
Abstract
Cardiovascular problems are a major cause of morbidity and mortality, mainly due to coronary artery disease and atherosclerosis, in type 2 diabetes mellitus. However, female gender is a protective factor in the development of, for example, atherosclerosis and hypertension. One of the female hormones, 17β-estradiol (E2), is known to protect against the cardiovascular injury resulting from endothelial dysfunction, but the mechanism by which it does so remains unknown. Our hypothesis was that E2-mediated activation of Akt and mitogen-activated protein kinase (MAPK), and the subsequent endothelial NO synthase (eNOS) phosphorylation, might protect the aorta in diabetic mellitus. The experimental type 2 diabetic model we employed to test that hypothesis (female mice given streptozotocin and nicotinamide) is here termed fDM. In fDM aortas, we examined the E2-induced relaxation response and the associated protein activities. In control (age-matched, nondiabetic) aortas, E2 induced a vascular relaxation response that was mediated via Akt/eNOS and mitogen-activated/ERK-activating kinase (MEK)/eNOS pathways. In fDM aortas (vs. control aortas), (a) the E2-induced relaxation was enhanced, (b) the mediation of the response was different (via Akt/eNOS and p38 MAPK/eNOS pathways), and (c) E2 stimulation increased p38 MAPK and eNOS phosphorylations, decreased MEK phosphorylation, but did not alter estrogen receptor activity. We infer that at least in fDM aortas, E2 has beneficial effects (enhanced vascular relaxation and protection) that are mediated through Akt activation and (compensating for reduced MEK activation) p38 MAPK activation, leading to enhanced eNOS phosphorylation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Premenopausal women have less cardiovascular disease and lower cardiovascular morbidity and mortality than men of the same age; however, these cardioprotective benefits disappear after the menopause [36]. It is widely thought that estrogen exerts protective effects on the cardiovascular system [4]. Diabetes mellitus is an important risk factor for cardiovascular diseases, and impaired vasorelaxation has been described in diabetic humans and in animal models of that disease [9, 11, 42, 46, 49, 51].
In this study, we employed a well-established type 2 diabetic mouse model. In 1998, Masiello and coworkers produced a new model of human type 2 diabetic mellitus in adult rats by giving nicotinamide and streptozotocin (STZ) together [31]. Our mouse model differs in that we give 1.5 g/kg nicotinamide shortly before 200 mg/kg STZ treatment. We previously reported that our model has an impaired glucose tolerance and impaired insulin responsiveness (vs. age-matched, nondiabetic controls), confirming its type 2 diabetic status [28, 47–49]. In most diabetic models and in human diabetes, a common finding is an impaired endothelium-dependent relaxation to acetylcholine (Ach). However, we previously reported that our diabetic model mice did not exhibit an impaired ACh-induced relaxation response, but did exhibit a significantly impaired Akt-dependent relaxation response [28]. Furthermore, we previously confirmed that the female diabetic mouse model we employed here similarly did not display an impaired ACh-induced relaxation response and that it has better preserved vascular functions via the Akt pathway, as well as greater nitric oxide (NO) production via augmented endothelial NO synthase (eNOS) activity and expression [48, 49]. Those findings are consistent with females having an inherent cardioprotective advantage. However, the precise mechanisms underlying this advantage are not understood.
Until recently, the cardioprotective effects of estrogen were attributed mainly to hormonal effects on serum lipid concentration and modification [24]. However, new roles for estrogens are emerging that may contribute to beneficial effects within the cardiovascular system. Estrogens induce vasodilation and inhibit both the response of vessels to injury and the development of atherosclerosis. Many of these estrogen-dependent cardiovascular-related effects occur rapidly after administration of estrogen and are not dependent on gene expression; such actions are commonly referred to as “nongenomic” effects. In contrast, long-term actions of estrogens, such as those that elicit cell proliferation and inhibit the response to vascular injury, require gene expression and are usually designated “genomic” functions [14].
It has been shown that a membrane-associated form of estrogen receptor (ER) mediates the rapid estrogen-dependent nongenomic functions [40]. Moreover, a subpopulation of ERα has been localized to endothelial cell caveolae, where they are coupled to eNOS in a functional signaling module [7]. eNOS, which is the primary physiological source of NO in the vascular system, is expressed in endothelial cells. It is thus possible that eNOS plays a critical role in the cardiovascular protection afforded by 17-β-estradiol (E2), which is one of the estrogens [12]. E2 is a key regulator and performs functions in a wide array of tissues, including those in the cardiovascular system [18, 19, 56]. There is compelling evidence that E2 activates distinct signaling pathways, such as Akt [14, 17] and mitogen-activated protein kinase (MAPK) [57], and that the engagement of membrane ER results in rapid endothelial NO production through an Akt pathway [16]. MAPK is actually a large family of serine/threonine kinases, and these are activated by a variety of extracellular stimuli [1, 21]. The MAPK superfamily consists of three well-characterized subfamilies: mitogen-activated/ERK-activating kinase (MEK), p38 MAPK, and c-Jun NH2-terminal kinases (JNK). Studies have shown that MAPK plays an important role in endothelial dysfunction [22, 26, 32]. Furthermore, activation of p38 MAPK has been suggested to be critical for cardioprotection [13, 20, 53]. As the protective properties of NO emerge, one proposal is that the cardiovascular protective effect of estrogen may be mediated through augmentation of endothelial NO production [6].
However, the molecular events underlying the NO-related rapid functions of E2 in diabetes remain elusive. We hypothesized that the beneficial vascular effects of E2 in diabetes mellitus are mediated via an Akt-dependent pathway and/or MAPK-dependent pathway and are exerted through regulation of eNOS activation.
