
Abstract
Intracellular calcium is a major coordinator of numerous aspects of cellular physiology, including muscle contractility and cell survival. In cardiac muscle, aberrant Ca2+ cycling has been implicated in a range of pathological conditions including cardiomyopathies and heart failure. The sarco(endo)plasmic reticulum Ca2+ transport adenosine triphosphatase (SERCA2a) and its regulator phospholamban (PLN) have a central role in modulating Ca2+ homeostasis and, therefore, cardiac function. Herein, we discuss the mechanisms through which SERCA2a and PLN control cardiomyocyte function in health and disease. Emphasis is placed on our newly identified PLN-binding partner HS-1-associated protein X-1 (HAX-1), which has an anti-apoptotic function and presents with numerous similarities to Bcl-2. Recent evidence indicates that proteins of the Bcl-2 family can influence ER Ca2+ content, a critical determinant of cellular sensitivity to apoptosis. The discovery of the PLN/HAX-1 interaction therefore unveils an important new link between Ca2+ homeostasis and cell survival, with significant therapeutic potential.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
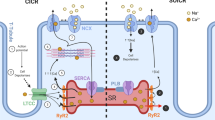
In cardiac muscle, regulation of intracellular Ca2+ homeostasis is mediated by the sarcoplasmic reticulum (SR), an intracellular membranous network surrounding the contractile machinery. Through its direct involvement in Ca2+ cycling, the SR has a critical role in controlling contraction and relaxation in cardiac muscle. During excitation–contraction coupling, Ca2+ entry through the L-type Ca2+ channel triggers the release of Ca2+ from SR Ca2+ stores via the ryanodine receptor, resulting in increased cytosolic Ca2+ levels and initiation of contraction [1–4]. During relaxation, cytosolic Ca2+ is partly sequestered back into the SR lumen by the sarco(endo)plasmic reticulum Ca2+ adenosine triphosphatase (ATPase; SERCA) pump and partly extruded to the external medium through the action of plasma-membrane Ca2+ ATPase (PMCA) and the Na+/Ca2+ exchanger (NCX; Fig. 1). In humans, about 70% of the cytosolic Ca2+ returns to the SR, while the rest is removed from the cell by the activities of NCX (28%) and PMCA (2%) [3]. As the SR represents the major source of Ca2+ store, the SR Ca2+ content and, therefore, the amount of Ca2+ available for release play a critical role in cardiac contractility.

Schematic representation of the major players implicated in excitation–contraction coupling of the heart. In response to membrane depolarization, Ca2+ entry through the L-type Ca2+ channel (LTCC) activates the ryanodine receptor (RyR) and triggers release of Ca2+ from sarcoplasmic reticulum (SR) Ca2+ stores. This results in increased cytosolic Ca2+ levels, which bind to myofibrils and initiate cardiac contraction. Reuptake of cytosolic Ca2+ to the SR lumen by the sarco(endo)plasmic reticulum Ca2+ ATPase (SERCA2a) pump and removal to the external medium through the action of plasma-membrane Ca2+ ATPase (PMCA) and the Na+/Ca2+ exchanger (NCX) results in cardiac relaxation. PLN phospholamban, JNT junction, TRI triadin, CSQ calsequestrin
Evidence from human and experimental studies has indicated that defects in SR Ca2+ handling are associated with attenuated contractility, which can progress to heart failure [5]. In particular, cardiac systolic dysfunction and impaired cardiac relaxation are common hallmarks of heart failure. At the cellular level, depressed relaxation reflects impaired removal of cytosolic Ca2+ and reduced cardiac SR loading. This could be due to decreased SERCA2 protein levels and/or increased inhibition by its regulatory protein PLN [6, 7]. Importantly, the progression of heart failure, which is marked by a decline in cardiac function, has been associated with cardiomyocyte loss through activation of the apoptotic pathways [8–10]. This occurs through a signaling cascade, which includes cytochrome c release from the mitochondria, activation of caspases, protein, and DNA degradation [8, 11, 12]. Loss of myocytes represents an important component of cardiac remodeling and contributes to the transition from an adaptive myocardial condition to end-stage heart failure [13].
This review focuses on the critical role of the SERCA2a/PLN complex in regulating SR Ca2+ cycling and cardiac contractility, and presents current knowledge on its detailed mode of function. An emphasis is given on the emerging role of HAX-1 in cardiac muscle, which through its interaction with PLN, could represent a link between Ca2+ homeostasis and cell survival. Deciphering the molecular mechanisms bridging Ca2+ cycling aberrations with apoptosis and, therefore, advanced stages of disease progression could unveil promising new therapeutic targets.
SERCA2a activity is critical in regulation of SR Ca2+ homeostasis
There are three different SERCA genes (human nomenclature ATP2A1-3), each encoding at least two different protein isoforms. Cardiac muscle expresses SERCA2a, a 110-kDa transmembrane protein that functions by transporting two Ca2+ from the cytosol to the SR lumen at the expense of adenosine triphosphate (ATP) hydrolysis [14–18]. As SERCA2a activity controls both the rate of cytosolic Ca2+ removal and the degree of SR Ca2+ load, it represents a fundamental determinant of both cardiac relaxation and contraction.
Transgenic animal models have been developed to define the functional role of the SERCA pump in Ca2+ homeostasis and cardiac physiology. Transgenic mice overexpressing SERCA2a by 1.2- or 1.5-fold exhibited increased SR Ca2+ transport and enhanced rates of cardiac contractility and relaxation [19–21]. No cardiac pathology was observed in these animals, suggesting that SERCA2a overexpression can be tolerated by the heart. On the other hand, absence of the SERCA2 gene is lethal, with the homozygous null (SERCA2−/−) mice dying early in development [22]. Heterozygous (SERCA2+/−) mice are viable, showing 35% decrease in SERCA2 protein levels as a result of the loss of one copy of SERCA2 allele. Although no cardiac pathology was exhibited at rest, reduction in SERCA2 levels in combination with an increased hemodynamic load resulted in an accelerated pathway to heart failure [23]. These mice show impaired intracellular Ca2+ homeostasis and decreased rates of cardiac contractile function, a finding that demonstrates the requirement for two functional copies of the SERCA2 gene for effective SR Ca2+ cycling and cardiac function [22, 24].
Mutation screening on specific SERCA2a genomic regions, corresponding to the PLN interacting region as well as the SERCA2a phosphorylation and nucleotide-binding domains, did not identify any genetic variants resulting in amino acid alterations in adult dilated cardiomyopathy patients [25]. This suggests that the SERCA2a sequence is highly conserved among individuals. Loss of function mutations in the SERCA2 gene are known to cause Darier’s disease, an autosomal dominant skin disorder [26–28]. Interestingly, however, these patients do not exhibit any cardiac pathology.
Although no SERCA2a mutations have been identified in heart disease patients to date, significant expression changes have been observed in failing hearts. Specifically, experimental models of myocardial failure were found to exhibit reduced SERCA2a messenger RNA (mRNA) and protein levels, suggesting an impairment in cytosolic Ca2+ removal, SR Ca2+ load and overall Ca2+ cycling [29–31]. Similarly, decreased SERCA2a expression and SR Ca2+ transport were observed in human failing hearts [32–37]. Taken together, these findings demonstrate a direct correlation between SERCA2a levels, SR Ca2+ transport, and heart failure.
SERCA2a-binding partners
While experimental evidence suggests that proteins involved in SR Ca2+ release, such as the ryanodine receptor, function as part of a macromolecular complex, the existence of such a protein complex in the regulation of SR Ca2+ uptake has only recently begun to emerge. In particular, SERCA2a has been found to interact with proteins of the SR lumen, such as histidine-rich calcium-binding protein [38] and calreticulin [39], while its cytosolic region has been shown to bind to S100A1 [40], acylphosphatase [41], and Bcl-2 (see section below) [42, 43]. Furthermore, PLN and sarcolipin have been found to bind to the cytosolic and/or transmembrane domains of SERCA2a, with accumulating evidence suggesting that these interactions lead to inhibition of the pump’s affinity for Ca2+ [18, 44]. PLN has proven to be a major regulator of SERCA2a activity, and so far, it is the only SERCA2a-associated protein directly involved in cardiac disease development, including heart failure.
PLN is a major regulator of SERCA2a activity
PLN is a 52-amino-acid transmembrane protein of the SR that is expressed mainly in cardiac but also in smooth and slow-twitch skeletal muscles [45–47]. Based on its protein sequence, PLN appears to contain three domains, namely a largely helical cytosolic domain IA (amino acids 1–20), an unstructured domain IB (amino acids 21–30), and a domain II (amino acids 31–52) that forms a transmembrane helix [45, 48]. Detailed cross-linking and site-directed mutagenesis studies have demonstrated that residues in both the cytoplasmic (IA and IB domains) and the transmembrane portions of PLN can interact directly with SERCA2a [49–54]. PLN can be phosphorylated at serine 16 by the cAMP-dependent protein kinase (PKA) and threonine 17 by the Ca2+–calmodulin-dependent protein kinase (CaMKII) [55, 56]. Moreover, it has been shown that PLN exists in both pentameric and monomeric forms, where the monomer is considered to be the functionally active unit, while the pentamer may act as a reservoir [57, 58].
