris Bioscience, Bristol, UK); U46619 and 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002) (Cayman Chemical, Ann Arbor, MI, USA); 2-(2-(1-(2-(dimethylamino)acetyl)-5-methoxyindolin-6-ylamino)-7H pyrrolo[2,3-d] pyrimidin-4-ylamino)-6-fluoro-N-methylbenzamide (GSK1838705A) (AdooQ Bioscience, Irvine, CA, USA); and dimethyl sulfoxide (DMSO) (Wako Pure Chemical Industries, Osaka, Japan). The antibody sources were as follows: phospho-PDK1 (Ser241), PDK1, and insulin receptor β subunit (IR β) (Cell Signaling, Beverly, CA, USA); p-MYPT1 (Thr853) (Santa Cruz Biotechnology, Santa Cruz, CA, USA); 5-HT2A receptor (ImmunoStar, Hudson, WI, USA); and MYPT1, ROCK1, and ROCK2 (BD Biosciences, San Jose, CA, USA).
Animals and experimental design
Male Wistar rats were obtained at the age of 4–8 weeks (JLA, INC., Tokyo, Japan). All animals were allowed a standard laboratory diet (MF; Oriental Yeast Industry, Tokyo, Japan) and water ad libitum in a controlled environment (room temperature 21–22 °C, humidity 50 ± 5 %) until the rats were 9–17 weeks old (body weight 0.3–0.6 kg). This study was approved by the Hoshi University Animal Care and Use Committee, and all studies were conducted in accordance with “Guide for the Care and Use of Laboratory Animals” adopted by the Committee on the Care and Use of Laboratory Animals of Hoshi University (which is accredited by the Ministry of Education, Culture, Sports, Science, and Technology, Japan).
Arterial isolation and organ culture procedure
In all experiments, non-fasted rats were anesthetized with isoflurane (initially at 5 % and then maintained at 2.5 %) via a nose cone for surgical procedures and euthanized by thoracotomy and exsanguination via cardiac puncture. After euthanasia, common carotid arteries (diameter approx. 1 mm) were isolated under sterile conditions and placed in an ice-cold, oxygenated, modified Krebs-Henseleit solution (KHS). Each artery was carefully cleaned and cut into rings. Some segments were placed in 300 μl of low-glucose (5.5 mM) or high-glucose (25 mM) Dulbecco’s modified Eagle medium (DMEM: Gibco BRL, Grand Island, NY, USA) supplemented with 1 % penicillin streptomycin (Gibco BRL) and 1 % fetal bovine serum (FBS: Biological Industries, Kibbutz Beit Kaemek, Israel) in the absence or presence of insulin or PS48 (a PDK1 activator [19]). To investigate the effects of various signaling pathways on prolonged insulin treatment in carotid arteries, a given ring was incubated for 30 min in the appropriate drug-containing DMEM (viz., 1 × 10−6 M GSK1838705A [an IR inhibitor [56]], 1 × 10−5 M LY294002 [a PI3K inhibitor [40]], or 1 × 10−5 M GSK2334470 [PDK1 inhibitor [42]]), before insulin treatment, remaining present thereafter. Also, some rings treated these inhibitors alone (without insulin treatment). They were maintained at 37 °C in an atmosphere of 95 % air and 5 % CO2 for approximately 24 h.
Measurement of isometric force
Vascular isometric force was recorded as described in our previous papers [39, 40]. After organ culture, the arterial rings were placed in oxygenated, modified KHS. This solution consisted of (in mM) 118.0 NaCl, 4.7 KCl, 25.0 NaHCO3, 1.8 CaCl2, 1.2 NaH2PO4, 1.2 MgSO4, and 11.0 glucose. Isotonic high K+ solution was prepared by replacing NaCl with KCl. Ring segments (2 mm in length) were suspended via a pair of stainless steel pins in a well-oxygenated (95 % O2–5 % CO2) bath containing 5 ml of KHS at 37 °C. The rings were stretched until an optimal resting tension of 1.0 g was loaded and then allowed to equilibrate for at least 45 min. After stabilization, the contractile response to 80 mM KCl was measured. Force generation was monitored using an isometric transducer (model TB-611T; Nihon Kohden, Tokyo, Japan). For the contraction studies, 5-HT (1 × 10−9–3 × 10−5 M) or U46619 (10−10–10−7.5 M) was added cumulatively to the bath until a maximal response was achieved.
