
Abstract
This study elucidates the role of cell volume in contractions of endothelium-denuded vascular smooth muscle rings (VSMR) from the rat aorta. We observed that hyposmotic swelling as well as hyper- and isosmotic shrinkage led to VSMR contractions. Swelling-induced contractions were accompanied by activation of Ca2+ influx and were abolished by nifedipine and verapamil. In contrast, contractions of shrunken cells were insensitive to the presence of L-type channel inhibitors and occurred in the absence of Ca2+o. Thirty minutes preincubation with bumetanide, a potent Na+,K+,Cl− cotransport (NKCC) inhibitor, decreased Cl−i content, nifedipine-sensitive 45Ca uptake and contractions triggered by modest depolarization ([K+]o=36 mM). Elevation of [K+]o to 66 mM completely abolished the effect of bumetanide on these parameters. Bumetanide almost completely abrogated phenylephrine-induced contraction, partially suppressed contractions triggered by hyperosmotic shrinkage, but potentiated contractions of isosmotically shrunken VSMR. Our results suggest that bumetanide suppresses contraction of modestly depolarized cells via NKCC inhibition and Cl−i-mediated membrane hyperpolarization, whereas augmented contraction of isosmotically shrunken VSMR by bumetanide is a consequence of suppression of NKCC-mediated regulatory volume increase. The mechanism of bumetanide inhibition of contraction of phenylephrine-treated and hyperosmotically shrunken VSMR should be examined further.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Since the first observations on increased monovalent ion exchange in vascular smooth muscle rings (VSMR) [25] and red blood cells [51] from spontaneously hypertensive rats, several research teams have reported that, at least partially, these abnormalities are caused by enhanced electroneutral Na+,K+,2Cl−cotransport (NKCC) (for review see [41, 47]). Two isoforms of this carrier have been cloned from vertebrate cDNA libraries. A ubiquitous NKCC1 isoform has been detected in all types of cells studied so far, whereas expression of the renal-specific NKCC2 isoform is limited to the apical membrane of epithelial cells from the thick ascending limb and macula densa [37, 53].
The hypothesis on NKCC1 involvement in the pathogenesis of hypertension is based on three major observations. First, in erythrocytes from F2 hybrids of spontaneously hypertensive and normotensive rats, NKCC activity correlated positively with blood pressure [10, 28]. Second, NKCC1−/− knockout mice exhibited decreased blood pressure as well as attenuated vascular tone and constriction of VSMR treated with the α-adrenoceptor agonist phenylephrine (PE) [16, 35]. Third, NKCC inhibition with bumetanide reduced contractions of rat VSMR and the guinea pig ureter triggered by K+o-induced depolarization, PE and electrical stimulation [4, 29]. Brown et al. were the first to suggest that NKCC contributes to the maintenance of vascular tone via adjustment of the transmembrane Cl− gradient [11]. Indeed, bumetanide decreased [Cl−]i, hyperpolarized vascular smooth muscle cells (VSMC) [15] and abolished differences in these parameters between normotensive and DOCA-salt-hypertensive rats [11].
In experiments on bone marrow transplantation from progenitors to X-ray-irradiated F1 hybrids of spontaneously hypertensive and normotensive rats, Bianchi et al. showed that enhanced NKCC detected in this experimental model of primary hypertension is genetically determined rather than a consequence of the hypertensive milieu [10]. On the other hand, other researchers detected augmented NKCC in VSMR from rodents with secondary hypertension [15, 24] and enhanced content of endogenous NKCC inhibitor in the plasma of salt-sensitive rats [5], suggesting that the carrier is subjected to cell-type-specific regulation by diverse stimuli. Indeed, no systematic involvement of Ca2+, cAMP and cGMP has been detected in studies of NKCC in human and rat erythrocytes [17], whereas in VSMR and cultured VSMC, this carrier was activated by Ca2+i-raising vasoconstrictors, including angiotensin II, and was inhibited by cAMP- and cGMP-mediated vasodilators [3, 4, 42, 49, 55, 57]. Opposing regulation by vasodilators was documented in a study of Na+-independent K+,Cl− cotransport (KCC) [2], i.e. another member of the Cl−-coupled cotransporter superfamily contributing to outwardly directed Cl− transport [1].
Similarly to “ying-yang” regulation by vasodilators, NKCC and KCC are also affected reciprocally by cell volume modulation. Indeed, shrinkage-induced activation of inwardly directed NKCC, Na+/H+ exchange, and Na+-coupled transport of organic osmolytes contributes to regulatory volume increase (RVI), whereas swelling-induced and outwardly directed KCC, K+ and anion channels provide regulatory volume decrease (RVD) [20, 31, 36]. It is important to underline that cell shrinkage activates NKCC1 in all types of nucleated cells studied so far [39]. Keeping this in mind, we employed hyperosmotic and isosmotic shrinkage to further explore the role of NKCC in VSMC contraction. The results of our study were reported in part at the 13th European Meeting on Hypertension [7].
Materials and methods
Preparation of VSMR
Endothelium-denuded VSMR were obtained from the thoracic aorta of 11- to 13-week-old Wistar rats euthanized under deep intraperitoneal anesthesia with sodium pentobarbital (Nembutal, 70 mg/kg) in accordance with institutional animal care guidelines. The isolated aorta was placed in physiologically balanced salt solution (PSS). Connective tissue and fat were taken out with scissors, whereas the endothelium was removed by careful rotation of a wooden manipulator inside the VSMR lumen just before the experiments. The 2- to 3-mm VSMR were either used immediately or stored at 4°C for up to 24 h. In preliminary experiments, we documented that 24 h storage did not affect VSMR contractile responses.
Isometric VSMR contraction
VSMR were mounted in 1-ml baths with stainless steel hooks inserted into the vascular ring orifice. One hook was fastened to a mechanical force transducer MX2B (Tomsk, Russia) with silk thread; the other served as an anchor. The tissues were bathed in PSS, buffered with TRIS at pH 7.4 (37°C) and bubbled with 95%O2/5% CO2 at a volumetric speed of ~1 ml/min. To control the contractile response, the VSMR were equilibrated for 1 h at tension of 0.5–1 g and exposed to K+o-induced depolarization caused by isosmotic substitution of 30 mM NaCl with KCl. The VSMR were then exposed to modified PSS containing either altered ions concentration or sucrose in the presence or absence of the test compounds. Isometric changes in VSMR tension were recorded with an XY recorder (Carl Zeiss, Jena, Germany).