Methods
Reagents
STZ, nicotinamide, E2 (17-β-estradiol), N G-nitro-l-arginine (L-NNA), and monoclonal β-actin antibody were all purchased from Sigma Chemical Co. (St. Louis, MO, USA). Sodium nitroprusside (SNP) was from Wako (Osaka, Japan), while ACh was from Daiichi Pharmaceuticals (Tokyo, Japan). Akt inhibitor (=1L-6-hydroxymethyl-chiro-inositol 2-[(R)-2-O-methyl-3-O-octadecylcarbonate]), PD98059, SB203580, SP600125, and PP2 were from CALBIOCHEM (La Jolla, CA, USA). E2 was dissolved in dimethyl sulfoxide. All other agents were dissolved in saline. All concentrations are expressed as the final molar concentration of the base in the organ bath. Horseradish peroxide (HRP)-linked secondary anti-mouse or anti-rabbit antibody was purchased from Promega (Madison, WI, USA). Antibodies against Akt, phosphorylated Akt at Ser473, phosphorylated eNOS at Ser1177, p38 MAPK, phosphorylated p38 MAPK at Thr180/Tyr182, MEK1/2, phosphorylated MEK1/2 at Ser221, ERα, and phosphorylated ERα at Ser118 were obtained from Cell Signaling Technology (Danvers, MA, USA), while the antibody against eNOS was from BD Bioscience (San Jose, CA, USA).
Animals and experimental design
Type 2 diabetic mice were generated as previously described [28, 47–49], females being used on this occasion. Female Institute of Cancer Research control (nondiabetic) mice were obtained at 5 weeks of age. Diabetes was induced by intraperitoneal injection of nicotinamide (1.5 g/kg body weight) in saline 15 min before an injection via the tail vein of STZ (200 mg/kg) dissolved in a citrate buffer [28, 47–49]. Age-matched control mice received the vehicles by the above routes. Mice were housed in temperature-controlled cages under a 12-h light–dark cycle and given free access to water and normal chow. At 12 weeks after the above administrations, the mice were euthanized with inhaled isoflurane and assigned to various experiments. The animal protocols were approved as conforming to the Guide for the Care and Use of Laboratory Animals by the issuing committee (the Committee on the Care and Use of Laboratory Animals of Hoshi University, which is accredited by the Ministry of Education, Culture, Sports, Science, and Technology, Japan).
Measurement of plasma parameters
Plasma parameters were measured as described previously [28, 32, 47–49]. Plasma samples were stored at −20 °C until analysis. Briefly, plasma glucose was determined by the use of a commercially available enzyme kit (Wako Chemical, Osaka, Japan). Plasma insulin was measured by enzyme immunoassay (Shibayagi, Gunma, Japan). Plasma E2 levels were measured by following the manufacturer’s instructions accompanying a commercially available E2 enzyme immunoassay (DRG, Marburg, Germany).
Measurement of isometric force
The thoracic aorta was carefully cleaned of all fat and connective tissue and rings approximately 2 mm long were cut from it. Each ring was suspended by a pair of stainless steel pins in a well-oxygenated (95 % O2–5 % CO2) bath containing modified Krebs–Henseleit solution (KHS) of the following composition (mM): 118.0 NaCl, 4.7 KCl, 25.0 NaHCO3, 1.8 CaCl2, 1.2 NaH2PO4, 1.2 MgSO4, and 11.0 glucose at 37 °C. The rings were stretched until a resting tension of 1.5 g was loaded, which was optimal for inducing the maximal contraction [28, 32, 47–49]. After 1 h equilibration, rings were contracted to a stable tension using prostaglandin F2α (PGF2α; 10−6–3 × 10−6M). Force generation was monitored by means of an isometric transducer (model TB-611T; Nihon Kohden, Tokyo, Japan). We plotted complete concentration–response relationships for ACh (10−9–10−5M), E2 (10−10–10−7M), and SNP (10−10–10−5M). Some rings were preincubated with Akt inhibitor (10−6M), L-NNA (10−4M), PD98059 (5 × 10−5M), SB203580 (10−6M), SP600125 (10−6M), or PP2 (10−5M) when the effects of inhibitors on the responses to the above relaxant agents were to be examined. To that end, one of these inhibitors was added to the bath 30 min before precontraction.
Measurement of NOx
The concentration of NOx (nitrite + nitrate) in the effluent from a given tissue was sampled and assayed by the method described previously (ENO20; Eicom, Kyoto, Japan) [47–49]. Each aorta was cut into rings 5 mm in length, and these were placed in 0.5 mL KHS at 37 °C. Effluent samples were collected on two occasions as follows: sample 1, for a 20-min period after application of 10−7M E2; sample 2, for a 20-min period without E2 stimulation. The amount of NOx was calculated as follows: agonist-stimulated NOx = [sample 1 − sample 2]. The concentration of nitrite plus nitrate in the KHS and the reliability of the reduction column were examined in each experiment.
Western blotting
Frozen aortic samples were pulverized and lysed with lysis buffer containing 50 mM Tris–HCl buffer (pH 7.5), 150 mM NaCl, 1 % Triton-X, and protease and phosphatase inhibitor cocktails (Complete Protease Inhibitor Cocktail and PhosSTOP; Roche Diagnostics, Indianapolis, IN, USA), as previously described [25–28, 32, 33, 37]. Protein assays were performed using a bicinchoninic acid protein assay reagent kit (Pierce). Protein (20 μg) was loaded into each lane for all western blots, which were performed as described previously [28, 47–49]. Antibodies against phospho-ERα (Ser118), total ERα, phospho-Akt (Ser473), total Akt, phospho-MEK1/2 (Ser221), total MEK1/2, phospho-p38 MAPK (Thr180/Tyr182), total p38 MAPK, phospho-eNOS (Ser1177), or total eNOS were diluted 1:1,000, whereas antibody against β-actin was diluted 1:5,000. Detection was achieved using a HRP-conjugated IgG followed by enhanced chemiluminescence. Band intensity was quantified by densitometry. Results were normalized to β-actin expression and expressed relative to control (vehicle-treated control).
Statistical analysis
All data are expressed as means ± SE. For comparisons between groups, data were analyzed using one- or two-way analysis of variance (ANOVA) with a post hoc Bonferroni test or Student’s t test. Western blot data were analyzed by a one-way ANOVA with a post hoc Dunnett’s test or by Student’s t test. Significance was set at P < 0.05.