Western blotting
Each carotid arterial ring was cultured with insulin (1 × 10−6 M) or vehicle (0.0001 N HCl) and PS48 (3 × 10−4 M) or vehicle (DMSO). After organ culture, the arterial rings were suspended via a pair of stainless steel pins in a well-oxygenated (95 % O2–5 % CO2) bath containing 5 ml of KHS at 37 °C, and then 3 × 10−5 M 5-HT was applied for 5 min. Next, carotid arterial rings were washed with ice-cold Ca2+-free solution containing sodium orthovanadate (1 × 10−3 M) and EDTA (5 × 10−3 M) and rapidly removed, after which they were freeze-clamped in liquid nitrogen and stored at −80 °C for Western blotting. For measurements of IRβ and 5-HT2A receptors, protein samples were obtained from freshly isolated carotid arteries and cultured with insulin (1 × 10−6 M) or vehicle (0.01 N HCl) for 24 h. After organ culture, carotid arterial rings were rapidly removed, and then, they were freeze-clamped in liquid nitrogen and stored at −80 °C for Western blotting. The protein levels of PDK1, phosphorylated PDK1, MYPT1, phosphorylated MYPT1, ROCK1, ROCK2, IRβ, and 5-HT2A receptor were quantified using immunoblotting procedures, essentially as described previously [34, 38–40]. Carotid arterial protein extracts (20 μg/lane) were applied to 7.5 or 10 % SDS-PAGE and transferred to polyvinylidene difluoride membranes. Blots were incubated with anti-PDK1 (58–68 kDa; 1:1000), anti-phospho-PDK1 (Ser241) (58–68 kDa; 1:1000), anti-MYPT1 (130 kDa; 1:1000), anti-phospho-MYPT1 (Thr853) (130 kDa; 1:200), anti-ROCK1 (∼160 kDa; 1:500), anti-ROCK2 (∼160 kDa; 1:500), anti-IRβ (95 kDa; 1:500), anti-5-HT2A receptor (53 kDa; 1:1000), and anti-β-actin (42 kDa; 1:5000) antibodies, with detection being achieved using a horseradish peroxidase-conjugated IgG followed by enhanced chemiluminescence. The resulting bands were analyzed using CS Analyzer 3.0 software (ATTO, Tokyo, Japan). The phosphorylation levels of PDK1 and MYPT1 were normalized by total PDK1 and total MYPT1, respectively, and then expressed as fold increase (relative to vehicle). The protein expressions of ROCK1, ROCK2, IRβ, and 5-HT2A receptors were normalized by β-actin and then expressed as fold increase (relative to vehicle).
Statistical analysis
The contractile force exerted by carotid arterial rings is expressed as a percentage of the 80-mM KCl-induced contraction. Concentration-response curves with agonists were fitted using a nonlinear interactive fitting program (GraphPad Prism 5.0; GraphPad Software Inc., San Diego, CA, USA). Data are expressed as the mean ± SE. Statistical evaluations were performed using Student’s t test for comparisons between two groups. Statistical analysis of the values of E max (the maximal effect generated by 5-HT) was performed using one-way ANOVA with Bonferroni’s post hoc test. Statistical evaluations of concentration-response curves were performed using two-way ANOVA with repeated measures followed by Bonferroni’s post hoc test. Values of P < 0.05 were considered significant.
Results
Effects of prolonged treatment with insulin on 5-HT-induced contractile responses
We firstly examined the effects of high concentration of insulin or glucose on 5-HT (1 × 10−7–3 × 10−5 M)-induced contraction. Exposure of organ-cultured carotid artery rings to 5-HT led to a concentration-dependent rise in tension in both insulin- (1 × 10−7 or 1 × 10−6 M) and vehicle-treated arteries, although the contractile force of 5-HT was greater in insulin-treated arteries than in the vehicle-treated arteries in both normal (Fig. 1a) and high glucose (Fig. 1b) conditions. The E max values of 5-HT-induced contractile response were significantly increased in insulin (1 × 10−6 M)-treated arteries in both normal and high glucose conditions (Fig. 1c). The reference contractions induced by 80 mM KCl were similar among all groups (Fig. 1d). On the other hand, the contractile force of another constrictor, U46619, was similar between insulin (1 × 10−7 M)-treated and vehicle-treated arteries under conditions of normal glucose (data not shown). These results suggested that high insulin but not high glucose could enhance 5-HT-induced contraction in rat carotid arteries. As glucose did not influence 5-HT-induced contraction in organ-cultured carotid arteries, all subsequent experiments were conducted in normal glucose DMEM for organ culture.

Effects of insulin treatment on 5-hydroxytryptamine (5-HT)-induced contraction in organ-cultured rat carotid arteries. Carotid arteries were preincubated with vehicle (0.0001 N HCl) or insulin (1 × 10−7 or 1 × 10−6 M) in normal- (5.5 mM glucose) (a) or high-glucose (25 mM) Dulbecco’s modified Eagle’s medium (b). E max of 5-HT-induced contraction (c). d The 80 mM KCl-induced contraction in carotid arteries cultured with vehicle or insulin in normal or high-glucose medium. Each data-point represents the mean ± SE from five experiments. a, b *P < 0.05, ***P < 0.001, vehicle vs. insulin 1 × 10−7 M, ### P < 0.001, vehicle vs. insulin 1 × 10−6 M. c *P < 0.05, vehicle vs. insulin 1 × 10−6 M in normal-glucose medium. # P < 0.05, vehicle vs. insulin 1 × 10−6 M in high-glucose medium
Effects of a selective IR antagonist and PI3K inhibitor on the insulin-induced enhancement of 5-HT-induced contraction
Because insulin signals were reported to involve the IR/IRS/PI3K pathway [43, 45, 50], in the second series of experiments, we examined whether 5-HT-induced contraction augmented by insulin could be suppressed by IR and PI3K inhibitors (Fig. 2). Cotreatment with the IR antagonist GSK1838705A (1 × 10−6 M) (Fig. 2a) or PI3K inhibitor LY294002 (1 × 10−5 M) (Fig. 2b) together with insulin (1 × 10−7 M) suppressed 5-HT-induced contraction in organ-cultured carotid arteries.