Cultured VSMC
The precise measurement of cell volume and inward ion fluxes in VSMR are complicated by a relatively large extracellular space, the presence of fibroblasts, and VSMC heterogeneity [18]. On the other hand, cultured VSMC rapidly downregulate the expression of several specific genes that define their contractile phenotype in vivo. Keeping this in mind, we used a low-density seeding strategy for VSMC derived from the aortae of Wistar-Kyoto (WKY) rats to select a cell line possessing the highest expression of smooth muscle-specific α-actin, SM22 protein and myosin light chain kinase. This cell line, WKY-7, also possessed the highest sensitivity to angiotensin II and endothelin-1, measured by mitogen-activated protein kinase ERK1/2 phosphorylation [14]. In the present study, WKY-7 cells were grown for 48–72 h in DMEM supplemented with fetal and newborn calf serum (10% of each), glutamine (2 mM), penicillin (100 U/ml), and streptomycin (100 µg/ml). To establish quiescence, VSMC were incubated before experiments during 48 h in the presence of 0.2% calf serum.
NKCC activity
NKCC activity was measured as a bumetanide-sensitive component of the 86Rb influx rate. WKY-7 cells seeded in 24-well plates were washed twice with 2-ml aliquots of PSS. The medium was then aspirated, and 0.25 ml of PSS with 1 μCi/ml 86Rb and 1 mM ouabain with or without bumetanide was added. After 5-min incubation at 37°C, isotope uptake was terminated by the addition of 2 ml of ice-cold medium W containing 100 mM MgCl2 and 10 mM HEPES-TRIS buffer (pH 7.4). Radioactivity of the incubation medium and cell lysate was measured with a liquid scintillation analyzer, and the rate of 86Rb influx (V, nmol per mg of protein per 5 min) was calculated as V=A/am, where A was the radioactivity of the samples (cpm), a was the specific radioactivity of K+ (86Rb) in the medium (cpm/nmol), and m was protein content measured with the modified Lowry method. For more details see [46].
Intracellular water volume in WKY-7 cells seeded in 12-well plates was measured as [14C]-urea available space according to a previously described protocol [46] and calculated as V=Ac/Amm, where Ac was the radioactivity of the cells after 30 min incubation with 2 μCi/ml [14C]-urea (dpm), Am was the radioactivity of the incubation medium (dmp/μl), and m was protein content in the cell lysate (mg).
Intracellular Cl− content in WKY-7 cells was measured as the steady-state distribution of 36Cl as described previously in detail [44]. To calculate [Cl−]i, the volume of intracellular water was quantified in parallel experiments with the protocol presented above. To measure Cl−i content in VSMR, the rings were subjected to 5 h preincubation in PSS containing 2 μCi/ml 36Cl; in some experiments, bumetanide and sucrose were added in the last 30 min of incubation. The VSMR were then rinsed in 3×50-ml aliquots of ice-cold PSS and solubilized in scintillation cocktail containing Triton X-100:toluene 1:2 (v/v), 4 g/l 2,5-diphenyl-1,3,4-oxadiazole (PPO) and 0.1 g/l 1,4-bis[5-phenyl-2-oxazolyl]-benzene; 2–2′-p-phenylene-bis[5-phenyloxazole] (POPOP). Intracellular Cl− content (nmol/mg) was calculated as [Cl−]i=A/am, where A was the radioactivity of the samples (cpm), a was the specific radioactivity of Cl− in the medium (cpm/nmol), and m was the VSMR wet weight (mg) measured before incubation.
Voltage-gated L-type Ca2+ channel activity was estimated as verapamil- or nifedipine-sensitive component of the 45Ca influx rate. WKY-7 cells seeded in 24-well plates were preincubated for 30 min with PSS. Then, this medium was aspirated, and 0.25 ml of PSS containing 0.1 mM CaCl2 with or without nifedipine and verapamil was added. Isotope uptake was initiated by the addition of 0.25 ml of the same PSS with 3 μCi/ml 45Ca, and terminated in 5 min as indicated above. To induce cell depolarization, KCl in isotope-containing medium was increased up to 60 mM by equimolar substitution of NaCl. For more details see [45].
Solutions and chemicals
PSS contained 120.4 mM NaCl, 5.9 mM KCl, 2.5 mM CaCl2, 1.2 mM MgCl2, 5.5 mM glucose, and 15 mM TRIS. Ca2+-free PSS contained 0.5 mM EGTA and 3.6 mM MgCl2. To avoid the impact of Cl−/HCO3 exchange on intracellular Cl− handling, bicarbonate-free PSS was employed in all experiments, and pH was adjusted to 7.4 by the addition of 0.1 N HCl. PSS osmolality was increased by the addition of 50–300 mM sucrose as a cell-impermeable osmolyte. In Cl−-free PSS, KCl, NaCl and MgCl2 were substituted with Na-gluconate, K-gluconate and MgSO4, respectively, whereas CaCl2 was omitted. Previously, it was shown that the absence of Ca2+o does not affect volume-dependent regulation of ouabain-resistant 86Rb fluxes in VSMC [43]. Chemicals were obtained from Sigma (St. Louis, Mo., USA) with the exception of EGTA, POPOP, PPO (Serva, Heidelberg, Germany), verapamil (Orion, Helsinki, Finland) and cell culture medium (Gibco BRL, Gaithersburg, Mo., USA). Radiochemicals were from New England Nuclear (Boston, Mass.) and Amersham (Mississauga, Ont.). Stock solutions of bumetanide were prepared in DMSO, whereas nifedipine was dissolved in 70% ethanol. Neither DMSO nor ethanol at a final concentration of 0.1% (v/v), respectively, affected the parameters measured.
Statistics
The data, presented as mean±SE, were analyzed by Student’s t-test or the t-test for dependent samples, as appropriate. Significance was defined as P<0.05.
Results
Cell volume modulation in hypo- and isosmotic media
Ten minutes incubation of WKY-7 cells was sufficient to establish the steady-state distribution of [14C]-urea between the extra- and intracellular compartment (Fig. 1A, curve 1). In the presence of 150 mM sucrose, VSMC volume was decreased by ~20% in the first 5 min, and did not change significantly during the next 40 min of incubation (Fig. 1a, curve 2). Figure 1b shows the dependence of cell volume on sucrose added to isosmotic PSS at concentrations from 50 to 150 mM.