Results
Characteristics of the type 2 diabetic mouse model
The experimental model employed here (mice given STZ and partially protected with a suitable dose of nicotinamide) has been in use for about a decade [28, 31, 38, 47–49]. In this model, the diabetic syndrome shares a number of features with human type 2 diabetes. It is characterized by stable moderate hyperglycemia, glucose intolerance, and altered but still significant glucose-stimulated insulin secretion. However, there are little published data about the female version of this model [48, 49]. As shown in Table 1, the non-fasting plasma glucose levels were significantly higher in nicotinamide + STZ-induced female diabetic mice (fDM) than in the controls. Body weight, plasma insulin levels, and plasma E2 levels were not different between the controls and fDM (Table 1). The concentrations of E2 given by cumulative administration to induce the aortic relaxation response were much higher than the normal circulating levels shown in Table 1. Our experiments were, of course, performed in vitro, making such comparisons somewhat problematic. Possibly, the results described below may be relevant to whether excessive circulating E2 might contribute to cardiovascular protection via activated E2-induced signal transduction. Furthermore, others have examined the vascular relaxation responses induced by high concentrations of E2 [15]. Our use of such high concentrations of E2 allows comparisons between our results and previous ones. Moreover, we thought that it was an acceptable way of examining the vasoactive effect of estradiol and an optimal way to gain insights into the potential importance of the acute effects of estradiol for cardiovascular health.
Aortic relaxations
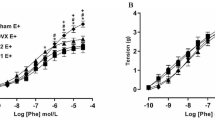
When the PGF2α (1 × 10−6–3 × 10−6M)-induced contraction had reached a plateau, ACh, SNP, or E2 was added cumulatively. ACh induced a concentration-dependent relaxation that was not different between aortic rings from fDM and controls (Fig. 1a). The E2-induced relaxation was significantly stronger in fDM than in the controls (Fig. 1b). The SNP-induced endothelium-independent relaxation was not different between fDM and controls (Fig. 1c). Further, the E2-induced and ACh-induced relaxation responses in both fDM and control aortic rings were severely impaired or abolished by treatment with the NOS inhibitor, L-NNA (Fig. 2a, c). The ACh-induced relaxation was not altered by treatment with an Akt inhibitor in rings from fDM or controls, but the E2-induced relaxation was severely impaired by such treatment (Fig. 2b, d). This result suggests that Akt/eNOS signaling is involved in the mediation of the E2-induced relaxation response, but that it is not the only signaling pathway involved.

Concentration–response curves for agonist-induced relaxations (a–c) of aortic rings from control and fDM. Relaxations induced by: a ACh; b E2; c SNP. Values are mean ± SE; n = 6; **P < 0.01 vs. control

Effects of L-NNA (a, c) and Akt inhibitor (b, d) on E2 (a, b)-induced and ACh (c, d)-induced relaxations of aortic rings. In each case, aortas were isolated from fDM and control. Aortic rings were preincubated with L-NNA (10−4 M; 30 min) or Akt inhibitor (10−6M; 30 min). Values are mean ± SE; n = 6. *P < 0.05, **P < 0.01, or ***P < 0.001 vs. control. ###P < 0.001 vs. fDM
Effects of various inhibitors on E2-induced aortic responses
There is compelling evidence that E2 activates MAPK [23, 45, 57]. We therefore investigated the role of the MAPK signaling pathway in modulating vascular responses under the particular pathological conditions present in our type 2 diabetes model. In the controls, the E2-induced relaxation was (a) not changed by pretreatment with the p38 MAPK inhibitor SB203580, the JNK inhibitor SP600125, or the Src inhibitor PP2, but (b) significantly attenuated by pretreatment with the MEK inhibitor PD98059 (Fig. 3a). These results suggest that in the nondiabetic controls, the E2-induced relaxation response is mediated via the MEK pathway as well as via the Akt pathway. Intriguingly, in fDM, the E2-induced aortic relaxation was not changed by pretreatment with PD98059 (Fig. 3b), but instead it was significantly reduced by pretreatment with SB203580 (Fig. 3c). The ACh-induced relaxation response was not changed by pretreatment with those two inhibitors in either fDM or control (Fig. 4). These results suggest (a) that only in fDM does p38 MAPK contribute to the E2-induced aortic relaxation and that in the controls, this relaxation is mediated via the MEK pathway, and (b) that underlying the augmented E2-induced response observed in fDM was a reduced operation of the MEK pathway and an increased operation of the p38 MAPK pathway (vs. the controls).

Effects of various inhibitors on E2-induced relaxations of aortic rings. In each case, aortas were isolated from fDM and control. Aortic rings were preincubated with PD98059 (5 × 10−5M; 30 min), SB203580 (10−6M; 30 min), SP600125 (10−6M; 30 min), or PP2 (10−6M; 30 min). Values are mean ± SE; n = 6. ***P < 0.001 vs. control. ##P < 0.01 vs. fDM

Effects of PD98059 and SB203580 on ACh-induced relaxations of aortic rings from fDM. Aortic rings were preincubated with PD98059 (5 × 10−5M; 30 min) (a) or SB203580 (10−6M; 30 min) (b). Values are mean ± SE; n = 6
Measurement of aortic NOx release and eNOS activity
Since Akt and MAPK play important roles in eNOS activation/NO production, we measured NOx release from the aorta (Fig. 5). ACh (10−7M)-stimulated NOx production was not different between controls and fDM. In contrast, the effect of E2 (10−7M) on NOx production was greater in fDM than in controls. These data are consistent with those obtained for vascular relaxation responses. The E2-stimulated NOx level was decreased by preincubation with Akt inhibitor in both fDM and control. In contrast, although preincubation with PD98059 decreased the E2-stimulated NOx level in control, it did not alter it in fDM. The reverse was true with SB203580: preincubation with this inhibitor did not alter the E2-stimulated NOx level in control, but it significantly decreased it in fDM. These data are consistent with those described above for the effects of those inhibitors on E2-induced relaxation responses.