PLN can interact with SERCA2a and inhibit its activity, thus preventing Ca2+ entry to the SR through SERCA2a [47]. Specifically, PLN acts by lowering the apparent affinity of SERCA2a for Ca2+ but has little or no effect on the maximal velocity rate (V max) at saturating Ca2+ and ATP concentrations [59]. At low Ca2+ concentrations, PLN interacts and reversibly inhibits the affinity of SERCA2a for Ca2+, while elevations in Ca2+ concentration lead to dissociation of the SERCA2a/PLN protein complex, an effect that may be due to Ca2+-induced conformational changes of SERCA2a [18]. The phosphorylation state of PLN has also been shown to affect SERCA2a activity. In its dephosphorylated state, PLN interacts with SERCA2a and inhibits the enzyme’s affinity for Ca2+. However, upon β-adrenergic stimulation, phosphorylation of PLN relieves this inhibitory effect on SERCA2a, leading to enhanced SR Ca2+ transport [60, 61]. These findings indicate the crucial functional role of PLN in regulating SERCA2a activity in a Ca2+ or phosphorylation-dependent manner and further suggest its involvement as a key determinant of β-adrenergic stimulation in the heart, with potential applications in pharmacologic therapeutic approaches.
Lessons on PLN function from transgenic animal models
The importance of PLN in cardiac physiology is further emphasized by studies on genetically modified mouse models. Ablation of PLN was associated with an increased affinity of SERCA2a for Ca2+, increased SR Ca2+ uptake, and increased contractile parameters, which resulted in an overall hypercontractile cardiac function that persisted throughout aging [62, 63]. Interestingly, the highly stimulated function of PLN null hearts could be minimally stimulated by β-adrenergic agonists [64, 65]. Similarly, heterozygous (PLN+/−) mice with 60% reduction in PLN levels exhibited significant increases in contractile parameters, although these increases were lower than those exhibited by the PLN null (PLN−/−) mice [66]. Conversely, two- and fourfold overexpression of PLN in mouse heart resulted in decreased SERCA2a affinity for Ca2+, decreased contractile parameters, and depressed left ventricular function. Stimulation with β-adrenergic agonists restored the contractile parameters to levels similar to wild type by alleviating the inhibitory effects of PLN [6, 46]. Taken together, these animal model studies have demonstrated the fundamental role of PLN in SR Ca2+ cycling and cardiac function under basal and β-adrenergic stimulated conditions.
In addition to PLN-deficient or PLN-overexpressing mouse models, transgenic mice overexpressing specific PLN mutants have allowed the in vivo examination of the importance of specific amino acid residues in PLN function and elucidation of their role in SR Ca2+ cycling. Chronic inhibition of SERCA2a was observed upon overexpression of superinhibitory PLN mutants (Asn27Ala, Leu37Ala, Ile40Ala, and Val49Gly), which have been shown to alter the ratio of PLN monomers and pentamers, thus affecting SERCA2a inhibition [58, 67–69]. All transgenic animals exhibited depressed SERCA2a function, decreased Ca2+ kinetics, and impaired contractility. As a result of SERCA2a chronic inhibition, some of these models developed significant left ventricular hypertrophy, which progressed to cardiac dysfunction and heart failure [58, 68, 69]. Therefore, these findings suggest that specific amino acids are critical for PLN function, as changes at these residues can cause alterations in SR Ca2+ handling and subsequently lead to cardiac remodeling and progression to heart failure.
Detailed evaluation of the functional significance of PLN dual-site phosphorylation at Ser16 and Thr17 was accomplished, following the generation of transgenic animals expressing phosphorylation-site-specific PLN mutants [70–73]. While Ser16 phosphorylation may be a prerequisite for Thr17 phosphorylation, it has been suggested that Ser16 phosphorylation may mediate the β-agonist response, and Thr17 may play an important role in frequency-dependent increase of cardiac contraction and relaxation.
Collectively, the findings from the numerous transgenic animal models have provided evidence for the pivotal role of PLN in the regulation of SR Ca2+ homeostasis and suggest its potential use as a promising therapeutic target for heart disease.
PLN mutations lead to cardiomyopathy and heart failure
Identification of PLN mutations in patients with cardiomyopathy and heart failure highlight the critical role of PLN and SR Ca2+ cycling in controlling cardiac function and provide clues on the molecular mechanisms underlying disease pathogenesis.
To date, six different PLN genetic variations have been reported [74–79]. Two of these mutations (R9C and R14Del) are characterized by gain-of-function, causing chronic inhibition of SERCA2a activity. Substitution of arginine by cysteine at amino acid position 9 (R9C) was linked with autosomal dominant inheritance of dilated cardiomyopathy and heart failure in a large American family [74]. The functional consequences of this mutation were evaluated by cellular and biochemical studies performed in a heterologous cell culture system, through the analysis of a generated transgenic mouse model and also by studies on human cardiac tissue obtained from explanted hearts. A significant reduction in the levels of PLN phosphorylation was observed, resulting in impaired cardiomyocyte Ca2+ handling and cardiac function. Overexpression of the R9C mutation prevented cAMP-dependent PKA phosphorylation of wild-type PLN by local trapping of PKA in a stable mutant PLN–PKA complex and thus minimizing the PKA-mediated relief of PLN inhibitory function on SERCA2a. While the R9C mutation itself was not a strong inhibitor of SERCA2a, its ability to block PLN phosphorylation caused a dominant effect, leading to chronic inhibition of SERCA2a [74]. The other gain-of-function mutation is a heterozygous deletion of arginine at amino acid residue 14 (R14del), which was associated with inherited dilated cardiomyopathy and premature death [75]. Transgenic mice overexpressing this PLN mutation recapitulated the human dilated cardiomyopathy phenotype and resulted in premature death. In vitro and in vivo assessment of the functional consequences of the mutation determined a dominant effect of R14del, which could not be reversed upon PKA phosphorylation, therefore resulting in chronic inhibition of SERCA2a activity. Through this chronic inhibition, both PLN mutations impair SR Ca2+ cycling, leading to cardiac dysfunction and heart failure.
A loss-of-function human PLN mutation, resulting in the substitution of a leucine residue at position 39 with a premature stop codon (L39X), was identified in two families with hereditary heart failure [76]. Adenoviral overexpression of PLN L39X mutation in rat cardiomyocytes did not have an effect on SERCA2a activity. Moreover, overexpression of PLN L39X mutant in HEK293 cells demonstrated that the mutant protein was unstable, a finding that was also verified in the explanted cardiac tissue of a heart failure patient where no PLN protein was detected. This indicates the existence of a naturally occurring “PLN null mutation” associated with lack of inhibition on SERCA2a activity. However, in contrast to the hypercontractile phenotype observed in the PLN null mouse, deficiency of PLN in human hearts results in significantly impaired cardiac function and leads to the development of heart failure. A possible explanation for this is that PLN may be of paramount importance in humans, as it is required to maintain a high Ca2+ reserve for proper cardiac function through human life [80]. The critical role of PLN in the human heart was further emphasized by the strong inhibitory function elicited through the expression of the “human PLN” in the mouse null heart, which led to cardiac remodeling [80].
In addition to the above changes, three genetic variations within the upstream noncoding region of the gene (A to G at −77 bp, C to G at −42 bp, or A to C at −36 bp) have also been reported in patients with cardiomyopathy. Functional analysis of the effect of these changes in cultured cells determined alterations in the transcriptional activity of the PLN promoter, suggesting that they may lead to alterations in SR Ca2+ homeostasis and disease pathogenesis [77–79].
Taken together, these studies emphasize the importance of PLN in regulating SERCA2a activity and, in this way, SR Ca2+ homeostasis and cardiac function. Importantly, chronic inhibition or absence of PLN activity has been directly associated with heart failure.
PLN interacts with the anti-apoptotic protein HAX-1
The role of PLN in SR Ca2+ cycling has been carefully characterized. However, its function in other molecular pathways is only starting to come to light. Discovering these pathways would contribute toward the better understanding of heart failure pathogenesis and could reveal new therapeutic targets.
Recently, we reported the identification of HS-1-associated protein X-1 (HAX-1), a ∼35-kDa ubiquitously expressed mitochondrial protein with anti-apoptotic function, as a PLN-binding partner [81]. The minimal binding region of HAX-1 was mapped to a C-terminal fragment, encoding amino acids 203–245, whereas the PLN-binding region contained amino acids 16–22, a region that includes both the Ser16 and Thr17 phosphorylation sites. This region of PLN contains residues Ile18, Glu19, Met20, and Pro21, which are suggested to form a turn connecting the two α-helical stretches of the protein. This conformation may provide the necessary flexibility to the protein that could be important in the kinetics of monomer–pentamer formation, in PLN phosphorylation and dephosphorylation, as well as in its association with SERCA2a. It was therefore proposed that binding of HAX-1 to this region of PLN may represent a regulatory mechanism on any of these reactions and could provide the means for controlling the conformation and activity of PLN [81]. Similarly to the SERCA2/PLN interaction, binding of HAX-1 to PLN was found to be diminished upon phosphorylation of PLN by cAMP-dependent protein kinase and increasing Ca2+ concentrations, thus indicating that HAX-1 may regulate the functional properties of PLN in the heart. Through this association, HAX-1 could therefore have an important role on SR Ca2+ cycling.