Effect of an insulin receptor (IR) antagonist, phosphoinositide 3-kinase (PI3K) inhibitor, or 3-phosphoinositide-dependent protein kinase 1 (PDK1) inhibitor on the enhancement of 5-hydroxytryptamine (5-HT)-induced contraction in organ-cultured rat carotid arteries exposed to insulin. Concentration-response curves for 5-HT-induced contraction in insulin (1 × 10−7 M for 24 h)-treated carotid arteries in the absence or presence of GSK1838705A (IR inhibitor, 1 × 10−6 M) (a), LY294002 (PI3K inhibitor, 1 × 10−5 M) (b), or GSK2334470 (PDK1 inhibitor, 1 × 10−5 M). d Concentration-response curves for 5-HT-induced contraction in carotid arterial rings treated with vehicle (DMSO), GSK1838705A (1 × 10−6 M), LY294002 (1 × 10−5 M), or GSK2334470 (1 × 10−5 M) for 22–24 h. Data are the means ± SE from seven (a), nine (b, c), or six (d) experiments: **P < 0.01, ***P < 0.001 vs. vehicle
Effects of a selective PDK1 inhibitor on 5-HT-induced contraction enhanced by insulin
PDK1 was reported to be an immediate downstream effector of PI3K and a master kinase in various responses [2, 10, 54, 64, 69]. Next, to continue our investigation of the role of PDK1 on 5-HT-induced contraction augmented by insulin, we assessed the effect of PDK1 inhibition on the 5-HT-induced contraction of cultured carotid arteries in the presence of insulin (Fig. 2c). Treatment with the PDK1 inhibitor GSK2334470 (1 × 10−5 M) suppressed 5-HT-induced contraction in organ-cultured carotid artery in the presence of insulin (1 × 10−7 M).
When carotid rings were treated with each inhibitor alone (without insulin treatment) for 24 h, GSK1838705A (1 × 10−6 M), LY294002 (1 × 10−5 M), or GSK2334470 (1 × 10−5 M) did not influence 5-HT-induced contractions (Fig. 2d).
Evaluations of PDK1, MYPT1, ROCK1, and ROCK2 protein expression in the absence or presence of insulin in cultured carotid arteries stimulated with 5-HT
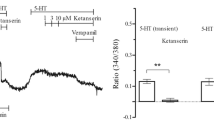
Using pharmacological approaches, enhanced 5-HT-induced contraction was observed in insulin-treated arteries, and this was suppressed by inhibiting IR/PI3K/PDK1 signaling. To investigate the possible mechanisms underlying the alterations in 5-HT-induced contraction in organ-cultured carotid arteries in the presence of high insulin concentrations, we investigated whether the activity of PDK1 and MYPT1, which is the regulatory subunit of myosin light chain phosphatase and the phosphorylation of which at Thr853 stimulated by constrictors in a Rho kinase-dependent manner, affects contractile responses [17, 47, 60, 65]. Moreover, we assessed that the expression of ROCK1/2 was altered in carotid arteries subjected to prolonged insulin treatment with 5-HT stimulation. The 5-HT-stimulated carotid arterial expression of phosphorylated PDK1 (Fig. 3a) and MYPT1 (Fig. 3b) was significantly greater in the insulin-treated group (vs. vehicle). Conversely, the protein expression of ROCK1 (Fig. 4a) and ROCK2 (Fig. 4b) was not significantly different between insulin and vehicle treatment.

Effects of prolonged treatment with insulin on the phosphorylation of 3-phosphoinositide-dependent protein kinase 1 (PDK1) and myosin phosphatase target subunit 1 (MYPT1) in organ-cultured rat carotid arteries. Western blots for p-PDK1/PDK1 (a) and p-MYPT1/MYPT1 (b) in organ-cultured carotid arteries exposed to insulin (1 × 10−6 M). After organ-cultured carotid arteries were stimulated with 5-hydroxytryptamine (3 × 10−5 M) for 5 min. Representative Western blots (upper panels). Bands were quantified as described in the “Methods” (lower panels). Results were shown as the fold increase relative to control (vehicle). Equal protein loading was confirmed using β-actin antibody. Each column represents the mean ± SE from six (a) or eight (b) experiments. *P < 0.05, **P < 0.01 vs. vehicle

Effects of prolonged treatment with insulin on the expression of ROCK in organ-cultured rat carotid arteries. Western blots for ROCKs in organ-cultured carotid arteries exposed to insulin (1 × 10−6 M for 24 h). After organ-cultured carotid arteries were stimulated with 5-hydroxytryptamine (3 × 10−5 M) for 5 min. a Representative Western blots. b, c Bands of ROCK1 (b) and ROCK2 (c) were quantified as described in the “Methods.” Results are shown as the fold increase relative to control (vehicle). Each column represents the mean ± SE from six experiments
Effects of prolonged PDK1 activation on 5-HT-induced contraction and the activities of PDK1 and MYPT1 in organ-cultured carotid arteries
Next, to assess the direct relationship between PDK1 and contraction in response to 5-HT in carotid arteries, we investigated the effect of prolonged PDK1 activator treatment on 5-HT-induced contraction (Fig. 5) and the phosphorylation of MYPT1 and PDK1 (Fig. 6). Prolonged treatment with a PDK1 activator (PS48; 3 × 10−4 M for 24 h) [19, 58] enhanced 5-HT-induced contraction (Fig. 5a). This enhancement of 5-HT-induced contraction induced by PS48 was suppressed by pretreatment with GSK2334470 (1 × 10−5 M) but not by LY294002 (1 × 10−5 M) (Fig. 5b). Moreover, the 5-HT-induced contraction in carotid arteries cultured with (PS48 3 × 10−4 M) was not further modified by cotreatment with insulin (1 × 10−7 M) (Fig. 5c). The phosphorylation of MYPT1 upon 5-HT stimulation was significantly greater in PS48-treated arteries than in vehicle-treated arteries (Fig. 6a). Predictably, the expression of phosphorylated PDK1 was significantly greater in the PS48-treated group (vs. vehicle) (Fig. 6b).