Analysis of NOx production under E2 stimulation. a NOx production under ACh or E2 stimulation in aortas isolated from control and fDM. Aortic rings were treated with 10−7M ACh or 10−7M E2 for 20 min. b Aortic rings from control and fDM were treated with E2 alone (10−7M; for 20 min), E2 + Akt inhibitor (10−6M), E2 + PD98059 (5 × 10−5M), or E2 + SB203580 (10−6M). Values are mean ± SE; n = 6. *P < 0.05 or ***P < 0.001 vs. E2-stimulated control. ###P < 0.001 vs. E2-stimulated fDM
The precise molecular mechanism by which E2 regulates eNOS activity remains unknown. The finding that eNOS can be phosphorylated at Ser1177 supports the important role of phosphorylation in regulating eNOS activity [12, 13, 26–28, 33, 37, 47, 48]. So, to evaluate whether the diabetes-related augmentation of the aortic response to E2 seen in fDM might be mediated via the MAPK/eNOS and/or the Akt/eNOS pathway, we measured eNOS activity by examining the expression of phosphorylated eNOS (at Ser1177) using western blotting. Use of anti-eNOS antibody allowed detection of an immunoreactive protein with a molecular weight of 140 kDa. The total eNOS protein expression level did not differ between fDM and control (Fig. 6a). In the basal (unstimulated) condition, the phospho-eNOS level did not differ between fDM and control (Fig. 6b). In aortas stimulated with E2, the expression level of phospho-eNOS was significantly increased in both fDM and control (vs. the non-stimulated group). Pretreatment with Akt inhibitor decreased the E2-stimulated phospho-eNOS level in both fDM and control. Importantly, pretreatment with PD98059 decreased the E2-stimulated phospho-eNOS expression level in control (vs. E2-stimulated control), but had no such effect in fDM. As in the NOx experiment, the reverse was true when SB203580 was used: it did not alter the E2-stimulated phospho-eNOS expression level in the controls (vs. E2-stimulated control), but it significantly decreased it in fDM (Fig. 6b). Thus, the results obtained for the phosphorylation level of eNOS were in essence similar to those obtained for NO production, suggesting that the interaction of E2 with aortic eNOS, which has been directly correlated with favorable cardioprotective effects, involves phosphorylation of eNOS. Moreover, these findings are consistent with the eNOS activation being downstream of Akt, MEK, and p38 MAPK.

Total eNOS expression (a) together with eNOS phosphorylation at Ser1177 under E2-stimulated conditions (b). Data are shown for aortic rings from control and fDM. a Top representative western blots. A single band at 140 kDa was observed. Bottom Bands quantified by scanning densitometry. b E2-stimulated eNOS phosphorylation. Aortic strips were treated with vehicle (basal), E2 alone (10−7M; for 20 min), E2 + Akt inhibitor (10−6M), E2 + PD98059 (5 × 10−5M), or E2 + SB203580 (10−6M). Values are mean ± SE; n = 6. **P < 0.01 vs. E2-stimulated control. ###P < 0.001 vs. E2-stimulated fDM
Activation of Akt, MEK, and p38 MAPK in the aorta
We looked for differences in Akt, MEK, and p38 MAPK activations by examining the expressions of phosphorylated Akt (at Ser473), phosphorylated MEK1/2 (at Ser221), and phosphorylated p38 MAPK (at Thr180/Tyr182), respectively, using western blotting. In the unstimulated condition, the Ser473 Akt phosphorylation level, the Ser221 MEK1/2 phosphorylation level, and the Thr180/Tyr182 phosphorylation level were not different between control and fDM (data not shown). The E2-stimulated Akt phosphorylation level was not different between control and fDM (Fig. 7e). Although pretreatment with PD98059 or SB203580 did not alter E2-stimulated Akt phosphorylation in either group, pretreatment with Akt inhibitor markedly decreased the E2-induced stimulation of Akt phosphorylation in both groups (Fig. 7e), suggesting that MAPK is downstream of Akt or acts via a pathway completely different from Akt. Moreover, under E2 stimulation, the MEK1/2 phosphorylation level was significantly lower in fDM than in control (Fig. 7f). In the controls, pretreatment with Akt inhibitor or SB203580 did not alter E2-stimulated MEK1/2 phosphorylation, but pretreatment with PD98059 significantly decreased it (Fig. 7f). So, we suggest that E2-induced eNOS activation/NO production is mediated not only via the Akt pathway, but also via a MAPK pathway (viz., MEK1/2 in Control, but p38 MAPK in fDM). Basically, the present findings suggest that E2 increases NO production via Akt/eNOS signaling and MEK/eNOS signaling in the controls, but via Akt/eNOS signaling and p38 MAPK/eNOS signaling in fDM. As shown in Fig. 7g, the E2-stimulated aortic phospho-p38 MAPK level was significantly higher in fDM than in control (Fig. 7g). In fDM, the E2-stimulated p38 MAPK phosphorylation level was not changed by pretreatment with the Akt inhibitor or PD98059, although pretreatment with SB203580 greatly reduced it (Fig. 7g). Thus, the results obtained for the phosphorylation levels of Akt, MEK, and p38 MAPK were consistent with those obtained for eNOS expression and NO production. Moreover, these findings suggest that the Akt/eNOS pathway and the p38 MAPK/eNOS pathway (in particular, activation of p38 MAPK) are upstream of the enhancement of the E2-stimulated aortic relaxation response seen in fDM.

Total Akt (b), MEK1/2 (c), and p38 MAPK (d) expressions together with Akt phosphorylation at Ser473, MEK1/2 phosphorylation at Ser221, and p38 MAPK phosphorylation at Thr180/Tyr182 under E2-stimulated conditions (e, f, and g, respectively). Data are shown for aortic rings from control and fDM. a Representative western blots. b–g Bands quantified by scanning densitometry. b–d Total protein expressions. e–g E2-stimulated protein phosphorylations. Aortic rings were treated with E2 alone (10−7M; 20 min), E2 + Akt inhibitor (10−6M), E2 + PD98059 (5 × 10−5M), or E2 + SB203580 (10−6M). Values are mean ± SE; n = 6. *P < 0.05 or **P < 0.01 vs. E2-stimulated control. ###P < 0.001 vs. E2-stimulated fDM (with no inhibitor)
ER activation in the aorta
The E2-mediated relaxation of the aorta is associated with the ERα subtype. Here, we examined total ER expression and activity in aortas by western blotting. Use of anti-ER antibody allowed detection of an immunoreactive protein with a molecular weight of 66 kDa. The total expression of this protein was not different between fDM and control (Fig. 8a, c).