HAX-1 was originally identified to interact with HS1, a protein with proposed involvement in B cell signal transduction in hemopoietic cells [82]. Subsequently, HAX-1 has been found to interact with a number of cytoskeletal and viral proteins (Table 1). Although the biological significance of these interactions remains unclear, the existence of multiple interacting proteins for HAX-1 indicates its involvement in multiple cellular pathways. Immunofluorescence microscopy studies have localized HAX-1 to the mitochondria, endoplasmic reticulum, and the nuclear envelope [82, 85, 86, 88, 89, 95, 96]. We previously reported a preferential mitochondrial localization of HAX-1 after transient transfections in HEK293 cells. Interestingly, upon cotransfection with PLN, HAX-1 underwent cellular redistribution and colocalized with PLN at the ER [81]. A similar finding was also reported in the presence of the HAX-1 interacting protein Vpr. Specifically, coexpression of HAX-1 with the predominantly nuclear localized protein Vpr resulted in the codistribution of the two proteins in cytoplasmic bodies outside the nucleus and mitochondria [86]. It was therefore proposed that the subcellular localization and functional properties of HAX-1 may vary among different tissues, depending on which interacting partners are available [81].
Based on its weak sequence similarity to Nip3 and its homology to Bcl-2 domains BH1 and BH2, HAX-1 was initially proposed to be involved in promoting cell survival. Its anti-apoptotic function was supported by experimental evidence after the identification of its interaction with K15 Kaposis’s sarcoma, Omi/HtrA2 protease, and Vpr [85, 86, 97]. Studies in Jurkat, HEK293, or Hela cell lines demonstrated that HAX-1 overexpression provides protection against Fas treatment, γ-irradiation, serum deprivation, Bax overexpression, or hypoxia/reoxygenation-induced cell death [81, 82, 85, 96]. Furthermore, HAX-1 was found to be overexpressed in psoriasis, a chronic inflammatory disease characterized by increased proliferation and diminished susceptibility to apoptosis, implicating HAX-1 in the regulation of cell viability [101].
Even though HAX-1 is highly expressed in skeletal and cardiac muscles, its role in muscle function has recently been unraveled through the identification of its interactions with caspase-9 [96] and PLN [81]. The PLN/HAX-1 interaction was suggested to play a role in modulating Ca2+ cycling in the regulation of cardiac contractility. Importantly, this complex may regulate SR/ER Ca2+ homeostasis, which could affect mitochondrial Ca2+ redistribution and in turn influence mitochondrial Ca2+ accumulation and initiation of the apoptotic cell death signaling cascade [102]. In the presence of PLN, HAX-1 exhibited an enhanced anti-apoptotic effect after hypoxia/reoxygenation-induced cell death [81]. This finding further emphasizes the critical functional role of the PLN/HAX-1 interaction, which represents a new link between Ca2+ handling and cell survival.
The identification of the HAX-1/caspase-9 interaction provided new evidence linking HAX-1 with the apoptotic pathway. Specifically, adenoviral overexpression of HAX-1 in rat cardiomyocytes resulted in significant attenuation of hypoxia/reoxygenation-induced cell death. This was shown to be mediated through the direct interaction of HAX-1 with caspase-9, probably occurring within the mitochondria, which in turn led to inhibition of caspase-9 processing and subsequent inhibition of caspase-3 activation [96]. Based on its interaction with caspase-9, an initiator caspase of apoptosis, HAX-1 was proposed to act during early stages of apoptosis.
Collectively, these findings highlight the involvement of HAX-1 in SR/ER Ca2+ cycling, mediated through PLN, but also implicate HAX-1 in cell survival via inhibition of the caspase-9 apoptotic pathway. This critical new link between Ca2+ cycling and apoptosis could hold important clues for understanding the association of SERCA2a and PLN with heart failure.
HAX-1 and disease
HAX-1 deficiency was recently associated with autosomal recessive severe congenital neutropenia (Kostmann disease) [103]. Severe congenital neutropenia represents a primary immunodefiency syndrome comprising of a genetically heterogeneous group of disorders, which is characterized by low peripheral blood neutrophils and myeloid maturation arrest [104]. Three different nonsense HAX-1 mutations (W44X, R86X and Q190X) have been identified so far in different populations [103, 105, 106]. Analysis of the functional significance of the W44X mutation in cells from affected individuals determined a critical role for HAX-1 in the maintenance of the inner mitochondrial membrane potential. Specifically, deficiency of this protein caused destabilization of membrane potential, leading to increased apoptosis in myeloid cells and disease progression [103]. Given the recently identified involvement of HAX-1 in cardiac muscle, an important biological question yet to be answered is whether these patients present cardiac functional defects.
Although there have been no reports directly associating HAX-1 with heart disease, its potential involvement in Ca2+ homeostasis through PLN interaction makes it an interesting candidate for future genetic studies.
HAX-1 and the Bcl-2 family of proteins
Regulation of SR Ca2+ homeostasis is of vital importance for cardiac cell survival, as defects in SR Ca2+ cycling have been associated with cardiac dysfunction, heart failure, and death. The identification of HAX-1, a mitochondrial protein with anti-apoptotic function, as a binding partner of PLN indicates the direct association between a mitochondrial and SR protein, and provides new insights into key players and mechanisms modulating Ca2+ cycling in cardiac contractility and cell death. Accumulating evidence has unraveled a novel aspect of the Bcl-2 anti-apoptotic protein family in promoting cell survival through regulation of ER Ca2+ homeostasis. Interestingly, HAX-1 presents a number of structural and functional similarities to Bcl-2. Therefore, while the precise function of the anti-apoptotic protein HAX-1 in cardiac muscle is slowly beginning to emerge, its similarity to Bcl-2 implicates HAX-1 in the regulation of cell survival. This can give rise to novel hypotheses regarding the molecular mechanisms of HAX-1 function and potentially uncover new links between Ca2+ cycling and apoptosis, especially in cardiac disease.
In mammals, the Bcl-2 gene family consists of at least 12 members [107], encoding proteins with pro- or anti-apoptotic function that exert a key role in the regulation of apoptosis. These proteins are important in controlling the mitochondrial pathway of apoptosis and are critical in the decision toward initiation of the cellular death cascade, which leads to release of pro-apoptotic factors, caspase activation, and commitment to apoptosis. Bcl-2, the founding member of this family, is an anti-apoptotic protein that provides protection against various apoptosis-inducing agents by maintaining the outer mitochondrial membrane integrity and, in this way, preventing the release of pro-apoptotic factors. A characteristic feature of the Bcl-2 protein family is the presence of at least one of the four conserved Bcl-2 homology (BH) domains, which are thought to be important in mediating protein interactions between family members [108, 109]. Since HAX-1 presents structural similarity to Bcl-2 due to its sequence homology to the Bcl-2 domains BH1 and BH2, it becomes intriguing to speculate that these two mitochondrial proteins might participate in similar molecular pathways.
The Bcl-2 protein family and its role in ER Ca2+ homeostasis
Mitochondria are central players in the initiation of apoptosis, and numerous studies have focused on their role in regulating the molecular pathways leading to cell death. Recent experimental evidence has demonstrated the critical role of ER Ca2+ content in determining cellular sensitivity to apoptotic stimuli. In particular, increased ER Ca2+ load was associated with sensitization to apoptosis, while decreased ER Ca2+ levels are thought to provide protection against apoptotic stimuli [110, 111]. A communication between the ER and the mitochondria was proposed to represent a critical mechanism in determining cell fate, hence defining a functional role for the ER as a new gateway to apoptosis [110]. The spatial organization and proximity of ER and mitochondrial membranes has been shown to result in the existence of close contact sites between the two organelles [102, 112–114]. This may facilitate Ca2+ movement between the two organelles, further pinpointing their critical relationship in defining cellular fate. The direct communication between ER and mitochondria could be of particular importance in tissues such as cardiac muscle, which require accurate and tight regulation of Ca2+ homeostasis coupled to bioenergetics on a beat-to-beat basis.
Over the past few years, it has become evident that anti-apoptotic proteins may be important in promoting cell survival by regulating homeostasis of additional organelles. Studies on Bcl-2 have determined that, in addition to its mitochondrial localization, it is also present in other membrane cellular compartments including the ER and the outer nuclear membrane [115–117]. Several other proteins of the Bcl-2 family, such as BAX, BAK, and Bcl-XL, also localize to the ER [118–120]. While the function of Bcl-2 at these extra-mitochondrial sites remains unclear, experimental evidence suggests that it is most likely related to cell survival.