Prolonged treatment with a 3-phosphoinositide-dependent protein kinase 1 (PDK1) activator augments 5-hydroxytryptamine (5-HT)-induced contraction in rat carotid arteries. Effects of a PDK1 activator (PS48: 3 × 10−4 M for 24 h) on 5-HT-induced contraction in organ-cultured rat carotid arteries (a). Effects of GSK2334470 (1 × 10−5 M) and LY294002 (1 × 10−5 M) on the concentration-response curves for 5-HT in the presence of PS48 (3 × 10−4 M) in organ-cultured rat carotid arteries (b). c Effects of coincubation with PS48 (3 × 10−4 M) and insulin (1 × 10−7 M) for 22–24 h on the 5-HT-induced contraction. The data represent the mean ± SE from four to six experiments. a **P < 0.01, ***P < 0.001 vs. vehicle. b ***P < 0.001, vehicle vs. GSK2334470

Effects of prolonged treatment with PS48 (3 × 10−4 M) on the phosphorylation of myosin phosphatase target subunit 1 (MYPT1) and 3-phosphoinositide-dependent protein kinase 1 (PDK1) in organ-cultured rat carotid arteries stimulated with 5-hydroxytryptamine (3 × 10−5 M for 5 min). Representative Western blots for p-MYPT1/MYPT1 (a) and p-PDK1/PDK1 (b) (upper panels). Bands were quantified as described in the “Methods” (lower panels). Results are shown as the fold increase relative to control (vehicle). Equal protein loading was confirmed using β-actin antibody. Each column represents the mean ± SE from eight experiments. *P < 0.05 vs. vehicle
Expression of IRβ and 5-HT2A receptors
Finally, to investigate the possible mechanisms underlying the above amplifier effects of insulin on 5-HT-induced contractions in carotid arteries, we finally examined the protein expression of the IRβ and 5-HT2A receptors (Fig. 7). Although the protein expressions of IRβ were not significantly altered between fresh and vehicle-cultured carotid arteries, the protein expressions of IRβ were significantly decreased in the insulin (1 × 10−6 M)-treated group compared to vehicle-treated group (Fig. 7b) in cultured carotid arteries. Conversely, the protein expressions of the 5-HT2A receptor were similar for the three groups (Fig. 7c).

Western blots for IRβ and 5-HT2A receptor expressions in freshly isolated carotid arteries and vehicle- (0.01 N HCl) or insulin (1 × 10−6 M for 24 h)-cultured carotid arteries. a Representative Western blot is shown. b, c Corresponding densitometric analysis showing the expressions of IRβ (b) and 5-HT2A receptor (c). Results are shown as the fold increase relative to control (vehicle). Each column represents the mean ± SE from eight experiments. *P < 0.05 vs. vehicle
Discussion
In the present study, we examined whether high insulin concentrations cause increased vascular contractile responses to 5-HT. The major findings of the present study are that prolonged exposure to high insulin levels but not high glucose levels in rat carotid arteries enhances 5-HT-induced contraction and the increase in 5-HT-induced contraction induced by high insulin concentrations is due to the activation of IR, PI3K, and PDK1 pathways (Fig. 8). It was also found that PDK1 activation leads to increased 5-HT-induced contraction similarly as exposure to high insulin levels in rat carotid arteries.

Summary of the present results. Prolonged exposure of insulin increases 5-HT-induced contraction in rat carotid arteries via the activation of IR/PI3K/PDK1 pathway
Alterations of contractile responses to endogenous ligands were often observed in the arteries of long-term models of type 2 diabetes [33–35, 37–39, 46, 57]. Other researchers [12, 19, 44, 48] and our group [36, 40] found that 5-HT-induced contraction was increased in arteries from type 2 diabetic animal models. In addition, we suggested that the enhancement of 5-HT-induced contraction was attributable to various signal transduction pathways including Src, Rho kinase, MAPKs, and PI3K pathways in type 2 diabetic arteries [36, 40]. The major hallmarks of type 2 diabetes are hyperglycemia and hyperinsulinemia. In the present study, we illustrated that prolonged exposure to high insulin levels could augment 5-HT-induced contraction in rat carotid arteries, whereas high-glucose treatment did not influence this contraction. On the other hand, high insulin could not alter other constrictor U46619 (TP agonist)-induced contraction. Therefore, these results suggest that high insulin levels act as a specific amplifier of 5-HT-induced contraction and may speculate that insulin affects specific signal-transduction cascade to contract vascular smooth muscle upon stimulation of each G protein-coupled receptor.
Among the 5-HT receptor subtypes, it has been reported that 5-HT-induced arterial contractions were mainly mediated by 5-HT2A receptors [67]. There was an increase in arterial contractions in response to some endogenous agonists in vascular diseases which may be associated with increase in their receptor expression. For example, Edvinsson’s group clearly demonstrated that 5-HT-induced vasocontractions were associated with upregulation of 5-HT receptors (such as 5-HT2A), in mesenteric arteries using an organ culture system [7, 32]. In the present study, we found that the protein expression of 5-HT2A receptors did not change among freshly isolated and cultured (vehicle and insulin for 24 h) groups, suggesting that the extent of altered receptor expression (via upregulation and downregulation) did not influence the enhancement effects of insulin on 5-HT-induced contractions. Moreover, we and others previously observed that the increase in 5-HT-induced contractions in diabetic arteries was attributable to an increase in intracellular signaling rather than its receptor expression [36, 40, 48]. Therefore, we speculate that the site(s) of action resulted in enhancement of 5-HT-induced contraction by insulin that may be related to downstream molecules.