Total estrogen receptor (ER) expression under basal conditions, together with ER phosphorylation at Ser118 under E2-stimulated conditions. Data are shown for aortic rings from control and fDM. a Representative western blots. b, c Bands quantified by scanning densitometry. b E2 (10−7M; for 20 min)-stimulated ER phosphorylation. c Total protein expression. Values are mean ± SE; n = 6
Finally, we examined the aortic expressions of phosphorylated ER (at Ser118) under E2 stimulation. The basal level of this phosphorylated ER was similar between fDM and control (data not shown). Under E2 stimulation, the phosphorylation of ER was likewise not different between fDM and control (Fig. 8a, b).
Discussion
Understanding the pathophysiology involved in female diabetic mellitus is important, as is understanding how E2 mitigates any consequent cardiovascular disease. In the present study, we report the mechanistic details of how E2 protects against vascular dysfunction in female diabetes through Akt- and p38 MAPK-dependent eNOS regulation. Specifically, in aortas from a female mouse model of type 2 diabetes (fDM) (vs. nondiabetic controls), we found that (a) E2-induced vascular relaxation and (b) NO production were markedly increased and that (c) eNOS phosphorylation under E2 stimulation was not decreased (actually, it tended to be increased). Moreover, responses (a), (b), and (c) were inhibited if the MEK-specific inhibitor PD98059 (in control aortas) or the p38 MAPK-specific inhibitor SB203580 (in diabetic aortas) were applied before the E2. Our findings indicate that the above alterations were accompanied by a decrease in MEK activation and an increase in p38 MAPK activation in fDM. At present, we can only speculate that it is beneficial that E2-induced vascular relaxation is greater in fDM than in control, and likewise that the MAPK signaling pathway is shifted from the MEK1/2 signaling pathway to the p38 MAPK signaling pathway in the fDM aorta. Our reasoning is that the increased activation of p38 MAPK seems to occur as compensation (possibly, overcompensation) for the decreased activation of MEK1/2 (i.e., if the latter reduction were not compensated for by a shift to an alternative pathway, harm might result). Several potential mechanisms have been proposed for the beneficial effects of E2 on organ function in cardiovascular disease and diabetes [18, 19, 39, 56]. The present study, examining the signal transduction mechanism in female diabetic mice for the first time, suggests that E2-mediated cardiovascular protection may be exerted through a compensatory pathway (that is, the p38 MAPK/eNOS pathway), rather than through the MEK/eNOS pathway, in diabetes.
A major finding made in the present study is that in the fDM aortas, the E2-induced relaxation response was markedly increased vs. the control aortas. The rapid (i.e., “nongenomic”; see “Introduction”) vasodilator effect of E2 is at least partially related to activation of eNOS and increased NO production [16, 43, 44]. As just indicated, NO is regulated by the amount of eNOS protein in the aorta and by its activity or phosphorylation level. Our results demonstrate that the aortic expression of eNOS protein was not different between fDM and control and that its phosphorylation level was increased under E2 stimulation in both groups. In line with our findings, others have already shown that E2 facilitates both the expression of eNOS and its activity in endothelial cells, effects that are E2-specific because they are inhibited by ER blockade [2]. Nonetheless, the present data are the first to demonstrate an enhancement of eNOS phosphorylation by E2 in the mouse aorta in a female diabetic model. In addition, L-NNA-induced inhibition of NOS, including eNOS, caused attenuation, but not complete abolition, of the E2-induced relaxation response. Thus, it is feasible that the role of the eNOS activation induced by E2 in aortas from not only the controls, but also from fDM, is to act as an early protective trigger.
The rapid actions of estrogen in the vasculature appear to play a major role in mediating the protective properties of that steroid hormone [35, 40, 41]. The action of E2 involves short-term activation of downstream signaling systems such as eNOS [54]. Furthermore, engagement of membrane ERs results in rapid endothelial NO release through an Akt-dependent pathway [16]. In the present report, we have strong evidence that E2 induces vascular relaxation by eNOS phosphorylation through an Akt-dependent pathway. However, Akt phosphorylation under E2 stimulation was not different between the controls and fDM.
Most kinases, such as MAPKs, as well as Akt, phosphorylate eNOS upon a physical association with the enzyme, either directly or via their binding to an adaptor protein [5]. However, whether these kinases play any role in affecting vascular function in diabetes remains to be established. A novel observation presented here is that E2 rapidly activated eNOS in an ER/MEK-dependent manner. Interestingly, in fDM, the E2-induced relaxation response was not altered by pretreatment with PD98059, and the E2-induced MEK phosphorylation level was significantly lower than in the controls. Furthermore, to our knowledge, we are the first to present evidence that the ERα is phosphorylated under E2 stimulation and, moreover, that this response is not different between control and fDM. We speculate that in fDM, a certain change has taken place downstream of the ER/MAPK (MEK)/eNOS pathway, possibly not in the ER/Akt/eNOS signaling pathway. A previous study indicated that following heart ischemia–reperfusion, there is a differential association of p38 MAPK with different types of caveolin-induced signals (survival or death signals) [10]. We demonstrate here a new differential involvement of p38 MAPK in signaling mechanisms. Our data show that the E2-induced relaxation response was attenuated by p38 MAPK inhibition only in fDM and that p38 MAPK phosphorylation under E2 stimulation was greater in fDM than in control. Indeed, the existing evidence as to the roles performed by p38 MAPK in the diabetic aorta is complex and controversial. Some studies have suggested that activation of p38 MAPK is beneficial [1, 20, 29], but others have indicated that its activation is detrimental so that in cardiovascular disease its inhibition is protective [8, 19, 30, 34, 52]. Recently, Toque et al. reported that in a mouse model of hypertension, there was overactivation of p38 MAPK, leading to enhanced eNOS activation [50]. In this context, additional findings demonstrate that the acute activation of p38 MAPK induced by E2 is protective and reduces dysfunction in cardiovascular disease [23]. Our data demonstrate that in fDM, E2 activated p38 MAPK, and also that blocking MEK activation partially attenuated the enhanced vasorelaxation (and possibly, tissue protection) afforded by E2 in the aorta only in fDM. Other studies have shown that in addition to p38, other signaling molecules, such as Akt, play roles in estrogen-mediated protective effects in the endothelial cell [14, 17]. However, whether they are up- or down-stream of p38 MAPK, or indeed are regulated independently by E2, remains unclear at present. At present, we think that there may be interactions between Akt and MEK1/2 in the controls, but between Akt and p38 MAPK in fDM, as well as a compensatory p38 MAPK activation as a result of decreased MEK1/2 activation in fDM. However, further studies will be needed to clarify the relationship between p38 MAPK and other pathways, such as Akt and MEK.