The use of specific organelle-targeted mutants showing restricted subcellular localization has been very important on deciphering the role of Bcl-2 at the ER membrane and thus further defining its contribution to regulation of cellular apoptosis. An ER-localized Bcl-2 mutant (Bcl-2-cb5), which was generated by exchanging the C-terminal tail of wild-type Bcl-2 with the corresponding sequence from an ER-specific isoform of cytochrome b5, has been shown to protect against a variety of apoptotic-stimuli including serum starvation, ionizing radiation, brefeldin A, ceramide, and staurosporine [121–124]. Moreover, this ER-localized Bcl-2 mutant can inhibit the loss of mitochondrial membrane potential [124] and cytochrome c release [123], suggesting that Bcl-2-cb5 can indirectly protect the mitochondria. These studies indicate that Bcl-2 does not need to be associated with mitochondria to inhibit cytochrome c release and have implicated Bcl-2 in an apoptotic crosstalk between the ER and the mitochondria. Similarly to Bcl-2, HAX-1 can localize at the mitochondria or the ER, exhibiting anti-apoptotic properties in both sites [81]. Thus, HAX-1 could represent a new mediator of the ER–mitochondria crosstalk, with direct implications in cell death decisions.
Considering the critical function of ER in Ca2+ homeostasis, the role of Bcl-2 as a regulator of ER Ca2+ levels has been evaluated. In human breast epithelial cells and mouse lymphoma cells, Bcl-2 overexpression was shown to result in increased ER Ca2+ concentration, a finding which was proposed to correlate with maintenance of cell growth and viability [43, 125]. On the other hand, studies in HeLa cells [126], human prostate cancer cells [127], and HEK293 cells [128] identified decreased ER Ca2+ levels as the result of Bcl-2 overexpression. This decrease in ER Ca2+ concentration was proposed to enhance cell survival by reducing the amount of Ca2+ available for release and subsequent uptake by the mitochondria after apoptotic stimulus. The above discrepancies on the effect of Bcl-2 overexpression could be due to differences between cells lines, Bcl-2 expression levels, and Ca2+ measurement methodologies [108].
The anti-apoptotic protein Bcl-XL has also been found to modulate ER Ca2+ levels and cell survival, further implicating anti-apoptotic proteins in the regulation of Ca2+ homeostasis. Specifically, Bcl-XL overexpression in a murine T-cell line caused a dose-dependent decrease in the expression levels of type 1 inositol 1,4,5-triphosphate receptor (IP3R) and thus in the amount of receptor-mediated Ca2+ released after IP3 stimulation [129, 130]. The identification of an interaction between Bcl-XL and IP3R has provided additional evidence for the involvement of Bcl-XL in the regulation of ER Ca2+ homeostasis, leading to enhanced cellular bioenergetics and preserved survival [131].
In addition to anti-apoptotic proteins, the pro-apoptotic proteins BAX and BAK have also been shown to modulate ER Ca2+ content, to affect the amount of Ca2+ taken up by the mitochondria and subsequently to induce apoptosis [119, 132]. Analysis of mouse embryonic fibroblasts (MEFs) from Bax and Bak double-knockout mice has provided valuable information on the role of these apoptotic proteins as regulators of ER Ca2+ homeostasis. A decreased ER Ca2+ content in Bax and Bak double-knockout MEF cells resulted in attenuated Ca2+ uptake by the mitochondria after ER Ca2+ release and consequent increased resistance to ceramide and arachidonic-acid-induced cell death [133]. Overexpression of BAX or SERCA corrected the ER Ca2+ imbalance in these cells and restored apoptotic response to cell death stimuli, thus demonstrating the involvement of BAX in modulating ER Ca2+ homeostasis and promoting cell death. The decreased levels of ER Ca2+ in Bax and Bak double-knockout MEF cells were associated with increased passive leak of Ca2+ and hyperphosphorylated state of inositol triphosphate receptor type 1 (IP3R-1) [134]. These findings suggest a role for BAX and BAK in both ER and mitochondria, with the two proteins potentially providing a Ca2+-dependent crosstalk between the two organelles.
Taken together, these studies provide significant evidence on the involvement of Bcl-2 family proteins in the regulation of ER Ca2+ homeostasis, with direct effects on ER Ca2+ content and cell survival. In parallel to these proteins, HAX-1 is highly likely to play a similar role. Specifically, HAX-1 interacts with PLN, the major regulator of SERCA2a activity, and exhibits anti-apoptotic properties when localized to the ER, thus implicating it in the regulation of ER/SR Ca2+ homeostasis and cell survival (Fig. 2).

HAX-1 is a promising new link between Ca2+ cycling and cell survival. During cardiac contraction, part of the SR Ca2+ content that is released from RyR channels can be taken up by closely positioned mitochondria. Sequestration of Ca2+ to the SR by SERCA2a results in refilling of the SR. PLN exerts inhibitory effects on SERCA2a activity and thus represents a critical regulator of SR Ca2+ homeostasis. The anti-apoptotic protein HAX-1 interacts with PLN, an association directly implicating HAX-1 in the regulation of SR Ca2+ uptake to promote cell survival. If SR Ca2+ uptake is tightly regulated, then SR Ca2+ content is maintained at optimal levels, and subsequently, mitochondria take up optimal amounts of Ca2+ (thin arrows). This can positively regulate mitochondrial bioenergetics to support cell function and promote cell survival. Conversely, deregulation of SR Ca2+ uptake, as seen in heart failure, can affect the amount of SR Ca2+ available for release and subsequently cause mitochondrial Ca2+ overload (thick arrows). This would result in caspase activation, initiation of the apoptotic signaling cascade, and cell death. HAX-1 is emerging as a critical player in the regulation of SR Ca2+ homeostasis, with direct effects on SR Ca2+ content and cell survival
Bcl-2 interacts with SERCA and modulates its activity
Although the precise mechanism through which Bcl-2 regulates ER Ca2+ homeostasis is still unclear, experimental evidence supports a number of different alternatives [42, 43, 128, 135–137]. Of particular interest is the effect of Bcl-2 on SERCA activity, mediated through their direct association that parallels the PLN/HAX-1 binding.
Overexpression of Bcl-2 in human breast epithelial cells and prostate cancer cells was shown to influence SERCA protein expression, resulting in an increase or decrease of SERCA2 or SERCA2b, respectively. Although the underlying cause of this discrepancy has not been clarified, these findings implicate Bcl-2 in SERCA expression regulation and provide a possible explanation for the observed alterations in ER Ca2+ levels [43, 127]. Immunoprecipitation studies in DHL-4 human lymphoma cell line, MCF10A breast epithelial cells and rat skeletal muscle identified a direct interaction between Bcl-2 and SERCA1 or SERCA2 [42, 43]. Further in vitro analysis of this interaction determined that Bcl-2 inhibits SERCA activity in a time- and dose-dependent manner, and causes a conformational transition of SERCA, leading to partial unfolding of the protein [42]. It was recently proposed that SERCA inactivation by Bcl-2 occurs through displacement of SERCA from caveolae-related domains of the SR into a different membrane environment [138]. Although the binding sites involved in the interaction between Bcl-2 and SERCA have not been determined, current findings suggest that the occurrence of a tight association with the ATP-binding or the Ca2+-binding domains of SERCA is probably unlikely. It is possible, however, that a transient interaction of Bcl-2 with these domains may trigger transition of SERCA into an inactive conformation and may therefore release Bcl-2 from the protein complex [138].
Collectively, these findings reveal the critical role of Bcl-2 in modulating SERCA activity and ER Ca2+ levels. Furthermore, when considered jointly, the HAX-1 and Bcl-2 association with the SERCA/PLN complex may give rise to the hypothesis that there is a direct link between the SR Ca2+ uptake complex and apoptosis, with direct implications in cardiac function and disease development, such as heart failure.
Conclusion
Ca2+ cycling is a critical determinant of cardiomyocyte contractility and cardiac function. A major regulator of SR Ca2+ uptake is the SERCA2a/PLN complex, with aberrations in its function being directly associated with heart failure. HAX-1, the novel binding partner of PLN, presents with numerous structural and functional similarities to the Bcl-2 family and is therefore emerging as an intriguing new link between Ca2+ homeostasis and cell survival. Over the past few years, it has become apparent that a crucial aspect in the function of anti-apoptotic proteins of the Bcl-2 family includes regulation of ER Ca2+ homeostasis and protection of mitochondria from Ca2+ overload. Similarly, HAX-1 could be implicated in the promotion of cell survival by indirectly or even directly affecting SERCA2a activity. This putative role of HAX-1 as a mediator between SR Ca2+ content and cell survival needs to be further evaluated. Elucidating the precise pathways implicated in this process will be invaluable in understanding the pathogenetic mechanisms of heart failure and importantly could unveil promising new therapeutic targets.