It was reported that insulin acts on various target organs including the vasculature through the activation of IR and subsequently activates multiple signaling pathways [41]. Hyperstimulation of the IR alters the balance of these signaling pathways such as suppressing PI3K/Akt signaling and boosting MAPK signaling [24]. In the present study, the enhancement of 5-HT-induced contraction induced by prolonged insulin exposure was reduced by pretreatment with an IR antagonist and PI3K inhibitor. It was reported previously that PDK1 is one of the major downstream targets of PI3K upon insulin stimulation [2, 10, 28, 69]. We found that in carotid arteries subjected to prolonged exposure to insulin, (1) 5-HT-induced contraction was suppressed by pretreatment with a PDK1 inhibitor, and (2) the phosphorylation (activation) of PDK1 upon 5-HT stimulation was increased. Moreover, prolonged exposure to a PDK1 activator increased 5-HT-induced carotid arterial contraction similarly as high insulin levels. In addition, pretreatment with a PI3K inhibitor did not influence 5-HT-induced contraction in carotid arteries subjected to prolonged exposure to a PDK1 activator. Finally, no additive augmentation in 5-HT-induced contraction was seen in cotreatment with insulin and a PDK1 activator. These results strongly suggest that the IR/PI3K/PDK1 pathway contributes to increased 5-HT-induced contraction.
PDK1 has been implicated as a major hub of multiple signaling cascades in the regulation of various cellular processes [2, 6, 10, 15, 54, 64, 69]. In the present study, we focused on the relationship between PDK1 and MYPT1 phosphorylation because phosphorylation of MYPT1 at Thr853 by Rho kinase is a key process of vascular smooth muscle contraction [17, 47, 60, 65]. A novel, intriguing, and potentially important finding of the present study was that the linkage between PDK1 and MYPT1 phosphorylation is functionally present in rat carotid arteries. In the present study, we found that 5-HT-induced MYPT1 phosphorylation at Thr853 was increased by prolonged treatment with a PDK1 activator as well as high insulin exposure and treatment with insulin did not affect the expression of ROCK1 and ROCK2 upon 5-HT stimulation. These results suggest that the increased MYPT1 phosphorylation induced by insulin (and PDK1 activation) is independent of ROCK expression. There are few reports suggesting an interaction between PDK1 and Rho kinase. The colocalization of PDK1 and ROCK1 at the cell membrane and sustained RhoA/ROCK1 activation was observed in colorectal cancer cells [8]. Okada et al. [49] observed that chemotaxis induced by PDGF-D is mediated by the activation of the PDGF-ββ receptor/PI3K/PDK1/Akt/Rac1/ROCK pathway in malignant mesothelioma cells. Further investigations will be required on these points in the vasculature.
In conclusion, in the present study, we demonstrated that prolonged exposure of rat carotid arteries to high insulin levels enhanced 5-HT-induced contraction. Moreover, we for the first time demonstrated that PDK1 activation promotes 5-HT-induced vascular contraction. Further studies on PDK1 might contribute to the development of new pharmaceutical therapies for the prevention of type 2 diabetes-associated vascular diseases.
References
Abe H, Yamada N, Kamata K, Kuwaki T, Shimada M, Osuga J, Shionoiri F, Yahagi N, Kadowaki T, Tamemoto H, Ishibashi S, Yazaki Y, Makuuchi M (1998) Hypertension, hypertriglyceridemia, and impaired endothelium-dependent vascular dysfunction in mice lacking insulin receptor substrate-1. J Clin Invest 101:1784–1788
Alessi DR (2001) Discovery of PDK1, one of the missing links in insulin signal transduction. Colworth Medal Lecture. Biochem Soc Trans 29:1–14
Arencibia JM, Pastor-Flores D, Bauer AF, Schulze JO, Biondi RM (2013) AGC protein kinases: from structural mechanism of regulation to allosteric drug development for the treatment of human diseases. Biochim Biophys Acta 1834:1302–1321
Bender SB, Klabunde RE (2007) Altered role of smooth muscle endothelin receptors in coronary endothelin-1 and alpha1-adrenoceptor-mediated vasoconstriction in Type 2 diabetes. Am J Physiol Heart Circ Physiol 293:H2281–H2288
Bender SB, McGraw AP, Jaffe IZ, Sowers JR (2013) Mineralocorticoid receptor-mediated vascular insulin resistance: an early contributor to diabetes-related vascular disease? Diabetes 62:313–319
Calleja V, Laguerre M, de Las H-MG, Parker PJ, Requejo-Isidro J, Larijani B (2014) Acute regulation of PDK1 by a complex interplay of molecular switches. Biochem Soc Trans 42:1435–1440