The literature contains some conflicting results concerning the effects of SB203580 on p38 MAPK phosphorylation. Some studies have asserted that SB203580 does not inhibit the phosphorylation itself, but rather inhibits activities downstream of p38 MAPK [55], whereas other studies suggest an inhibitory effect of this agent on p38 MAPK phosphorylation [3]. This issue remains to be resolved. Finally, (a) in the controls and fDM, the ACh-induced aortic relaxation response is not mediated via Akt, MEK, or p38 MAPK pathways, whereas (b) the E2-induced aortic relaxation response is mediated via Akt and MEK (in control) or via Akt and p38 MAPK (in fDM). However, the mechanism by which MAPK is involved in eNOS activation remains uncertain. Moreover, as yet we have no insight into why or how “pathway selection” takes place in the control and fDM, and whether there is a switch that somehow selects between MEK activation and p38 MAPK activation.
In summary, the present results indicate that in fDM, E2 stimulation of the aorta activates the p38 MAPK pathway, possibly compensating, or overcompensating, for a reduced activation of the MEK pathway, and that eNOS phosphorylation is thereby increased, leading to enhanced vascular relaxation. These findings suggest that the enhanced vascular effect of E2 in female diabetes may be mediated via an Akt/p38 MAPK-dependent pathway, not via an Akt/MEK-dependent pathway, and exerted through increased phosphorylation of eNOS. Even so, since E2 can exert its effects in multiple ways, we do not consider activation of p38 MAPK to be the only relevant action of E2 under such conditions. Nonetheless, the present findings highlight a potentially important relationship between E2 and the p38 MAPK/eNOS pathway in female diabetes mellitus. Knowledge of this relationship may inform the development of new therapeutic and/or preventive modalities for the treatment of diabetic vascular dysfunction in females.
References
Anter E, Chen K, Shapira OM, Karas RH, Keaney JF Jr (2005) p38 mitogen-activated protein kinase activates eNOS in endothelial cells by an estrogen receptor alpha-dependent pathway in response to black tea polyphenols. Circ Res 96:1072–1078
Anter E, Thomas SR, Schulz E, Shapira OM, Vita JA, Keaney JF Jr (2004) Activation of endothelial nitric-oxide synthase by the p38 MAPK in response to black tea polyphenols. J Biol Chem 279:46637–46643
Bamba S, Andoh A, Yasui H, Makino J, Kim S, Fujiyama Y (2003) Regulation of IL-11 expression in intestinal myofibroblasts: role of c-Jun AP-1- and MAPK-dependent pathways. Am J Physiol Gastrointest Liver Physiol 285:G529–G538
Barrett-Connor E (1997) Sex differences in coronary heart disease. Why are women so superior? The 1995 Ancel Keys Lecture. Circulation 95:252–264
Butt E, Bernhardt M, Smolenski A, Kotsonis P, Frohlich LG, Sickmann A, Meyer HE, Lohmann SM, Schmidt HH (2000) Endothelial nitric-oxide synthase (type III) is activated and becomes calcium independent upon phosphorylation by cyclic nucleotide-dependent protein kinases. J Biol Chem 275:5179–5187
Caulin-Glaser T, Garci-Cardena G, Sarrel P, Sessa WC, Bender JR (1997) 17 beta-estradiol regulation of human endothelial cell basal nitric oxide release, independent of cytosolic Ca2+ mobilization. Circ Res 81:885–892
Chambliss KL, Yuhanna IS, Mineo C, Liu P, German Z, Sherman TS, Mendelsohn ME, Anderson RG, Shaul PW (2000) Estrogen receptor alpha and endothelial nitric oxide synthase are organized into a functional signaling module in caveolae. Circ Res 87:E44–E52
Clanachan AS, Jaswal JS, Gandhi M, Bottorff DA, Coughlin J, Finegan BA, Stone JC (2003) Effects of inhibition of myocardial extracellular-responsive kinase and p38 mitogen-activated protein kinase on mechanical function of rat hearts after prolonged hypothermic ischemia. Transplantation 75:173–180
Cohen RA (1995) The effect of nitric oxide and other endothelium-derived vasoactive substances in vascular disease. Prog Cardiovasc Dis 38:105–128
Das M, Cui J, Das DK (2007) Generation of survival signal by differential interaction of p38MAPKalpha and p38MAPKbeta with caveolin-1 and caveolin-3 in the adapted heart. J Mol Cell Cardiol 42:206–213
Eckel RH, Wassef M, Chait A, Sobel B, Barrett E, King G, Lopes-Virella M, Reusch J, Ruderman N, Steiner G, Vlassara H (2002) Prevention Conference VI: Diabetes and Cardiovascular Disease: Writing Group: pathogenesis of atherosclerosis in diabetes. Circulation 105:e138–e143
Fleming I, Busse R (2003) Molecular mechanisms involved in the regulation of the endothelial nitric oxide synthase. Am J Physiol Regul Integr Comp Physiol 284:R1–R12
Fujimoto H, Ohno M, Ayabe S, Kobayashi H, Ishizaka N, Kimura H, Yoshida K, Nagai R (2004) Carbon monoxide protects against cardiac ischemia–reperfusion injury in vivo via MAPK and Akt–eNOS pathways. Arterioscler Thromb Vasc Biol 24:1848–1853
Garban HJ, Marquez-Garban DC, Pietras RJ, Ignarro LJ (2005) Rapid nitric oxide-mediated S-nitrosylation of estrogen receptor: regulation of estrogen-dependent gene transcription. Proc Natl Acad Sci USA 102:2632–2636
Haas E, Meyer MR, Schurr U, Bhattacharya I, Minotti R, Nguyen HH, Heigl A, Lachat M, Genoni M, Barton M (2007) Differential effects of 17β-estradiol on function and expression of estrogen receptor α, estrogen receptor β, and GPR30 in arteries and veins of patients with atherosclerosis. Hypertension 49:1358–1363