Cell volume modulation in hyperosmotically (a, b) and isosmotically (c) shrunken vascular smooth muscle rings (VSMC). a Kinetics of cell volume modulation in hyperosmotic medium. WKY-7 cells were incubated for time intervals indicated on the x-axis in 0.5 ml physiologically balanced salt solution (PSS) containing 2 μCi/ml [14C]-urea. At the time point indicated by the arrow, 0.5 ml of PSS (1) or PSS containing 300 mM sucrose (2) was added. In both cases, the media contained 2 μCi/ml [14C]-urea. b Dependence of cell volume on medium osmolality. WKY-7 cells were incubated for 30 min in PSS containing 2 μCi/ml [14C]-urea and sucrose at concentrations indicated on the x-axis. c Kinetics of cell volume modulation under isosmotic shrinkage. The cells were incubated for 45 min in hyposmotic PSS containing 40 mM NaCl and 2 μCi/ml [14C]-urea. At the time point indicated by the arrow, this medium was aspirated, and isosmotic PSS (120.4 mM NaCl) with 2 μCi/ml [14C]-urea was added. Mean±SE from experiments performed in triplicate are shown
In additional experiments, we triggered the loss of intracellular osmolytes by preincubation of WKY-7 cells in hyposmotic medium, and then transferred them to isosmotic PSS. This approach is commonly taken to verify the role of isosmotic shrinkage in the regulation of cellular functions [40]. In contrast to hyperosmotic shrinkage, transfer from hypo- to isosmotic medium transiently decreased WKY-7 cell volume that was normalized in 25 min (Fig. 1c). This observation is consistent with previous studies revealed RVI in isosmotically-shrunken VSMC only [46, 48].
NKCC activity
In the presence of the Na+,K+-ATPase inhibitor ouabain, the addition of 10 μM bumetanide decreased the K+ (86Rb) uptake rate by threefold. Further elevation of this compound up to 100 μM did not significantly affect the rate of K+ influx (data not shown) that is consistent with ID50 values of ~1 μM reported in studies on the effect of bumetanide on NKCC1 activity in other cell types [21, 22, 53].
The elevation of medium osmolality by the addition of up to 150 mM of sucrose led to ca. threefold activation of NKCC, measured as a component of ouabain-resistant 86Rb influx inhibited by 10 μM bumetanide (Fig. 2a, curve 1). Suppression of NKCC detected at a sucrose concentration of 300 mM is probably caused by feedback inhibition of this carrier with Cl−i [37, 53]. We also noted that elevation of medium osmolality decreased the (ouabain+bumetanide)-resistant K+ influx rate from ~7 to 2 nmol mg protein−1 5 min−1 (Fig. 2a, curve 2). This effect was completely abolished in Cl−-free medium (Fig. 2a, curve 3). These results suggest that suppression of bumetanide-resistant K+ influx in hyperosmotic medium is caused by the inhibition of KCC reciprocally regulated by cell volume and detected in our previous studies [6, 43].

Volume-dependent modulation of Na+,K+,Cl− cotransport (NKCC) in VSMC. a Dependence of NKCC activity (1) and the bumetanide-resistant component of 86Rb influx (2 and 3) on medium osmolality. Curves 1, 2: cells were incubated for 5 min in PSS containing 1 mM ouabain, 1 μCi/ml 86Rb±10 μM bumetanide. Curve 3: cells were incubated for 5 min in Cl−-free PSS containing 1 mM ouabain, 1 μCi/ml 86Rb and 10 μM bumetanide. The concentration of sucrose added to control (1 and 2) or Cl−-free (3) PSS is indicated on the x-axis. b Kinetics of NKCC modulation in hyperosmotically (2) and isosmotically (3) shrunken cells. Cells were incubated for 45 min in isosmotic (1, 2) or hyposmotic (40 mM NaCl) (3) PSS. These media were then aspirated, and 0.25 ml of isosmotic PSS (1 and 3) or PSS containing 150 mM sucrose (2) was added. At the indicated time points, isotope uptake was initiated by the addition of 0.25-ml aliquots of the same media containing 1 mM ouabain, 1 μCi/ml 86Rb±10 μM bumetanide and terminated in the next 5 min. Mean±SE from experiments performed in quadruplicate are shown
The increment of NKCC detected after 5 min of 150 mM sucrose addition was preserved with up to 30 min of incubation in hyperosmotic medium (Fig. 2b, curve 2). In contrast, isosmotic shrinkage led to transient activation of this carrier that was completely abolished in 30 min of cell transfer from hypo- to isosmotic medium (Fig. 2b, curve 3).
Contractions of hyperosmotically and isosmotically shrunken VSMR: role of NKCC
Elevation of sucrose concentration from 50 to 200 mM led to dose-dependent VSMR contractions (Fig. 3, curve 2). The contractile response triggered by 150 mM sucrose (curve 3) remained stable for at least three subsequent additions made with 30-min washout intervals, and its amplitude with 20 min of sucrose addition was 51.8±9.0% (n=78) of the maximal contraction detected in the presence of 36 mM KCl (curve 1).

Isometric contractile force recording from vascular smooth muscle rings (VSMR) under hyperosmotic shrinkage. VSMR were subjected to K+o-induced depolarization (curve 1), subsequent elevation of sucrose concentration up to 300 mM (curve 2), and by the addition of 150 mM sucrose in the absence (curve 3) and presence (curve 4) of bumetanide. Bumetanide was added at a concentration of 10 μM 5 min before sucrose
Figure 4 shows representative tracing of the contractile response of depolarized and PE-treated VSMR in control and hyperosmotic medium. In these experiments, we observed that contractions triggered by equimolar substitution of 30 mM NaCl with KCl and by 1 μM PE were suppressed after 20 min of sucrose addition by 89.6±8.8% and 82.8±10.8% (n=6, P<0.00001), compared to contractions, triggered by these stimuli in isosmotic PSS.