Cao YX, He LC, Xu CB, Luo GG, Edvinsson L (2005) Enhanced transcription of contractile 5-hydroxytryptamine 2A receptors via extracellular signal-regulated kinase 1/2 after organ culture of rat mesenteric artery. Basic Clin Pharmacol Toxicol 96:282–288
Cartier-Michaud A, Malo M, Charriere-Bertrand C, Gadea G, Anguille C, Supiramaniam A, Lesne A, Delaplace F, Hutzler G, Roux P, Lawrence DA, Barlovatz-Meimon G (2012) Matrix-bound PAI-1 supports cell blebbing via RhoA/ROCK1 signaling. PLoS One 7:e32204
Chen X, Zhang Y, Wang Y, Li D, Zhang L, Wang K, Luo X, Yang Z, Wu Y, Liu J (2013) PDK1 regulates platelet activation and arterial thrombosis. Blood 121:3718–3726
Cho JY, Park J (2008) Contribution of natural inhibitors to the understanding of the PI3K/PDK1/PKB pathway in the insulin-mediated intracellular signaling cascade. Int J Mol Sci 9:2217–2230
DeMarco VG, Aroor AR, Sowers JR (2014) The pathophysiology of hypertension in patients with obesity. Nat Rev Endocrinol 10:364–376
Didion SP, Lynch CM, Faraci FM (2007) Cerebral vascular dysfunction in TallyHo mice: a new model of Type II diabetes. Am J Physiol Heart Circ Physiol 292:H1579–H1583
Forbes JM, Cooper ME (2013) Mechanisms of diabetic complications. Physiol Rev 93:137–188
Fraer M, Kilic F (2015) Serotonin: a different player in hypertension-associated thrombosis. Hypertension 65:942–948
Gagliardi PA, di Blasio L, Primo L (2015) PDK1: a signaling hub for cell migration and tumor invasion. Biochem Biophys Acta 1856:178–188
Gamoh S, Hisa H, Yamamoto R (2013) 5-hydroxytryptamine receptors as targets for drug therapies of vascular-related diseases. Biol Pharm Bull 36:1410–1415
Goulopoulou S, Webb RC (2014) Symphony of vascular contraction: how smooth muscle lose harmony to signal increased vascular resistance in hypertension. Hypertension 63:e33–39
Guo Z, Su W, Allen S, Pang H, Daugherty A, Smart E, Gong MC (2005) COX-2 up-regulation and vascular smooth muscle contractile hyperreactivity in spontaneous diabetic db/db mice. Cardiovasc Res 67:723–735
Hindie V, Stroba A, Zhang H, Lopez-Garcia LA, Idrissova L, Zeuzem S, Hirschberg D, Schaeffer F, Jorgensen TJ, Engel M, Alzari PM, Biondi RM (2009) Structure and allosteric effects of low-molecular-weight activators on the protein kinase PDK1. Nat Chem Biol 5:758–764
Ito K, Akazawa H, Tamagawa M, Furukawa K, Ogawa W, Yasuda N, Kudo Y, Liao CH, Yamamoto R, Sato T, Molkentin JD, Kasuga M, Noda T, Nakaya H, Komuro I (2009) PDK1 coordinates survival pathways and beta-adrenergic response in the heart. Proc Natl Acad Sci U S A 106:8689–8694
Iwabayashi M, Taniyama Y, Sanada F, Azuma J, Iekushi K, Kusunoki H, Chatterjee A, Okayama K, Rakugi H, Morishita R (2012) Role of serotonin in angiogenesis: induction of angiogenesis by sarpogrelate via endothelial 5-HT1B/Akt/eNOS pathway in diabetic mice. Atherosclerosis 220:337–342
Kanagy NL, Webb RC (1996) Increased responsiveness and decreased expression of G proteins in deoxycorticosterone hypertension. Hypertension 27:740–745
Kaumann AJ, Levy FO (2006) 5-hydroxytryptamine receptors in the human cardiovascular system. Pharmacol Ther 111:674–706
Kim JA, Montagnani M, Koh KK, Quon MJ (2006) Reciprocal relationships between insulin resistance and endothelial dysfunction: molecular and pathophysiological mechanisms. Circulation 113:1888–1904
Kobayashi T, Kaneda A, Kamata K (2003) Possible involvement of IGF-1 receptor and IGF-binding protein in insulin-induced enhancement of noradrenaline response in diabetic rat aorta. Br J Pharmacol 140:285–294
Kobayashi T, Matsumoto T, Kamata K (2005) IGF-1-induced enhancement of contractile response in organ-cultured aortae from diabetic rats is mediated by sustained thromboxane A2 release from endothelial cells. J Endocrinol 186:367–376
Kobayashi T, Nogami T, Taguchi K, Matsumoto T, Kamata K (2008) Diabetic state, high plasma insulin and angiotensin II combine to augment endothelin-1-induced vasoconstriction via ETA receptors and ERK. Br J Pharmacol 155:974–983
Kobayashi T, Taguchi K, Nemoto S, Nogami T, Matsumoto T, Kamata K (2009) Activation of the PDK-1/Akt/eNOS pathway involved in aortic endothelial function differs between hyperinsulinemic and insulin-deficient diabetic rats. Am J Physiol Heart Circ Physiol 297:H1767–H1775
Kobayashi T, Taguchi K, Takenouchi Y, Matsumoto T, Kamata K (2007) Insulin-induced impairment via peroxynitrite production of endothelium-dependent relaxation and sarco/endoplasmic reticulum Ca(2+)-ATPase function in aortas from diabetic rats. Free Radic Biol Med 43:431–443