Haynes MP, Sinha D, Russell KS, Collinge M, Fulton D, Morales-Ruiz M, Sessa WC, Bender JR (2000) Membrane estrogen receptor engagement activates endothelial nitric oxide synthase via the PI3-kinase–Akt pathway in human endothelial cells. Circ Res 87:677–682
Hisamoto K, Ohmichi M, Kurachi H, Hayakawa J, Kada Y, Nishio Y, Adachi K, Tasaka K, Miyoshi E, Fujiwara N, Taniguchi N, Murata Y (2001) Estrogen induces the Akt-dependent activation of endothelial nitric-oxide synthase in vascular endothelial cells. J Biol Chem 276:3459–3567
Hsieh YC, Yang S, Choudhry MA, Yu HP, Rue LW 3rd, Bland KI, Chaudry IH (2005) PGC-1 upregulation via estrogen receptors: a common mechanism of salutary effects of estrogen and flutamide on heart function after trauma-hemorrhage. Am J Physiol Heart Circ Physiol 289:H2665–H2672
Hsu JT, Kan WH, Hsieh CH, Choudhry MA, Schwacha MG, Bland KI, Chaudry IH (2007) Mechanism of estrogen-mediated attenuation of hepatic injury following trauma-hemorrhage: Akt-dependent HO-1 up-regulation. J Leukoc Biol 82:1019–1026
Jaswal JS, Gandhi M, Finegan BA, Dyck JR, Clanachan AS (2007) Inhibition of p38 MAPK and AMPK restores adenosine-induced cardioprotection in hearts stressed by antecedent ischemia by altering glucose utilization. Am J Physiol Heart Circ Physiol 293:H1107–H1114
Kan W, Zhao KS, Jiang Y, Yan W, Huang Q, Wang J, Qin Q, Huang X, Wang S (2004) Lung, spleen, and kidney are the major places for inducible nitric oxide synthase expression in endotoxic shock: role of p38 mitogen-activated protein kinase in signal transduction of inducible nitric oxide synthase expression. Shock 21:281–287
Kim B, Kim J, Bae YM, Cho SI, Kwon SC, Jung JY, Park JC, Ahn HY (2004) p38 mitogen-activated protein kinase contributes to the diminished aortic contraction by endothelin-1 in DOCA-salt hypertensive rats. Hypertension 43:1086–1091
Klinge CM, Blankenship KA, Risinger KE, Bhatnagar S, Noisin EL, Sumanasekera WK, Zhao L, Brey DM, Keynton RS (2005) Resveratrol and estradiol rapidly activate MAPK signaling through estrogen receptors alpha and beta in endothelial cells. J Biol Chem 280:7460–7468
Knopp RH, Zhu X, Bonet B, Bagatell C (1996) Effects of sex steroid hormones on lipoproteins, clotting, and the arterial wall. Semin Reprod Endocrinol 14:15–27
Kobayashi T, Matsumoto T, Ooishi K, Kamata K (2004) Differential expression of alpha2D-adrenoceptor and eNOS in aortas from early and later stages of diabetes in Goto–Kakizaki rats. Am J Physiol Heart Circ Physiol 287:H135–H143
Kobayashi T, Nogami T, Taguchi K, Matsumoto T, Kamata K (2008) Diabetic state, high plasma insulin and angiotensin II combine to augment endothelin-1-induced vasoconstriction via ETA receptors. Br J Pharmacol 155:974–983
Kobayashi T, Taguchi K, Nemoto S, Nogami T, Matsumoto T, Kamata K (2009) Activation of the PDK-1/Akt/eNOS pathway involved in aortic endothelial function differs between hyperinsulinemic and insulin-deficient diabetic rats. Am J Physiol Heart Circ Physiol 297:H1767–H1775
Kobayashi T, Taguchi K, Yasuhiro T, Matsumoto T, Kamata K (2004) Impairment of PI3-K/Akt pathway underlies attenuated endothelial function in aorta of type 2 diabetic mouse model. Hypertension 44:956–962
Kohmoto J, Nakao A, Stolz DB, Kaizu T, Tsung A, Ikeda A, Shimizu H, Tomiyama K, Sugimoto R, Choi AM, Billiar TR, Murase N, McCurry KR (2007) Carbon monoxide protects rat lung transplants from ischemia–reperfusion injury via a mechanism involving p38 MAPK pathway. Am J Transplant 7:2279–2790
Lu J, Shimpo H, Shimamoto A, Chong AJ, Hampton CR, Spring DJ, Yada M, Takao M, Onoda K, Yada I, Pohlman TH, Verrier ED (2004) Specific inhibition of p38 mitogen-activated protein kinase with FR167653 attenuates vascular proliferation in monocrotaline-induced pulmonary hypertension in rats. J Thorac Cardiovasc Surg 128:850–859
Masiello P, Broca C, Gross R, Roye M, Manteghetti M, Hillaire-Buys D, Novelli M, Ribes G (1998) Experimental NIDDM: development of a new model in adult rats administered streptozotocin and nicotinamide. Diabetes 47:224–229
Matsumoto T, Kobayashi T, Kamata K (2006) Mechanisms underlying lysophosphatidylcholine-induced potentiation of vascular contractions in the Otsuka Long–Evans Tokushima Fatty (OLETF) rat aorta. Br J Pharmacol 149:931–941
Matsumoto T, Noguchi E, Ishida K, Kobayashi T, Yamada N, Kamata K (2008) Metformin normalizes endothelial function by suppressing vasoconstrictor prostanoids in mesenteric arteries from OLETF rats, a model of type 2 diabetes. Am J Physiol Heart Circ Physiol 295:H1165–H1176
Meldrum DR, Dinarello CA, Cleveland JC Jr, Cain BS, Shames BD, Meng X, Harken AH (1998) Hydrogen peroxide induces tumor necrosis factor alpha-mediated cardiac injury by a P38 mitogen-activated protein kinase-dependent mechanism. Surgery 124:291–296
Mendelsohn ME, Karas RH (1999) The protective effects of estrogen on the cardiovascular system. N Engl J Med 340:1801–1811
Mondelsohn ME, Karas RH (2005) Molecular and cellular basis of cardiovascular gender differences. Science 308:1583–1587