References
Fabiato A, Fabiato F (1979) Calcium and cardiac excitation–contraction coupling. Annu Rev Physiol 41:473–84
Lederer WJ, Berlin JR, Cohen NM, Hadley RW, Bers DM, Cannell MB (1990) Excitation–contraction coupling in heart cells. Roles of the sodium–calcium exchange, the calcium current, and the sarcoplasmic reticulum. Ann NY Acad Sci 588:190–206
Bers DM (2002) Cardiac excitation–contraction coupling. Nature 415:198–205
Bers DM (2002) Calcium and cardiac rhythms: physiological and pathophysiological. Circ Res 90:14–17
Houser SR, Piacentino V III, Weisser J (2000) Abnormalities of calcium cycling in the hypertrophied and failing heart. J Mol Cell Cardiol 32:1595–1607
Dash R, Frank KF, Carr AN, Moravec CS, Kranias EG (2001) Gender influences on sarcoplasmic reticulum Ca2+-handling in failing human myocardium. J Mol Cell Cardiol 33:1345–1353
Hasenfuss G, Pieske B (2002) Calcium cycling in congestive heart failure. J Mol Cell Cardiol 34:951–969
Gustafsson AB, Gottlieb RA (2007) Bcl-2 family members and apoptosis, taken to heart. Am J Physiol Cell Physiol 292:C45–C51
Kang PM, Izumo S (2000) Apoptosis and heart failure: a critical review of the literature. Circ Res 86:1107–1113
Narula J, Kolodgie FD, Virmani R (2000) Apoptosis and cardiomyopathy. Curr Opin Cardiol 15:183–188
Gill C, Mestril R, Samali A (2002) Losing heart: the role of apoptosis in heart disease—a novel therapeutic target? FASEB J 16:135–146
Narula J, Haider N, Arbustini E, Chandrashekhar Y (2006) Mechanisms of disease: apoptosis in heart failure—seeing hope in death. Nat Clin Pract Cardiovasc Med 3:681–688
Khoynezhad A, Jalali Z, Tortolani AJ (2007) A synopsis of research in cardiac apoptosis and its application to congestive heart failure. Tex Heart Inst J 34:352–359
Lytton J, Zarain-Herzberg A, Periasamy M, MacLennan DH (1989) Molecular cloning of the mammalian smooth muscle sarco(endo)plasmic reticulum Ca2+-ATPase. J Biol Chem 264:7059–7065
Zarain-Herzberg A, MacLennan DH, Periasamy M (1990) Characterization of rabbit cardiac sarco(endo)plasmic reticulum Ca2(+)-ATPase gene. J Biol Chem 265:4670–4677
MacLennan DH, Rice WJ, Green NM (1997) The mechanism of Ca2+ transport by sarco(endo)plasmic reticulum Ca2+-ATPases. J Biol Chem 272:28815–28818
Campbell AM, Kessler PD, Sagara Y, Inesi G, Fambrough DM (1991) Nucleotide sequences of avian cardiac and brain SR/ER Ca(2+)-ATPases and functional comparisons with fast twitch Ca(2+)-ATPase. Calcium affinities and inhibitor effects. J Biol Chem 266:16050–16055
Asahi M, Nakayama H, Tada M, Otsu K (2003) Regulation of sarco(endo)plasmic reticulum Ca2+ adenosine triphosphatase by phospholamban and sarcolipin: implication for cardiac hypertrophy and failure. Trends Cardiovasc Med 13:152–157
He H, Giordano FJ, Hilal-Dandan R, Choi DJ, Rockman HA, McDonough PM, Bluhm WF, Meyer M, Sayen MR, Swanson E, Dillmann WH (1997) Overexpression of the rat sarcoplasmic reticulum Ca2+ ATPase gene in the heart of transgenic mice accelerates calcium transients and cardiac relaxation. J Clin Invest 100:380–389
Baker DL, Hashimoto K, Grupp IL, Ji Y, Reed T, Loukianov E, Grupp G, Bhagwhat A, Hoit B, Walsh R, Marban E, Periasamy M (1998) Targeted overexpression of the sarcoplasmic reticulum Ca2+-ATPase increases cardiac contractility in transgenic mouse hearts. Circ Res 83:1205–1214
Vetter R, Rehfeld U, Reissfelder C, Weiss W, Wagner KD, Gunther J, Hammes A, Tschope C, Dillmann W, Paul M (2002) Transgenic overexpression of the sarcoplasmic reticulum Ca2+ ATPase improves reticular Ca2+ handling in normal and diabetic rat hearts. FASEB J 16:1657–1659
Periasamy M, Reed TD, Liu LH, Ji Y, Loukianov E, Paul RJ, Nieman ML, Riddle T, Duffy JJ, Doetschman T, Lorenz JN, Shull GE (1999) Impaired cardiac performance in heterozygous mice with a null mutation in the sarco(endo)plasmic reticulum Ca2+-ATPase isoform 2 (SERCA2) gene. J Biol Chem 274:2556–2562
Schultz Jel J, Glascock BJ, Witt SA, Nieman ML, Nattamai KJ, Liu LH, Lorenz JN, Shull GE, Kimball TR, Periasamy M (2004) Accelerated onset of heart failure in mice during pressure overload with chronically decreased SERCA2 calcium pump activity. Am J Physiol Heart Circ Physiol 286:H1146–H1153
Ji Y, Lalli MJ, Babu GJ, Xu Y, Kirkpatrick DL, Liu LH, Chiamvimonvat N, Walsh RA, Shull GE, Periasamy M (2000) Disruption of a single copy of the SERCA2 gene results in altered Ca2+ homeostasis and cardiomyocyte function. J Biol Chem 275:38073–38080
Schmidt AG, Haghighi K, Frank B, Pater L, Dorn GW, Walsh RA, Kranias EG (2003) Polymorphic SERCA2a variants do not account for inter-individual differences in phospholamban–SERCA2a interactions in human heart failure. J Mol Cell Cardiol 35:867–870
Miyauchi Y, Daiho T, Yamasaki K, Takahashi H, Ishida-Yamamoto A, Danko S, Suzuki H, Iizuka H (2006) Comprehensive analysis of expression and function of 51 sarco(endo)plasmic reticulum Ca2+-ATPase mutants associated with Darier disease. J Biol Chem 281:22882–22895
Sakuntabhai A, Ruiz-Perez V, Carter S, Jacobsen N, Burge S, Monk S, Smith M, Munro CS, O'Donovan M, Craddock N, Kucherlapati R, Rees JL, Owen M, Lathrop GM, Monaco AP, Strachan T, Hovnanian A (1999) Mutations in ATP2A2, encoding a Ca2+ pump, cause Darier disease. Nat Genet 21:271–277
Ruiz-Perez VL, Carter SA, Healy E, Todd C, Rees JL, Steijlen PM, Carmichael AJ, Lewis HM, Hohl D, Itin P, Vahlquist A, Gobello T, Mazzanti C, Reggazini R, Nagy G, Munro CS, Strachan T (1999) ATP2A2 mutations in Darier’s disease: variant cutaneous phenotypes are associated with missense mutations, but neuropsychiatric features are independent of mutation class. Hum Mol Genet 8:1621–1630
Nagai R, Zarain-Herzberg A, Brandl CJ, Fujii J, Tada M, MacLennan DH, Alpert NR, Periasamy M (1989) Regulation of myocardial Ca2+-ATPase and phospholamban mRNA expression in response to pressure overload and thyroid hormone. Proc Natl Acad Sci USA 86:2966–2970
Zarain-Herzberg A, Afzal N, Elimban V, Dhalla NS (1996) Decreased expression of cardiac sarcoplasmic reticulum Ca(2+)-pump ATPase in congestive heart failure due to myocardial infarction. Mol Cell Biochem 163–164:285–290
O'Rourke B, Kass DA, Tomaselli GF, Kaab S, Tunin R, Marban E (1999) Mechanisms of altered excitation-contraction coupling in canine tachycardia-induced heart failure, I: experimental studies. Circ Res 84:562–570
Arai M, Alpert NR, MacLennan DH, Barton P, Periasamy M (1993) Alterations in sarcoplasmic reticulum gene expression in human heart failure. A possible mechanism for alterations in systolic and diastolic properties of the failing myocardium. Circ Res 72:463–469
Arai M, Matsui H, Periasamy M (1994) Sarcoplasmic reticulum gene expression in cardiac hypertrophy and heart failure. Circ Res 74:555–564
Hasenfuss G, Reinecke H, Studer R, Meyer M, Pieske B, Holtz J, Holubarsch C, Posival H, Just H, Drexler H (1994) Relation between myocardial function and expression of sarcoplasmic reticulum Ca(2+)-ATPase in failing and nonfailing human myocardium. Circ Res 75:434–442
Hasenfuss G (1998) Alterations of calcium-regulatory proteins in heart failure. Cardiovasc Res 37:279–289
Pieske B, Maier LS, Bers DM, Hasenfuss G (1999) Ca2+ handling and sarcoplasmic reticulum Ca2+ content in isolated failing and nonfailing human myocardium. Circ Res 85:38–46
Meyer M, Schillinger W, Pieske B, Holubarsch C, Heilmann C, Posival H, Kuwajima G, Mikoshiba K, Just H, Hasenfuss G et al (1995) Alterations of sarcoplasmic reticulum proteins in failing human dilated cardiomyopathy. Circulation 92:778–784