Kubota T, Kubota N, Moroi M, Terauchi Y, Kobayashi T, Kamata K, Suzuki R, Tobe K, Namiki A, Aizawa S, Nagai R, Kadowaki T, Yamaguchi T (2003) Lack of insulin receptor substrate-2 causes progressive neointima formation in response to vessel injury. Circulation 107:3073–3080
Laakso M, Kuusisto J (2014) Insulin resistance and hyperglycaemia in cardiovascular disease development. Nat Rev Endocrinol 10:293–302
Luo G, Xu CB, Cao YX, Edvinsson L (2004) Transcriptional up-regulation in expression of 5-hydroxytryptamine2A and transcriptional down-regulation of angiotensin II type 1 receptors during organ culture of rat mesenteric artery. Basic Clin Pharmacol Toxicol 95:280–287
Matsumoto T, Goulopoulou S, Taguchi K, Tostes RC, Kobayashi T (2015) Constrictor prostanoids and uridine adenosine tetraphosphate: vascular mediators and therapeutic targets in hypertension and diabetes. Br J Pharmacol 172:3980–4001
Matsumoto T, Ishida K, Nakayama N, Kobayashi T, Kamata K (2009) Involvement of NO and MEK/ERK pathway in enhancement of endothelin-1-induced mesenteric artery contraction in later-stage type 2 diabetic Goto-Kakizaki rat. Am J Physiol Heart Circ Physiol 296:H1388–H1397
Matsumoto T, Kakami M, Noguchi E, Kobayashi T, Kamata K (2007) Imbalance between endothelium-derived relaxing and contracting factors in mesenteric arteries from aged OLETF rats, a model of Type 2 diabetes. Am J Physiol Heart Circ Physiol 293:H1480–H1490
Matsumoto T, Kobayashi T, Ishida K, Taguchi K, Kamata K (2010) Enhancement of mesenteric artery contraction to 5-HT depends on Rho kinase and Src kinase pathways in the ob/ob mouse model of type 2 diabetes. Br J Pharmacol 160:1092–1104
Matsumoto T, Noguchi E, Ishida K, Kobayashi T, Yamada N, Kamata K (2008) Metformin normalizes endothelial function by suppressing vasoconstrictor prostanoids in mesenteric arteries from OLETF rats, a model of type 2 diabetes. Am J Physiol Heart Circ Physiol 295:H1165–H1176
Matsumoto T, Watanabe S, Kawamura R, Taguchi K, Kobayashi T (2014) Enhanced uridine adenosine tetraphosphate-induced contraction in renal artery from type 2 diabetic Goto-Kakizaki rats due to activated cyclooxygenase/thromboxane receptor axis. Pflugers Arch 466:331–342
Matsumoto T, Watanabe S, Kawamura R, Taguchi K, Kobayashi T (2014) Epigallocatechin gallate attenuates ET-1-induced contraction in carotid artery from type 2 diabetic OLETF rat at chronic stage of disease. Life Sci 118:200–205
Matsumoto T, Watanabe S, Taguchi K, Kobayashi T (2014) Mechanisms underlying increased serotonin-induced contraction in carotid arteries from chronic type 2 diabetic Goto-Kakizaki rats. Pharmacol Res 87:123–132
Muniyappa R, Montagnani M, Koh KK, Quon MJ (2007) Cardiovascular actions of insulin. Endocr Rev 28:463–491
Najafov A, Sommer EM, Axten JM, Deyoung MP, Alessi DR (2011) Characterization of GSK2334470, a novel and highly specific inhibitor of PDK1. Biochem J 433:357–369
Nakazawa T, Chiba T, Kaneko E, Yui K, Yoshida M, Shimokado K (2005) Insulin signaling in arteries prevents smooth muscle apoptosis. Arterioscler Thromb Vasc Biol 25:760–765
Nelson PM, Harrod JS, Lamping KG (2012) 5HT(2A) and 5HT(2B) receptors contribute to serotonin-induced vascular dysfunction in diabetes. Exp Diabetes Res 2012:398406
Nemoto S, Matsumoto T, Taguchi K, Kobayashi T (2014) Relationships among protein tyrosine phosphatase 1B, angiotensin II, and insulin-mediated aortic responses in type 2 diabetic Goto-Kakizaki rats. Atherosclerosis 233:64–71
Nemoto S, Taguchi K, Matsumoto T, Kamata K, Kobayashi T (2012) Pravastatin normalizes ET-1-induced contraction in the aorta of type 2 diabetic OLETF rats by suppressing the KSR1/ERK complex. Am J Physiol Heart Circ Physiol 303:H893–H902
Nunes KP, Rigsby CS, Webb RC (2010) RhoA/Rho-kinase and vascular diseases: what is the link? Cell Mol Life Sci 67:3823–3836
Nuno DW, Harrod JS, Lamping KG (2009) Sex-dependent differences in Rho activation contribute to contractile dysfunction in type 2 diabetic mice. Am J Physiol Heart Circ Physiol 297:H1469–1477
Okada A, Yaguchi T, Kanno T, Gotoh A, Nakano T, Nishizaki T (2012) PDGF-D/PDGF-ββ receptor-regulated chemotaxis of malignant mesothelioma cells. Cell Physiol Biochem 29:241–250
Olivares-Reyes JA, Arellano-Plancarte A, Castillo-Hernandez JR (2009) Angiotensin II and the development of insulin resistance: implications for diabetes. Mol Cell Endocrinol 302:128–139
Ozaki H, Karaki H (2002) Organ culture as a useful method for studying the biology of blood vessels and other smooth muscle tissues. Jpn J Pharmacol 89:93–100
Paneni F, Beckman JA, Creager MA, Cosentino F (2013) Diabetes and vascular disease: pathophysiology, clinical concequences, and medical therapy: part I. Eur Heart J 34:2436–2443