Nemoto S, Kobayashi T, Taguchi K, Matsumoto T, Kamata K (2011) Losartan improves aortic endothelium-dependent relaxation via proline-rich tyrosine kinase 2/Src/Akt pathway in type 2 diabetic Goto–Kakizaki rats. Am J Physiol Heart Circ Physiol 301:H2383–H2394
Novelli M, Fabregat ME, Fernandez-Alvarez J, Gomis R, Masiello P (2001) Metabolic and functional studies on isolated islets in a new rat model of type 2 diabetes. Mol Cell Endocrinol 175:57–66
Perera M, Petrie JR, Hillier C, Small M, Sattar N, Connell JM, Lumsden MA (2002) Hormone replacement therapy can augment vascular relaxation in post-menopausal women with type 2 diabetes. Hum Reprod 17:497–502
Pietras RJ, Szego CM (1977) Specific binding sites for oestrogen at the outer surfaces of isolated endometrial cells. Nature 265:69–72
Pietras RJ, Szego CM (1980) Partial purification and characterization of oestrogen receptors in subfractions of hepatocyte plasma membranes. Biochem J 191:743–760
Piper GM (1998) Review of alterations in endothelial nitric oxide production in diabetes: protective role of arginine on endothelial dysfunction. Hypertension 31:1047–1060
Russell KS, Haynes MP, Sinha D, Clerisme E, Bender JR (2000) Human vascular endothelial cells contain membrane binding sites for estradiol, which mediate rapid intracellular signaling. Proc Natl Acad Sci USA 97:5930–5935
Russell KS, Haynes MP, Caulin-Glaser T, Rosneck J, Sessa WC, Bender JR (2000) Estrogen stimulates heat shock protein 90 binding to endothelial nitric oxide synthase in human vascular endothelial cells. Effects on calcium sensitivity and NO release. J Biol Chem 275:5026–5030
Seval Y, Cakmak H, Kayisli UA, Arici A (2006) Estrogen-mediated regulation of p38 mitogen-activated protein kinase in human endometrium. J Clin Endocrinol Metab 91:2349–2357
Sowers JR (2004) Insulin resistance and hypertension. Am J Physiol Heart Circ Physiol 286:H1597–H1602
Taguchi K, Kobayashi T, Takenouchi Y, Matsumoto T, Kamata K (2011) Angiotensin II causes endothelial dysfunction via the GRK2/Akt/eNOS pathway in aortas from type 2 diabetic model. Pharmacol Res 64:535–546
Taguchi K, Matsumoto T, Kamata K, Kobayashi T (2012) Akt/eNOS pathway activation in endothelium-dependent relaxation is preserved in aortas from female, but not from male, type 2 diabetic mice. Pharmacol Res 65:56–65
Takenouchi Y, Kobayashi T, Taguchi K, Matsumoto T, Kamata K (2009) Gender differences in endothelial function in aortas from type 2 diabetic model mice. J Pharmacol Sci 111:91–99
Toque HA, Romero MJ, Tostes RC, Shatanawi A, Chandra S, Carneiro ZN, Inscho EW, Webb RC, Caldwell RB, Caldwell RW (2010) p38 Mitogen-activated protein kinase (MAPK) increases arginase activity and contributes to endothelial dysfunction in corpora cavernosa from angiotensin-II-treated mice. J Sex Med 7:3857–3867
Vanhoutte PM, Shimokawa H, Tang EH, Feletou M (2009) Endothelial dysfunction and vascular disease. Acta Physiol 196:193–222
Wang M, Tsai BM, Turrentine MW, Mahomed Y, Brown JW, Meldrum DR (2005) p38 mitogen activated protein kinase mediates both death signaling and functional depression in the heart. Ann Thorac Surg 80:2235–2241
Weinbrenner C, Liu GS, Cohen MV, Downey JM (1997) Phosphorylation of tyrosine 182 of p38 mitogen-activated protein kinase correlates with the protection of preconditioning in the rabbit heart. J Mol Cell Cardiol 29:2383–2391
Wyckoff MH, Chambliss KL, Mineo C, Yuhanna IS, Mendelsohn ME, Mumby SM, Shaul PW (2001) Plasma membrane estrogen receptors are coupled to endothelial nitric-oxide synthase through Galpha(i). J Biol Chem 276:27071–27076
Young PR, McLaughlin MM, Kumar S, Kassis S, Doyle ML, McNulty D, Gallagher TF, Fisher S, McDonnell PC, Carr SA, Huddleston MJ, Seibel G, Porter TG, Livi GP, Adams JL, Lee JC (1997) Pyridinyl imidazole inhibitors of p38 mitogen-activated protein kinase bind in the ATP site. J Biol Chem 272:12116–12121
Yu HP, Shimizu T, Choudhry MA, Hsieh YC, Suzuki T, Bland KI, Chaudry IH (2006) Mechanism of cardioprotection following trauma-hemorrhagic shock by a selective estrogen receptor-beta agonist: up-regulation of cardiac heat shock factor-1 and heat shock proteins. J Mol Cell Cardiol 40:185–194
Yue Y, Qin Q, Cohen MV, Downey JM, Critz SD (2002) The relative order of mK(ATP) channels, free radicals and p38 MAPK in preconditioning’s protective pathway in rat heart. Cardiovasc Res 55:681–689
Acknowledgments
This study was supported in part by the Ministry of Education, Culture, Sports, Science and Technology, Japan and by the Science Research Promotion Fund from the Promotion and Mutual Aid Corporation for Private Schools of Japan.
Conflict of interest
The authors declare to have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
We dedicate this study to our collaborator Dr. Katsuo Kamata, who passed away on April 30, 2011. He participated in the research design.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 28 kb)
Rights and permissions
About this article
Cite this article
Taguchi, K., Morishige, A., Matsumoto, T. et al. Enhanced estradiol-induced vasorelaxation in aortas from type 2 diabetic mice may reflect a compensatory role of p38 MAPK-mediated eNOS activation. Pflugers Arch - Eur J Physiol 464, 205–215 (2012). https://doi.org/10.1007/s00424-012-1131-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-012-1131-x