Arvanitis DA, Vafiadaki E, Fan GC, Mitton BA, Gregory KN, Del Monte F, Kontrogianni-Konstantopoulos A, Sanoudou D, Kranias EG (2007) Histidine-rich Ca-binding protein interacts with sarcoplasmic reticulum Ca-ATPase. Am J Physiol Heart Circ Physiol 293:H1581–H1589
Ihara Y, Kageyama K, Kondo T (2005) Overexpression of calreticulin sensitizes SERCA2a to oxidative stress. Biochem Biophys Res Commun 329:1343–1349
Kiewitz R, Acklin C, Schafer BW, Maco B, Uhrik B, Wuytack F, Erne P, Heizmann CW (2003) Ca2+-dependent interaction of S100A1 with the sarcoplasmic reticulum Ca2+-ATPase2a and phospholamban in the human heart. Biochem Biophys Res Commun 306:550–557
Nediani C, Celli A, Fiorillo C, Ponziani V, Giannini L, Nassi P (2003) Acylphosphatase interferes with SERCA2a-PLN association. Biochem Biophys Res Commun 301:948–951
Dremina ES, Sharov VS, Kumar K, Zaidi A, Michaelis EK, Schoneich C (2004) Anti-apoptotic protein Bcl-2 interacts with and destabilizes the sarcoplasmic/endoplasmic reticulum Ca2+-ATPase (SERCA). Biochem J 383:361–370
Kuo TH, Kim HR, Zhu L, Yu Y, Lin HM, Tsang W (1998) Modulation of endoplasmic reticulum calcium pump by Bcl-2. Oncogene 17:1903–1910
Bhupathy P, Babu GJ, Periasamy M (2007) Sarcolipin and phospholamban as regulators of cardiac sarcoplasmic reticulum Ca2+ ATPase. J Mol Cell Cardiol 42:903–911
Simmerman HK, Jones LR (1998) Phospholamban: protein structure, mechanism of action, and role in cardiac function. Physiol Rev 78:921–947
Kadambi VJ, Kranias EG (1997) Phospholamban: a protein coming of age. Biochem Biophys Res Commun 239:1–5
MacLennan DH, Kranias EG (2003) Phospholamban: a crucial regulator of cardiac contractility. Nat Rev Mol Cell Biol 4:566–577
Fujii J, Ueno A, Kitano K, Tanaka S, Kadoma M, Tada M (1987) Complete complementary DNA-derived amino acid sequence of canine cardiac phospholamban. J Clin Invest 79:301–304
James P, Inui M, Tada M, Chiesi M, Carafoli E (1989) Nature and site of phospholamban regulation of the Ca2+ pump of sarcoplasmic reticulum. Nature 342:90–92
Toyofuku T, Kurzydlowski K, Tada M, MacLennan DH (1994) Amino acids Lys–Asp–Asp–Lys–Pro–Val402 in the Ca(2+)-ATPase of cardiac sarcoplasmic reticulum are critical for functional association with phospholamban. J Biol Chem 269:22929–22932
Toyofuku T, Kurzydlowski K, Tada M, MacLennan DH (1994) Amino acids Glu2 to Ile18 in the cytoplasmic domain of phospholamban are essential for functional association with the Ca(2+)-ATPase of sarcoplasmic reticulum. J Biol Chem 269:3088–3094
Sharma P, Patchell VB, Gao Y, Evans JS, Levine BA (2001) Cytoplasmic interactions between phospholamban residues 1–20 and the calcium-activated ATPase of the sarcoplasmic reticulum. Biochem J 335:699–706
Kimura Y, Asahi M, Kurzydlowski K, Tada M, MacLennan DH (1998) Phospholamban domain Ib mutations influence functional interactions with the Ca2+-ATPase isoform of cardiac sarcoplasmic reticulum. J Biol Chem 273:14238–14241
Chen Z, Stokes DL, Jones LR (2005) Role of leucine 31 of phospholamban in structural and functional interactions with the Ca2+ pump of cardiac sarcoplasmic reticulum. J Biol Chem 280:10530–10539
Morris GL, Cheng HC, Colyer J, Wang JH (1991) Phospholamban regulation of cardiac sarcoplasmic reticulum (Ca(2+)-Mg2+)-ATPase. Mechanism of regulation and site of monoclonal antibody interaction. J Biol Chem 266:11270–11275
Wegener AD, Simmerman HK, Lindemann JP, Jones LR (1989) Phospholamban phosphorylation in intact ventricles. Phosphorylation of serine 16 and threonine 17 in response to beta-adrenergic stimulation. J Biol Chem 264:11468–11474
Kimura Y, Kurzydlowski K, Tada M, MacLennan DH (1997) Phospholamban inhibitory function is activated by depolymerization. J Biol Chem 272:15061–15064
Zvaritch E, Backx PH, Jirik F, Kimura Y, de Leon S, Schmidt AG, Hoit BD, Lester JW, Kranias EG, MacLennan DH (2000) The transgenic expression of highly inhibitory monomeric forms of phospholamban in mouse heart impairs cardiac contractility. J Biol Chem 275:14985–14991
Cantilina T, Sagara Y, Inesi G, Jones LR (1993) Comparative studies of cardiac and skeletal sarcoplasmic reticulum ATPases. Effect of a phospholamban antibody on enzyme activation by Ca2+. J Biol Chem 268:17018–17025
Inui M, Chamberlain BK, Saito A, Fleischer S (1986) The nature of the modulation of Ca2+ transport as studied by reconstitution of cardiac sarcoplasmic reticulum. J Biol Chem 261:1794–1800
Tada M, Katz AM (1982) Phosphorylation of the sarcoplasmic reticulum and sarcolemma. Annu Rev Physiol 44:401–423
Slack JP, Grupp IL, Dash R, Holder D, Schmidt A, Gerst MJ, Tamura T, Tilgmann C, James PF, Johnson R, Gerdes AM, Kranias EG (2001) The enhanced contractility of the phospholamban-deficient mouse heart persists with aging. J Mol Cell Cardiol 33:1031–1040
Luo W, Grupp IL, Harrer J, Ponniah S, Grupp G, Duffy JJ, Doetschman T, Kranias EG (1994) Targeted ablation of the phospholamban gene is associated with markedly enhanced myocardial contractility and loss of beta-agonist stimulation. Circ Res 75:401–409
Kiss E, Edes I, Sato Y, Luo W, Liggett SB, Kranias EG (1997) Beta-adrenergic regulation of cAMP and protein phosphorylation in phospholamban-knockout mouse hearts. Am J Physiol 272:H785–H790
Lorenz JN, Kranias EG (1997) Regulatory effects of phospholamban on cardiac function in intact mice. Am J Physiol 273:H2826–H2831
Luo W, Wolska BM, Grupp IL, Harrer JM, Haghighi K, Ferguson DG, Slack JP, Grupp G, Doetschman T, Solaro RJ, Kranias EG (1996) Phospholamban gene dosage effects in the mammalian heart. Circ Res 78:839–847
Zhai J, Schmidt AG, Hoit BD, Kimura Y, MacLennan DH, Kranias EG (2000) Cardiac-specific overexpression of a superinhibitory pentameric phospholamban mutant enhances inhibition of cardiac function in vivo. J Biol Chem 275:10538–10544
Haghighi K, Schmidt AG, Hoit BD, Brittsan AG, Yatani A, Lester JW, Zhai J, Kimura Y, Dorn GW II, MacLennan DH, Kranias EG (2001) Superinhibition of sarcoplasmic reticulum function by phospholamban induces cardiac contractile failure. J Biol Chem 276:24145–24152
Schmidt AG, Zhai J, Carr AN, Gerst MJ, Lorenz JN, Pollesello P, Annila A, Hoit BD, Kranias EG (2002) Structural and functional implications of the phospholamban hinge domain: impaired SR Ca2+ uptake as a primary cause of heart failure. Cardiovasc Res 56:248–259
Luo W, Chu G, Sato Y, Zhou Z, Kadambi VJ, Kranias EG (1998) Transgenic approaches to define the functional role of dual site phospholamban phosphorylation. J Biol Chem 273:4734–4739
Kuschel M, Karczewski P, Hempel P, Schlegel WP, Krause EG, Bartel S (1999) Ser16 prevails over Thr17 phospholamban phosphorylation in the beta-adrenergic regulation of cardiac relaxation. Am J Physiol 276:H1625–H1633
Chu G, Lester JW, Young KB, Luo W, Zhai J, Kranias EG (2000) A single site (Ser16) phosphorylation in phospholamban is sufficient in mediating its maximal cardiac responses to beta -agonists. J Biol Chem 275:38938–38943
Zhao W, Uehara Y, Chu G, Song Q, Qian J, Young K, Kranias EG (2004) Threonine-17 phosphorylation of phospholamban: a key determinant of frequency-dependent increase of cardiac contractility. J Mol Cell Cardiol 37:607–612
Schmitt JP, Kamisago M, Asahi M, Li GH, Ahmad F, Mende U, Kranias EG, MacLennan DH, Seidman JG, Seidman CE (2003) Dilated cardiomyopathy and heart failure caused by a mutation in phospholamban. Science 299:1410–1413