Effect of hyperosmotic shrinkage on VSMR contractions triggered by K+o-depolarization (a) and phenylephrine (PE) (b)
To study the effect of isosmotic shrinkage on VSMR contraction, we applied a protocol developed with cultured cells (Fig. 1c). Application of the hyposmotic solution resulted in VSMR contractions that were completely abolished in 30–45 min (Fig. 5, curve 2). Subsequent transfer of VSMR to isosmotic medium led to transient contractions (curve 3) with a maximal amplitude of 21.6±8.7% (n=7) compared to K+o-induced contractions. Like hyperosmotic shrinkage, presumed isosmotic shrinkage of VSMR sharply attenuated contraction triggered by K+o depolarization and PE (Fig. 6).

Isometric contractile force recording from VSMR under isosmotic shrinkage. VSMR were subjected to K+o-induced depolarization (curve 1), reduced osmolality (PSS containing 40 mM NaCl, curve 2), and transfer from hypo- to isosmotic medium (PSS) in the absence (curve 3) or presence (curve 4) of 10 μM bumetanide . For more details see text

Effect of isosmotic shrinkage on VSMR contractions triggered by K+o-depolarization (a) and PE (b). Broken lines show contractions in the absence of stimuli mentioned above
Five minutes of pretreatment with 10 µM bumetanide led to 12% inhibition of sucrose-induced contractions (Fig. 3, curve 4). It should be underlined that in contrast to rapid inhibition of 86Rb influx detected with 10 μM bumetanide in WKY-7 cells (Fig. 2a), its action on sucrose-induced VSMR contractions increased with time (Fig. 7a) and reached 33% of inhibition after 30-min preincubation (n=4, P<0.005). In contrast to hyperosmotic shrinkage, both maximal amplitude and duration of contractions detected in isosmotically shrunken VSMR were potentiated rather than attenuated by bumetanide (Fig. 5, curve 4 versus 3, and Table 1).

Kinetics of inhibition of sucrose- (a), K+o- (b) and PE- (c) induced contractions by bumetanide. VSMR were preincubated with 10 (1), 50 (2) or 100 μM (3) bumetanide, and then contractions were triggered by the addition of 150 mM sucrose, 1 μM PE or equimolar substitution of NaCl with 30 mM KCl. The amplitude of contractions in the absence of bumetanide was taken as 100%. Mean±SE from 4–7 experiments are shown
Keeping in mind the distinct effect of bumetanide in hyperosmotically and isosmotically shrunken VSMR, we examined its action on contractions triggered by 1 μM PE and by depolarization in modest- (36 mM) and high- (66 mM) K+ medium. Similarly to hyperosmotic shrinkage, contractions triggered by [K+]o elevation up to 36 mM were slightly inhibited by 5 min incubation with 10 μM bumetanide, reaching 35±12% and 53±10% of inhibition after 30 min preincubation with 10 or 100 μM bumetanide, respectively (Fig. 7b). A more striking action of bumetanide was detected in the study of PE-induced contractions. In this case, 30 min preincubation with 10 or 100 μM bumetanide suppressed contractions by 42% (n=4, P<0.02) and 89% (n=9, P>0.0000001) (Fig. 7c). In contrast to PE (Fig. 8a) and modest elevation of [K+]o (Fig. 8B), contractions in high-K+ medium were completely insensitive to bumetanide (Fig. 8c), and after 30 min preincubation with this compound at 100 μM concentration the maximal amplitude was 96±6% of control values (n=6).

Effect of bumetanide on contractile force generation by VSMR induced by PE (a) or by depolarization in moderate- (b) or high-K+ (c) medium
Role of intracellular Cl−
Bumetanide did not affect cell volume in hyperosmotically shrunken VSMC, but suppressed RVI detected in VSMC subjected to isosmotic shrinkage (Table 2). Comparison of the sucrose effect on cell volume and Cl−i content shows that [Cl−]i elevation by ~40% was mainly caused by attenuation of cell volume rather than by NKCC activation. Indeed, NKCC inhibition with bumetanide decreased the sucrose-induced increment of [Cl−]i by only10–15%. After 15 min of isosmotic shrinkage, [Cl−]i was increased in the absence and presence of bumetanide by 75% and 30%, respectively, which was also consistent with the magnitude of volume modulation observed in control and bumetanide-treated, isosmotically shrunken VSMC (Table 2).
Long-term maintenance in culture affects diverse cellular functions, including the activity of ion transporters [9, 52]. Thus, in VSMC subjected to more than ten passages and in the absence of any stimuli, such as cell shrinkage, NKCC operates as a (Na+,Cl−)-dependent K+/K+ exchanger [44]. These data are consistent with the modest effect of bumetanide on [Cl−]i in cultured WKY-7 cell line (Table 2) and contrast with the 30–40% decrease of [Cl−]i in freshly isolated rat femoral arteries [12]. Keeping this in mind, we studied the effect of bumetanide on [Cl−]i in control and shrunken aortic rings. Table 3 shows that 30 min preincubation with 100 μM bumetanide decreased Cl−i content by ~40%, and completely abolished the rise of this parameter caused by 20 min hyperosmotic shrinkage in the presence of 150 mM sucrose.
Role of L-type Ca2+ channels and extracellular Ca2+
It is generally accepted that electrical excitation leads to VSMC contraction caused by Ca2+ influx via voltage-gated long-lasting (L-type) Ca2+ channels (for references see [30]). K+o-induced depolarization of WKY-7 cells caused two- to threefold elevation of the 45Ca influx rate. Neither 1 μM nifedipine nor 30 μM verapamil, potent inhibitors of L-type Ca2+ channels, significantly affected baseline Ca2+ uptake, but completely abolished the depolarization-induced increment of the Ca2+ influx with a ID50 of ~0.01 and 1 μM, respectively (data not shown). These values are consistent with data obtained for L-type Ca2+ channels by the patch-clamp technique [34]. Neither hyperosmotic shrinkage nor hyposmotic swelling affected nifedipine-resistant Ca2+ uptake in control and depolarized VSMC (Fig. 9). Hyposmotic swelling sharply increased nifedipine-sensitive Ca2+ influx under baseline conditions, whereas hyperosmotic shrinkage inhibited by ~40% nifedipine-sensitive Ca2+ influx triggered by K+o-induced depolarization (Fig. 9).