Porter KE, Riches K (2013) The vascular smooth muscle cell: a therapeutic target in Type 2 diabetes? Clin Sci 125:167–182
Pullen N, Dennis PB, Andjelkovic M, Dufner A, Kozma SC, Hemmings BA, Thomas S (1998) Phosphorylation and activation of p70s6k by PDK1. Science 279:707–710
Ramage AG, Villalon CM (2008) 5-hydroxytryptamine and cardiovascular regulation. Trends Pharmacol Sci 29:472–481
Sabbatini P, Korenchuk S, Rowand JL, Groy A, Liu Q, Leperi D, Atkins C, Dumble M, Yang J, Anderson K, Kruger RG, Gontarek RR, Maksimchuk KR, Suravajjala S, Lapierre RR, Shotwell JB, Wilson JW, Chamberlain SD, Rabindran SK, Kumar R (2009) GSK1838705A inhibits the insulin-like growth factor-1 receptor and anaplastic lymphoma kinase and shows antitumor activity in experimental models of human cancers. Mol Cancer Ther 8:2811–2820
Sachidanandam K, Elgebaly MM, Harris AK, Hutchinson JR, Mezzetti EM, Portik-Dobos V, Ergul A (2008) Effect of chronic and selective endothelin receptor antagonism on microvascular function in type 2 diabetes. Am J Physiol Heart Circ Physiol 294:H2743–H2749
Sadowsky JD, Burlingame MA, Wolan DW, McClendon CL, Jacobson MP, Wells JA (2011) Turning a protein kinase on or off from a single allosteric site via disulfide trapping. Proc Natl Acad Sci U S A 108:6056–6061
Scortegagna M, Lau E, Zhang T, Feng Y, Sereduk C, Yin H, De SK, Meeth K, Platt JT, Langdon CG, Halaban R, Pellecchia M, Davies MA, Brown K, Stern DF, Bosenberg M, Ronai ZA (2015) PDK1 and SGK3 contribute to the growth of BRAF-mutant Melanomas and are potential therapeutic targets. Cancer Res 75:1399–1412
Seok YM, Azam MA, Okamoto Y, Sato A, Yoshioka K, Maeda M, Kim I, Takuwa Y (2010) Enhanced Ca2 + −dependent activation of phosphoinositide 3-kinase class IIα isoform-Rho axis in blood vessels of spontaneously hypertensive rats. Hypertension 56:934–941
Sowers JR (2004) Insulin resistance and hypertension. Am J Physiol Heart Circ Physiol 286:H1597–H1602
Taguchi K, Matsumoto T, Kobayashi T (2015) G-protein-coupled receptor kinase 2 and endothelial dysfunction: molecular insights and pathophysiological mechanisms. J Smooth Muscle Res in press
Tawaramoto K, Kotani K, Hashiramoto M, Kanda Y, Nagare T, Sakaue H, Ogawa W, Emoto N, Yanagisawa M, Noda T, Kasuka M, Kaku K (2012) Ablation of 3-phosphoinositide-dependent protein kinase 1 (PDK1) in vascular endothelial cells enhances insulin sensitivity by reducing visceral fat and suppressing angiogenesis. Mol Endocrinol 26:95–109
Toker A, Newton AC (2000) Cellular signaling: pivoting around PDK-1. Cell 103:185–188
Wang Y, Yoshioka K, Azam MA, Takuwa N, Sakurada S, Kayaba Y, Sugimoto N, Inoki I, Kimura T, Kuwaki T, Takuwa Y (2006) Class II phosphoinositide 3-kinase alpha-isoform regulates Rho, myosin phosphatase and contraction in vascular smooth muscle. Biochem J 394:581–592
Watts SW, Davis RP (2011) 5-hydroxytryptamine receptors in systemic hypertension: an arterial focus. Cardiovasc Ther 29:54–67
Watts SW, Morrison SF, Davis RP, Barman SM (2012) Serotonin and blood pressure regulation. Pharmacol Rev 64:359–388
Weber DS, Taniyama Y, Rocic P, Seshiah PN, Dechert MA, Gerthoffer WT, Griendling KK (2004) Phosphoinositide-dependent kinase 1 and p21-activated protein kinase mediate reactive oxygen species-dependent regulation of platelet-derived growth factor-induced smooth muscle cell migration. Circ Res 94:1219–1226
Wick KL, Liu F (2001) A new molecular target of insulin action: regulating the pivotal PDK1. Curr Drug Targets Immune Endocr Metabol Disord 1:209–221
Yamada T, Katagiri H, Asano T, Tsuru H, Inukai K, Ono H, Kodama T, Kikuchi M, Oka Y (2002) Role of PDK1 in insulin-signaling pathway for glucose metabolism in 3T3-L1 adipocytes. Am J Physiol Endocrinol Metab 282:E1385–E1394
Acknowledgments
We thank T. Adachi, K. Matsubara, Y. Noishiki, Y. Kimoto, H. Higa, M. Nagata, M. Ando, and M. Iguchi for technical assistance. This study was supported in part by JSPS KAKENHI Grant Numbers 26460107, 15K21419, and 15K07975.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
No conflict of interest.
Additional information
Shun Watanabe and Takayuki Matsumoto contributed equally to this work.
Rights and permissions
About this article

Cite this article
Watanabe, S., Matsumoto, T., Oda, M. et al. Insulin augments serotonin-induced contraction via activation of the IR/PI3K/PDK1 pathway in the rat carotid artery. Pflugers Arch - Eur J Physiol 468, 667–677 (2016). https://doi.org/10.1007/s00424-015-1759-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-015-1759-4