Haghighi K, Kolokathis F, Gramolini AO, Waggoner JR, Pater L, Lynch RA, Fan GC, Tsiapras D, Parekh RR, Dorn GWn, Maclennan DH, Kremastinos DT, Kranias EG (2006) A mutation in the human phospholamban gene, deleting arginine 14, results in lethal, hereditary cardiomyopathy. Proc Natl Acad Sci USA 103:1388–1393
Haghighi K, Kolokathis F, Pater L, Lynch RA, Asahi M, Gramolini AO, Fan GC, Tsiapras D, Hahn HS, Adamopoulos S, Liggett SB, Dorn GWn, MacLennan DH, Kremastinos DT, Kranias EG (2003) Human phospholamban null results in lethal dilated cardiomyopathy revealing a critical difference between mouse and human. J Clin Invest 111:869–876
Haghighi K, Chen G, Sato Y, Fan GC, He S, Kolokathis F, Pater L, Paraskevaidis I, Jones WK, Dorn Ii GW, Th Kremastinos D, Kranias EG (2008) A human phospholamban promoter polymorphism in dilated cardiomyopathy alters transcriptional regulation by glucocorticoids. Hum Mutat (in press)
Minamisawa S, Sato Y, Tatsuguchi Y, Fujino T, Imamura S, Uetsuka Y, Nakazawa M, Matsuoka R (2003) Mutation of the phospholamban promoter associated with hypertrophic cardiomyopathy. Biochem Biophys Res Commun 304:1–4
Medin M, Hermida-Prieto M, Monserrat L, Laredo R, Rodriguez-Rey JC, Fernandez X, Castro-Beiras A (2007) Mutational screening of phospholamban gene in hypertrophic and idiopathic dilated cardiomyopathy and functional study of the PLN −42 C>G mutation. Eur J Heart Fail 9:37–43
Zhao W, Yuan Q, Qian J, Waggoner JR, Pathak A, Chu G, Mitton B, Sun X, Jin J, Braz JC, Hahn HS, Marreez Y, Syed F, Pollesello P, Annila A, Wang HS, Schultz Jel J, Molkentin JD, Liggett SB, Dorn GW II, Kranias EG (2006) The presence of Lys27 instead of Asn27 in human phospholamban promotes sarcoplasmic reticulum Ca2+-ATPase superinhibition and cardiac remodeling. Circulation 113:995–1004
Vafiadaki E, Sanoudou D, Arvanitis DA, Catino DH, Kranias EG, Kontrogianni-Konstantopoulos A (2007) Phospholamban interacts with HAX-1, a mitochondrial protein with anti-apoptotic function. J Mol Biol 367:65–79
Suzuki Y, Demoliere C, Kitamura D, Takeshita H, Deuschle U, Watanabe T (1997) HAX-1, a novel intracellular protein, localized on mitochondria, directly associates with HS1, a substrate of Src family of tyrosine kinases. J Immunol 158:2736–2744
Kawaguchi Y, Nakajima K, Igarashi M, Morita T, Tanaka M, Suzuki M, Yokoyama A, Matsuda G, Kato K, Kanamori M, Hirai K (2000) Interaction of Epstein–Barr virus nuclear antigen leader protein (EBNA-LP) with HS1-associated protein X-1: implication of cytoplasmic function of EBNA-LP. J Virol 74:10104–10111
Matsuda G, Nakajima K, Kawaguchi Y, Yamanashi Y, Hirai K (2003) Epstein–Barr virus (EBV) nuclear antigen leader protein (EBNA-LP) forms complexes with a cellular anti-apoptosis protein Bcl-2 or its EBV counterpart BHRF1 through HS1-associated protein X-1. Microbiol Immunol 47:91–99
Sharp TV, Wang HW, Koumi A, Hollyman D, Endo Y, Ye H, Du MQ, Boshoff C (2002) K15 protein of Kaposi’s sarcoma-associated herpesvirus is latently expressed and binds to HAX-1, a protein with antiapoptotic function. J Virol 76:802–816
Yedavalli VS, Shih HM, Chiang YP, Lu CY, Chang LY, Chen MY, Chuang CY, Dayton AI, Jeang KT, Huang LM (2005) Human immunodeficiency virus type 1 Vpr interacts with antiapoptotic mitochondrial protein HAX-1. J Virol 79:13735–13746
Modem S, Reddy TR (2008) An anti-apoptotic protein, Hax-1, inhibits the HIV-1 rev function by altering its sub-cellular localization. J Cell Physiol 214:14–19
Dufva M, Olsson M, Rymo L (2001) Epstein–Barr virus nuclear antigen 5 interacts with HAX-1, a possible component of the B-cell receptor signalling pathway. J Gen Virol 82:1581–1587
Gallagher AR, Cedzich A, Gretz N, Somlo S, Witzgall R (2000) The polycystic kidney disease protein PKD2 interacts with Hax-1, a protein associated with the actin cytoskeleton. Proc Natl Acad Sci USA 97:4017–4022
Radhika V, Onesime D, Ha JH, Dhanasekaran N (2004) Galpha13 stimulates cell migration through cortactin-interacting protein Hax-1. J Biol Chem 279:49406–49413
Ramsay AG, Keppler MD, Jazayeri M, Thomas GJ, Parsons M, Violette S, Weinreb P, Hart IR, Marshall JF (2007) HS1-associated protein X-1 regulates carcinoma cell migration and invasion via clathrin-mediated endocytosis of integrin alphavbeta6. Cancer Res 67:5275–5284
Yin H, Morioka H, Towle CA, Vidal M, Watanabe T, Weissbach L (2001) Evidence that HAX-1 is an interleukin-1 alpha N-terminal binding protein. Cytokine 15:122–137
Kawaguchi Y, Nishimagi E, Tochimoto A, Kawamoto M, Katsumata Y, Soejima M, Kanno T, Kamatani N, Hara M (2006) Intracellular IL-1{alpha}-binding proteins contribute to biological functions of endogenous IL-1{alpha} in systemic sclerosis fibroblasts. Proc Natl Acad Sci USA 103:14501–14506
Ortiz DF, Moseley J, Calderon G, Swift AL, Li S, Arias IM (2004) Identification of HAX-1 as a protein that binds bile salt export protein and regulates its abundance in the apical membrane of Madin–Darby canine kidney cells. J Biol Chem 279:32761–32770
Kasashima K, Ohta E, Kagawa Y, Endo H (2006) The pleiotropic human prohibitin 2: mitochondrial functions and estrogen receptor-dependent nuclear translocation. J Biol Chem 281:36401–36410
Han Y, Chen Y-S, Liu Z, Bodyak N, Rigor D, Bisping E, Pu WT, Kang PM (2006) Overexpression of HAX-1 protects cardiac myocytes from apoptosis through caspase-9 inhibition. Circ Res 99:415–423
Cilenti L, Soundarapandian MM, Kyriazis GA, Stratico V, Singh S, Gupta S, Bonventre JV, Alnemri ES, Zervos AS (2004) Regulation of HAX-1 anti-apoptotic protein by Omi/HtrA2 protease during cell death. J Biol Chem 279:50295–50301
Sarnowska E, Grzybowska EA, Sobczak K, Konopinski R, Wilczynska A, Szwarc M, Sarnowski TJ, Krzyzosiak WJ, Siedlecki JA (2007) Hairpin structure within the 3′UTR of DNA polymerase beta mRNA acts as a post-transcriptional regulatory element and interacts with Hax-1. Nucleic Acids Res 35:5499–5510
Al-Maghrebi M, Brule H, Padkina M, Allen C, Holmes WM, Zehner ZE (2002) The 3′ untranslated region of human vimentin mRNA interacts with protein complexes containing eEF-1gamma and HAX-1. Nucleic Acids Res 30:5017–5028
Lauriat TL, Dracheva S, Kremerskothen J, Duning K, Haroutunian V, Buxbaum JD, Hyde TM, Kleinman JE, Alison ML (2006) Characterization of KIAA0513, a novel signaling molecule that interacts with modulators of neuroplasticity, apoptosis, and the cytoskeleton. Brain Res 1121:1–11
Mirmohammadsadegh A, Tartler U, Michel G, Baer A, Walz M, Wolf R, Ruzicka T, Hengge UR (2003) HAX-1, identified by differential display reverse transcription polymerase chain reaction, is overexpressed in lesional psoriasis. J Invest Dermatol 120:1045–1051
Pizzo P, Pozzan T (2007) Mitochondria-endoplasmic reticulum choreography: structure and signaling dynamics. Trends Cell Biol 17:511–517
Klein C, Grudzien M, Appaswamy G, Germeshausen M, Sandrock I, Schaffer AA, Rathinam C, Boztug K, Schwinzer B, Rezaei N, Bohn G, Melin M, Carlsson G, Fadeel B, Dahl N, Palmblad J, Henter JI, Zeidler C, Grimbacher B, Welte K (2007) HAX1 deficiency causes autosomal recessive severe congenital neutropenia (Kostmann disease). Nat Genet 39:86–92
Bohn G, Welte K, Klein C (2007) Severe congenital neutropenia: new genes explain an old disease. Curr Opin Rheumatol 19:644–650
Matsubara K, Imai K, Okada S, Miki M, Ishikawa N, Tsumura M, Kato T, Ohara O, Nonoyama S, Kobayashi M (2007) Severe developmental delay and epilepsy in a Japanese patient with severe congenital neutropenia due to HAX1 deficiency. Haematologica 92:e123–e125