Effect of hyperosmotic shrinkage and hyposmotic swelling on baseline (a) and depolarization-induced (b) 45Ca uptake by WKY-7 cells. Cells were incubated for 5 min with 45Ca (2–3 μCi/ml) in control (~6 mM KCl, 120 mM NaCl) or high-K+ PSS (~43 mM KCl, 83 mM NaCl) containing 0.1 mM CaCl2 with or without 1 μM nifedipine. To induced shrinkage, 150 mM sucrose was added. To induce swelling, NaCl concentration was decreased by 60 mM. Mean±SE from three experiments performed in triplicate are shown
Keeping in mind the K+o-dependent effect of bumetanide on VSMR contractions (Fig. 8), we studied 45Ca uptake at three different K+o concentrations. Thirty minutes of preincubation with 10 μM bumetanide did not alter 45Ca uptake at [K+]o=6 and 60 mM, whereas at modest depolarization ([K+]o=30 mM) the nifedipine-sensitive component of 45Ca uptake was decreased in bumetanide-treated VSMC by twofold (Table 4).
Consistent with previous data [19], both verapamil and nifedipine almost completely inhibited depolarization-induced VSMR contractions and partially suppressed the contractions triggered by PE (Table 5). We also observed sharp inhibition by L-type channel blockers of VSMR contractions in hypotonic medium. In contrast, neither verapamil nor nifedipine affected hyperosmotically and isosmotically shrunken VSMR contractions (Table 5).
To further examine the role of Ca2+, we subjected VSMR to 1 h preincubation in Ca2+-free solution containing the Ca2+ chelator EGTA. In contrast to almost complete inhibition of K+o- and PE-induced contractions by Ca2+ depletion documented in previous studies [19] and confirmed in our experiments, this procedure attenuated the amplitude of contractions of hyperosmotically and isosmotically shrunken VSMR by ~50 and 35%, respectively (Table 6).
To determine whether or not the lack of complete suppression of shrinkage-induced contractions in Ca2+-free medium is caused by increased sensitivity of the contractile machinery to Ca2+, we compared the effect of extracellular Ca2+ on depolarization-induced contractions of control and shrunken VSMR. The addition of 1 mM CaCl2 led to the development of full-scale contraction of VSMR subjected to depolarization in Ca2+-free medium (Fig. 10). In contrast, [Ca2+]o elevation up to 10 mM did not trigger contractions of hyperosmotically shrunken VSMR subjected to subsequent depolarization (Fig. 10). Negative results were also obtained in isosmotically shrunken VSMR (Fig. 11).

Effect of extracellular Ca2+ on depolarization-induced contractions of control and hyperosmotically shrunken VSMR

Effect of extracellular Ca2+ on depolarization-induced contractions of control and isosmotically shrunken VSMR
Discussion
The data presented here drew us to a number of conclusions: (1) both cell shrinkage and swelling lead to VSMR contractions; (2) contractions triggered by shrinkage are not mediated by activation of L-type Ca2+ channels and are at least partially Ca2+o-independent. In contrast, contractions of swollen VSMR are accompanied by elevated Ca2+ uptake and abolished by L-type Ca2+ channel inhibitors; (3) both hyperosmotic and isosmotic shrinkage inhibits contractions triggered by K+o-induced depolarization and PE; (4) NKCC inhibition with bumetanide decreases [Cl−]i and contractions triggered by modest depolarization as well as contractions triggered by PE and hyperosmotic shrinkage, but potentiated contractions of isosmotically shrunken VSMR; (5) suppression by bumetanide of contractions evoked by modest [K+]o elevation is caused by partial inactivation of L-type Ca2+ channels, probably because of Cl−i-mediated membrane hyperpolarization; and (6) in isosmotically shrunken cells, NKCC inhibition enhances contractions via RVI suppression. Data supporting these conclusions are considered below.
Cell volume modulation is sufficient to evoke VSMR contractions
Several lines of evidence indicate that VSMR contractions detected in anisosmotic media are caused by cell volume modulation rather than by altered medium osmolality per se. First, the magnitude of VSMR contractions triggered by sucrose at concentrations from 50 to 150 mM (Fig. 3) was in proportion to cell volume reduction measured as the volume of [14C]-available space (Fig. 1b). Second, the volume of hyposmotically swollen VSMC was rapidly normalized by RVD occurring via activation of K+ and Cl− channels [6]. These data are consistent with our results showing slight cell volume elevation after 45 min incubation of VSMC in hyposmotic medium (3.03±0.16 versus 2.85±0.18 µl/mg protein in control; n=3) and transient VSMR contractions (Fig. 5, curve 2). Third, sustained versus transient contractions detected in sucrose-treated (Fig. 3) and isosmotically shrunken VSMR (Fig. 5) are consistent with the dynamics of cell volume modulation observed under these conditions (Fig. 1a, c). Fourth, the transient kinetics of volume decrease in isosmotically shrunken VSMC are caused by NKCC-mediated RVI [46]. Both cell volume decrease (Table 2) and force generation in isosmotically shrunken VSMR were prolonged under NKCC inhibition with bumetanide (Table 2 and Fig. 5).
Contractions of shrunken and swollen VSMR: distinct impact of Ca2+-mediated signaling
Despite the monotonous impact on force generation, the cellular mechanisms underlying the contractile responses of shrunken and swollen VSMR are essentially different. Indeed, similarly to K+o-induced depolarization, hyposmotic swelling of VSMC augmented 45Ca influx inhibited by nifedipine (Fig. 9). Moreover, both nifedipine and verapamil abolished contractions of swollen VSMR (Table 5). These results strongly suggest that swelling-induced VSMR contractions were mediated by L-type Ca2+ channel activation (Fig. 12). In contrast, contractions triggered by hyper- and isosmotic shrinkage were resistant to L-type Ca2+ channels blockers (Table 5). Contractions of shrunken VSMR were also preserved in the absence of Ca2+o and depletion of intracellular Ca2+ stores by incubation in Ca2+-free, EGTA-containing medium, i.e. conditions abolishing K+o- and PE-induced contractions (Table 6).