Rezaei N, Moin M, Pourpak Z, Ramyar A, Izadyar M, Chavoshzadeh Z, Sherkat R, Aghamohammadi A, Yeganeh M, Mahmoudi M, Mahjoub F, Germeshausen M, Grudzien M, Horwitz MS, Klein C, Farhoudi A (2007) The clinical, immunohematological, and molecular study of Iranian patients with severe congenital neutropenia. J Clin Immunol 27:525–533
Youle RJ, Strasser A (2008) The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol 9:47–59
Annis MG, Yethon JA, Leber B, Andrews DW (2004) There is more to life and death than mitochondria: Bcl-2 proteins at the endoplasmic reticulum. Biochim Biophys Acta 1644:115–123
Cory S, Adams JM (2002) The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer 2:647–656
Demaurex N, Distelhorst C (2003) Cell biology. Apoptosis—the calcium connection. Science 300:65–67
Pinton P, Ferrari D, Rapizzi E, Di Virgilio F, Pozzan T, Rizzuto R (2001) The Ca2+ concentration of the endoplasmic reticulum is a key determinant of ceramide-induced apoptosis: significance for the molecular mechanism of Bcl-2 action. Embo J 20:2690–2701
Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, Tuft RA, Pozzan T (1998) Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 280:1763–1766
Szabadkai G, Rizzuto R (2004) Participation of endoplasmic reticulum and mitochondrial calcium handling in apoptosis: more than just neighborhood? FEBS Lett 567:111–115
Franzini-Armstrong C (2007) ER-mitochondria communication. How privileged? Physiology (Bethesda) 22:261–268
Akao Y, Otsuki Y, Kataoka S, Ito Y, Tsujimoto Y (1994) Multiple subcellular localization of bcl-2: detection in nuclear outer membrane, endoplasmic reticulum membrane, and mitochondrial membranes. Cancer Res 54:2468–2471
Krajewski S, Tanaka S, Takayama S, Schibler MJ, Fenton W, Reed JC (1993) Investigation of the subcellular distribution of the bcl-2 oncoprotein: residence in the nuclear envelope, endoplasmic reticulum, and outer mitochondrial membranes. Cancer Res 53:4701–4714
Lithgow T, van Driel R, Bertram JF, Strasser A (1994) The protein product of the oncogene bcl-2 is a component of the nuclear envelope, the endoplasmic reticulum, and the outer mitochondrial membrane. Cell Growth Differ 5:411–417
Zong WX, Li C, Hatzivassiliou G, Lindsten T, Yu QC, Yuan J, Thompson CB (2003) Bax and Bak can localize to the endoplasmic reticulum to initiate apoptosis. J Cell Biol 162:59–69
Nutt LK, Pataer A, Pahler J, Fang B, Roth J, McConkey DJ, Swisher SG (2002) Bax and Bak promote apoptosis by modulating endoplasmic reticular and mitochondrial Ca2+ stores. J Biol Chem 277:9219–9225
Hsu YT, Wolter KG, Youle RJ (1997) Cytosol-to-membrane redistribution of Bax and Bcl-X(L) during apoptosis. Proc Natl Acad Sci USA 94:3668–3672
Zhu W, Cowie A, Wasfy GW, Penn LZ, Leber B, Andrews DW (1996) Bcl-2 mutants with restricted subcellular location reveal spatially distinct pathways for apoptosis in different cell types. EMBO J 15:4130–4141
Rudner J, Lepple-Wienhues A, Budach W, Berschauer J, Friedrich B, Wesselborg S, Schulze-Osthoff K, Belka C (2001) Wild-type, mitochondrial and ER-restricted Bcl-2 inhibit DNA damage-induced apoptosis but do not affect death receptor-induced apoptosis. J Cell Sci 114:4161–4172
Hacki J, Egger L, Monney L, Conus S, Rosse T, Fellay I, Borner C (2000) Apoptotic crosstalk between the endoplasmic reticulum and mitochondria controlled by Bcl-2. Oncogene 19:2286–2295
Annis MG, Zamzami N, Zhu W, Penn LZ, Kroemer G, Leber B, Andrews DW (2001) Endoplasmic reticulum localized Bcl-2 prevents apoptosis when redistribution of cytochrome c is a late event. Oncogene 20:1939–1952
He H, Lam M, McCormick TS, Distelhorst CW (1997) Maintenance of calcium homeostasis in the endoplasmic reticulum by Bcl-2. J Cell Biol 138:1219–1228
Pinton P, Ferrari D, Magalhaes P, Schulze-Osthoff K, Di Virgilio F, Pozzan T, Rizzuto R (2000) Reduced loading of intracellular Ca(2+) stores and downregulation of capacitative Ca(2+) influx in Bcl-2-overexpressing cells. J Cell Biol 148:857–862
Vanden Abeele F, Skryma R, Shuba Y, Van Coppenolle F, Slomianny C, Roudbaraki M, Mauroy B, Wuytack F, Prevarskaya N (2002) Bcl-2-dependent modulation of Ca(2+) homeostasis and store-operated channels in prostate cancer cells. Cancer Cell 1:169–179
Foyouzi-Youssefi R, Arnaudeau S, Borner C, Kelley WL, Tschopp J, Lew DP, Demaurex N, Krause KH (2000) Bcl-2 decreases the free Ca2+ concentration within the endoplasmic reticulum. Proc Natl Acad Sci USA 97:5723–5728
Li C, Fox CJ, Master SR, Bindokas VP, Chodosh LA, Thompson CB (2002) Bcl-X(L) affects Ca(2+) homeostasis by altering expression of inositol 1,4,5-trisphosphate receptors. Proc Natl Acad Sci USA 99:9830–9835
Li C, Wang X, Vais H, Thompson CB, Foskett JK, White C (2007) Apoptosis regulation by Bcl-x(L) modulation of mammalian inositol 1,4,5-trisphosphate receptor channel isoform gating. Proc Natl Acad Sci USA 104:12565–12570
White C, Li C, Yang J, Petrenko NB, Madesh M, Thompson CB, Foskett JK (2005) The endoplasmic reticulum gateway to apoptosis by Bcl-X(L) modulation of the InsP3R. Nat Cell Biol 7:1021–1028
Nutt LK, Chandra J, Pataer A, Fang B, Roth JA, Swisher SG, O'Neil RG, McConkey DJ (2002) Bax-mediated Ca2+ mobilization promotes cytochrome c release during apoptosis. J Biol Chem 277:20301–20308
Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD, Pozzan T, Korsmeyer SJ (2003) BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science 300:135–139
Oakes SA, Scorrano L, Opferman JT, Bassik MC, Nishino M, Pozzan T, Korsmeyer SJ (2005) Proapoptotic BAX and BAK regulate the type 1 inositol trisphosphate receptor and calcium leak from the endoplasmic reticulum. Proc Natl Acad Sci USA 102:105–110
Palmer AE, Jin C, Reed JC, Tsien RY (2004) Bcl-2-mediated alterations in endoplasmic reticulum Ca2+ analyzed with an improved genetically encoded fluorescent sensor. Proc Natl Acad Sci USA 101:17404–17409
Chen R, Valencia I, Zhong F, McColl KS, Roderick HL, Bootman MD, Berridge MJ, Conway SJ, Holmes AB, Mignery GA, Velez P, Distelhorst CW (2004) Bcl-2 functionally interacts with inositol 1,4,5-trisphosphate receptors to regulate calcium release from the ER in response to inositol 1,4,5-trisphosphate. J Cell Biol 166:193–203
Rong Y, Distelhorst CW (2008) Bcl-2 protein family members: versatile regulators of calcium signaling in cell survival and apoptosis. Annu Rev Physiol 70:73–91
Dremina ES, Sharov VS, Schoneich C (2006) Displacement of SERCA from SR lipid caveolae-related domains by Bcl-2: a possible mechanism for SERCA inactivation. Biochemistry 45:175–184
Acknowledgment
This work was supported by research funds from the Biomedical Research Foundation, Academy of Athens; the John F. Kostopoulos Foundation; the Hellenic Cardiological Society; NIH HL26057, HL64018, and HL77101; the Leducq Foundation Trans-Antlantic alliance; and by the European Union Sixth Framework Program for Research and Technological Development, “Life Sciences, Genomics and Biotechnology for Health,” Valapodyn, contract #LSHG-CT-2006-037277.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Vafiadaki, E., Papalouka, V., Arvanitis, D.A. et al. The role of SERCA2a/PLN complex, Ca2+ homeostasis, and anti-apoptotic proteins in determining cell fate. Pflugers Arch - Eur J Physiol 457, 687–700 (2009). https://doi.org/10.1007/s00424-008-0506-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-008-0506-5