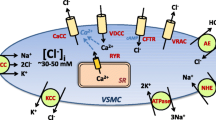
Cross-talk of cell volume, Na+,K+,Cl−-cotransport, intracellular Cl−i and Ca2+ in the regulation of vascular smooth muscle contraction. 1 NKCC; 2 L-type Ca2+ channels; 3 calcium release-activated Ca2+ channels; 4 Ca2+-activated anion channels; Em electrical membrane potential; MLCK myosin light chain kinase; RVI regulatory volume increase; bum, ver, nif bumetanide, verapamil and nifedipine, respectively. +, − activatory and inhibitory stimuli; ? unknown intermediates of signal transduction. For more details see text
In additional experiments, we observed that hyperosmotic shrinkage sharply attenuates rather than enhances contractions triggered by K+o-depolarization and PE (Fig. 4). This finding is consistent with data obtained on VSMR shrunken in the presence of 100 mM urea [58]. Inhibition of K+o- and PE-induced contractions was also seen in isosmotically shrunken VSMR (Fig. 6). Importantly, 30 min preincubation in hyposmotic medium preceding isosmotic shrinkage increased the magnitude of K+o-induced contractions (148.2±28.7 versus 100% in control; n=9, P<0.05) and did not influence contractions triggered by PE (100±14.4%, n=6). Viewed collectively, these results demonstrate the distinct mechanism of contractions triggered by cell shrinkage and physiological stimuli.
Several attractive hypotheses have been generated to explain the Ca2+-independent mechanism of excitation-contraction coupling detected in shrunken VSMR. Thus, Kravtsov and co-workers reported that K+o-induced contractions in Mg2+-free medium are not mediated by Ca2+-influx and occur in the presence of EDTA [30]. It should be underlined, however, that in contrast to shrunken VSMR, K+o-induced contractions in Mg2+-free medium were suppressed by L-type channel blockers [30]. It was shown that Ca2+o-independent VSMC contractions triggered by activators of protein kinase C, tyrosine and Rho-associated kinases were mediated by calcium sensitization of the contractile machinery [8, 23, 38]. Both myosin light chain phosphorylation and Rho kinase-dependent myosin II translocation have been detected in hyperosmotically shrunken cells [26, 27, 50, 54, 56]. However, in contrast to K+o- and PE-induced contractions, contractions of Ca2+-depleted, shrunken VSMR were not evoked by the addition of Ca2+ (Figs. 10, 11). Keeping in mind that the total content of macromolecules in shrunken cells is increased and that modest elevation of this parameter affects diverse intracellular proteins, including cytoskeleton organization [13, 33], the role of macromolecular crowding in contractions of shrunken VSMR deserves further investigation.
Role of NKCC and intracellular Cl−
Unlike the dominant contribution of K+ permeability to resting membrane potential in skeletal and cardiac muscles, the PK/PCl ratio in VSMC varies from 0.8 to 0.5 [12]. These data led Brown et al. to suggest that NKCC contributes to vascular tone via the maintenance of [Cl− ]i [11]. Later on, it was shown that NKCC inhibition with bumetanide decreases [Cl−]i, and hyperpolarizes the sarcolemma in freshly isolated VSMR [15]. These data suggest that NKCC inhibition will decrease VSMC contractions triggered by modest depolarization, but will not affect contractions in sharply depolarized cells (Fig. 12). Indeed, 30 min pretreatment with bumetanide reduced the increment of nifedipine-sensitive Ca2+ uptake and VSMR contractions evoked by 30 mM KCl but did not affect these parameters at [K+]o=60 mM (Table 4, Fig. 8). It may be assumed that the lack of effect of bumetanide in high-K+ medium is caused by partial inactivation of NKCC due to reduction of [Na+]o from 120 to 66 mM. However, the high affinity of VSMC NKCC for Na+o, documented in our previous study (K0.5~25 mM) [44] did not support this assumption.
We observed that at 100 μM concentration bumetanide almost completely blocked the contractions triggered by PE (Fig. 8). Akar et al. reported that at 10 μM and 20 min preincubation, bumetanide inhibited VSMR contractions triggered by 1 μM PE by only 5–10% [4]. Several explanations for this controversy might be proposed. First, complete and rapid inhibition of NKCC, detected in cultured VSMC in the presence of 10 μM bumetanide (Fig. 2), does not mean that this concentration will lead to rapid hyperpolarization. Indeed, the effect of bumetanide on K+o-induced contractions was increased with concentration and time (Fig. 7b). We also detected that 30 min of incubation with 10 or 100 μM bumetanide decreased [Cl−]i by 18% and 38%, respectively (Table 3). Second, the above-mentioned discrepancy is caused by differences between rat strains. Indeed, the PE EC50 of 10−6.9 M, detected in VSMR from rats in our study (data not shown), was an order of magnitude higher than values reported by Akar et al. [4]. Third, at high concentrations, bumetanide affects VSMR contractions independently of NKCC inhibition. This assumption makes sense in analyzing the mechanisms of almost complete inhibition of PE-induced contractions after 30 min preincubation with 100 μM bumetanide (Fig. 8a). However, keeping in mind the lack of effect of bumetanide on contractions triggered by high [K+]o (Fig. 8c), upstream PE-specific intermediates of signaling, such as α-adrenoceptors, phospholipase C, calcium release-activated Ca2+ channels, rather than downstream elements of the contractile machinery, should be examined as potential targets for high-ceiling diuretics. Fourth, at high concentrations loop diuretics partially suppress KCC [32]. Recent data suggest that KCC activation contributes to vasodilatory action of nitric oxide derivatives [1]. The role of this carrier in modulation of VSMR contraction caused by 100 μM bumetanide should be examined further.
Rapid elevation of [Cl−]i in sucrose-treated VSMC cells is a direct consequence of cell shrinkage. In contrast, NKCC activation is a major determinant of [Cl−]i elevation in isosmotically shrunken cells subjected to preliminary Cl− depletion in hyposmotic medium. A key role of NKCC in [Cl−]i adjustment is consistent with a rapid decline of this parameter in isosmotically shrunken VSMC treated with bumetanide (Table 2). Thus, it can be suggested that shrinkage-induced contraction is caused by [Cl−]i elevation and membrane depolarization. It should be underlined, however, that the lack of involvement of L-type Ca2+ channels in contractions of isosmotically and hyperosmotically shrunken VSMR (Table 6) contradicts this assumption. Moreover, we observed that inhibition of NKCC providing inwardly directed Cl− transport potentiates rather than inhibits the contraction of isosmotically shrunken VSMR (Fig. 5). Prolongation of isosmotically shrunken VSMR contractions by bumetanide is probably caused by suppression of NKCC-mediated RVI (Figs. 1c, 2c, Table 2). Two hypotheses can be offered to explain the partial inhibition with bumetanide of nifedipine-insensitive contraction of hyperosmotically shrunken VSMR (Fig. 7A). First, hyperpolarization developing under NKCC inhibition suppressed the activity of inwardly directed Ca2+ transporters distinct from L-type channels. It should be mentioned, however, that contractions of shrunken VSMR were only partially suppressed in Ca2+-free medium (Table 6). Second, [Cl−]i modulation affects intracellular proteins involved in assembly of the contractile machinery independently of the regulation of membrane potential.
In conclusion, our results show that both swelling and shrinkage lead to VSMR contractions. Swelling-induced contractions are caused by activation of L-type Ca2+ channels. In contrast, contractions of shrunken VSMR are insensitive to the presence of L-type channel inhibitors and occur in the absence of Ca2+o. Augmented contractions detected in isosmotically shrunken VSMR in the presence of bumetanide are probably caused by suppression of NKCC-mediated RVI. The mechanism of bumetanide-sensitive, Ca2+-independent contractions in hyperosmotically shrunken VSMR deserves further investigations.
References
Adragna N, White RE, Orlov SN, Lauf PK (2000) K-Cl cotransport in vascular smooth muscle and erythrocytes: possible implication in vasodilation. Am J Physiol 278:C381–C390
Adragna N, Di Fulvio M, Lauf PK (2004) Regulation of K-Cl cotransport: from function to genes. J Membr Biol (in press)
Akar F, Skinner E, Klein JD, Jena M, Paul RJ, O’Neill WC (1999) Vasoconstrictors and nitrovasodilators reciprocally regulate the Na+-K+-2Cl− cotransporter in rat aorta. Am J Physiol 276:C1383–C1390
Akar F, Jiang G, Paul RJ, O’Neill WC (2001) Contractile regulation of the Na+-K+-2Cl− cotransporter in vascular smooth muscle. Am J Physiol 281:C579–C584
Alvarez-Guerra M, Nazaret C, Garay RP (1998) The erythrocyte Na,K,Cl cotransporter and its circulating inhibitor in Dahl salt-sensitive rats. J Hypertens 16:1499–1504
Anfinogenova YJ, Rodriguez X, Grygorczyk R, Adragna N, Lauf PK, Hamet P, Orlov SN (2001) Swelling-induced K+ fluxes in vascular smooth muscle cells are mediated by charybdotoxin-sensitive K+ channels. Cell Physiol Biochem 11:295–310
Anfinogenova YJ, Kilin AA, Kovalev IV, Baskakov MB, Orlov SN (2003) Vascular smooth muscle contraction in hyperosmotic medium: role of Ca2+, anion channels and cell volume-sensitive Na+,K+,Cl− cotransport. J Hypertens 21:S101
Barandier C, Ming X-F, Yang Z (2003) Small G proteins as novel therapeutic targets in cardiovascular medicine. News Physiol Sci 18:18–22
Berk BC, Vallega G, Muslin AJ, Gordon HM, Canessa M, Alexander RW (1989) Spontaneously hypertensive rat vascular smooth muscle cells in culture exhibit increased growth and Na+/H+ exchange. J Clin Invest 83:822–829
Bianchi G, Ferrari P, Trizio P, Ferrandi M, Torielli L, Barber BR, Polli E (1985) Red blood cell abnormalities and spontaneous hypertension in rats. A genetically determined link. Hypertension 7:319–325
Brown RA, Chipperfield AR, Davis JPL, Harper AA (1999) Increased (Na+K+Cl−) cotransport in rat arterial smooth muscle in deoxycorticosterone (DOCA)/salt-induced hypertension. J Vasc Res 36:492–501
Chipperfield AR, Harper AA (2001) Chloride in smooth muscle. Prog Biophys Mol Biol 74:175–221
Cuneo P, Margi E, Verzola A, Grazi E (1992) “Macromolecular crowding” is a primary factor in the organization of the cytoskeleton. Biochem J 281:507–512
Davis A, Hogarth K, Fernandes D, Solway J, Niu J, Kolenko V, Browning D, Miano JM, Orlov SN, Dulin NO (2003) Functional significance of protein kinase A (PKA) activation by endothelin-1 and ATP: negative regulation of SRF-dependent gene expression by PKA. Cell Signal 15:597–604
Davis JPL, Chipperfield AR, Harper AA (1993) Accumulation of intracellular chloride by (Na-K-Cl) cotransport in rat arterial smooth muscle is enhanced in deoxycorticosterone acetate (DOCA)/salt hypertension. J Mol Cell Cardiol 25:233–237
Flagella M, Clarke LL, Miller ML, Erway LC, Giannella RA, Andriga A, Gawenis LR, Kramer J, Duffy JJ, Doetschman T, Lorenz JN, Yamoah EN, Cardell EL, Shull GE (1999) Mice lacking the basolateral Na-K-2Cl cotransporter have impaired epithelial chloride secretion and are profoundly deaf. J Biol Chem 274:26946–26955
Garay RP (1982) Inhibition of the Na+/K+ cotransport system by cyclic AMP and intracellular calcium in human red cells. Biochim Biophys Acta 688:786–792
Glukhova MA, Frid MG, Koteliansky VE (1994) Phenotypic changes of human aortic smooth muscle cells during development and in adult. J Atheroscler Thromb 1 [Suppl 1]:S47–S49
Godfraind T (1994) Calcium antagonists and vasodilation. Pharmacol Ther 64:37–75
Hoffmann EK, Simonsen LO (1989) Membrane mechanisms in volume and pH regulation in vertebrate cells. Physiol Rev 69:315–382
Isenring P, Forbush III B (1997) Ion and bumetanide binding by the Na-K-Cl cotransporter. Importance of transmembrane domains. J Biol Chem 272:24556–24562
Isenring P, Jacoby SC, Payne JA, Forbush BI (1998) Comparison of Na-K-Cl cotransporters: NKCC1, NKCC2 and HEK cell Na-K-Cl cotransporter. J Biol Chem 273:11295–11301
Janssen LJ, Lu-Chao H, Netherton S (2001) Excitation-contraction coupling in pulmonary vascular smooth muscle involves tyrosine kinase and Rho-kinase. Am J Physiol 280:L666–L674
Jiang G, Cobbs S, Klein JD, O’Neill WC (2003) Aldosterone regulates the Na-K-Cl cotransporter in vascular smooth muscle. Hypertension 41:1131–1135
Jones AW (1973) Altered ion transport in vascular smooth muscle from spontaneously hypertensive rats. Influence of aldosterone, norepinephrine and angiotensin. Circ Res 33:563–572
Klein JD, O’Neill WC (1993) Myosin light chain phosphorylation in endothelial cells is regulated by cell volume and correlates with volume-regulatory Na-K-2Cl cotransport (abstract). J Gen Physiol 102:18a
Klein JD, O’Neill WC (1995) Volume-sensitive myosin phosphorylation in vascular endothelial cells: correlation with Na-K-2Cl cotransport. Am J Physiol 269:C1524–C1531
Kotelevtsev YuV, Orlov SN, Pokudin NI, Agnaev VM, Postnov YuV (1987) Genetic analysis of inheritance of Na+,K+ cotransport, calcium level in erythrocytes and blood pressure in F2 hybrids of spontaneously hypertensive and normotensive rats. Bull Exp Biol Med 103:456–458
Kovalev IV, Baskakov MB, Anfinogenova YJ, Borodin YL, Kilin AA, Minochenko IL, Popov AG, Kapilevich LV, Medvedev MA, Orlov SN (2003) Effect of Na+,K+,2Cl− cotransport inhibitor bumetanide on electrical and contractile activity of smooth muscle cells in guinea pig ureter. Bull Exp Biol Med 136:145–149
Kravtsov GM, Bruce IC, Wong TK, Kwan CY (2003) A new view of K+-induced contraction in rat aorta: the role of Ca2+ binding. Pflugers Arch 446:529–540
Lang F, Busch G, Ritter M, Volkl H, Waldegger S, Gulbins E, Haussinger D (1998) Functional significance of cell volume regulatory mechanisms. Physiol Rev 78:247–306
Lauf PK, Adragna NC (2000) K-Cl cotransport: properties and molecular mechanism. Cell Physiol Biochem 10:341–354
Madden TL, Herzfeld J (1993) Crowding-induced organization of cytoskeletal elements. I. Spontaneous demixing of cytoplasmic proteins and model filaments to form filament bundles. Biophys J 65:1147–1154
McDonald TF, Pelzer S, Trautwein W, Pelzer DJ (1994) Regulation and modulation of calcium channels in cardiac, skeletal, and smooth muscle cells. Physiol Rev 74:365–512
Meyer JW, Flagella M, Sutliff RL, Lorenz JN, Nieman ML, Weber GS, Paul RJ, Shull GE (2002) Decreased blood pressure and vascular smooth muscle tone in mice lacking basolateral Na+-K+-2Cl− cotransporter. Am J Physiol 283:H1846–H1855
Mongin AA, Orlov SN (2001) Mechanisms of cell volume regulation and possible nature of the cell volume sensor. Pathophysiology 8:77–88
Mount DB, Delpire E, Gamba G, Hall AE, Poch E, Hoover RS, Hebert SC (1999) The electroneutral cation-chloride cotransporters. J Exp Biol 201:2091–2102
Nakao F, Kobayashi S, Mogami K, Mizukami Y, Shirao S, Miwa S, Todoroki-Ikeda N, Ito M, Matsuzaki M (2002) Involvement of Src family protein tyrosine kinases in Ca2+ sensitization of coronary artery contraction mediated by a sphindosylphosphorylcholine-Rho-kinase pathway. Circ Res 91:953–960
O’Neill WC (1999) Physiological significance of volume-regulated transporters. Am J Physiol 276:C995–C1011
O’Neill WC, Klein JD (1994) Regulation of vascular endothelial cell volume by Na-K-2Cl cotransport. Am J Physiol 262:C436–C444
Orlov SN (2003) Hypertension. In: Bernhardt I, Ellory JC (eds) Red cell membrane transport in health and disease. Springer, Berlin Heidelberg New York, pp 587–602
Orlov SN, Resink TJ, Bernhardt J, Buhler FR (1992) Na+-K+ pump and Na+-K+ co-transport in cultured vascular smooth muscle cells from spontaneously hypertensive rats: baseline activity and regulation. J Hypertens 10:733–740
Orlov SN, Resink TJ, Bernhardt J, Buhler FR (1992) Volume-dependent regulation of sodium and potassium fluxes in cultured vascular smooth muscle cells: dependence on medium osmolality and regulation by signalling systems. J Membr Biol 126:199–210
Orlov SN, Tremblay J, Hamet P (1996) Bumetanide-sensitive ion fluxes in vascular smooth muscle cells: lack of functional Na+,K+,2Cl− cotransport. J Membr Biol 153:125–135
Orlov SN, Tremblay J, Hamet P (1996) cAMP signaling inhibits dihydropyridine-sensitive Ca2+ influx in vascular smooth muscle cells. Hypertension 27:774–780
Orlov SN, Tremblay J, Hamet P (1996) Cell volume in vascular smooth muscle is regulated by bumetanide-sensitive ion transport Am J Physiol 270:C1388–C1397
Orlov SN, Adragna N, Adarichev VA, Hamet P (1999) Genetic and biochemical determinants of abnormal monovalent ion transport in primary hypertension. Am J Physiol 276:C511–C536
Orlov SN, Pchejetski D, Taurin S, Thorin-Trescases N, Maximov GV, Pshezhetsky AV, Rubin AB, Hamet P (2004) Apoptosis in serum-deprived vascular smooth muscle cells: evidence for cell volume-independent mechanism. Apoptosis 9:55–66
Owen NE, Ridge KM (1989) Mechanism of angiotensin II stimulation of Na-K-Cl cotransport of vascular smooth muscle cells. Am J Physiol 257:C629–C636
Pedersen SF, Beisner KH, Hougaard C, Willumsen BM, Lambert IH, Hoffmann EK (2002) Rho family GTP binding proteins are involved in the regulatory volume decrease in NIH3T3 mouse fibroblasts. J Physiol (Lond) 541:779–796
Postnov YuV, Orlov SN, Gulak PV, Shevchenko AS (1976) Altered permeability of the erythrocyte membrane for sodium and potassium in spontaneously hypertensive rats. Pflugers Arch 365:257–263
Raat NJ, Hartog A, van Os CH, Bindels RJ (1994) Regulation of Na+-K+-2Cl− cotransport in rabbit proximal tubule primary culture. Am J Physiol 267:F63–F69
Russell JM (2000) Sodium-potassium-chloride cotransport. Physiol Rev 80:212–276
Shrode LD, Klein JD, O’Neill WC, Putnam RW (1995) Shrinkage-induced activation of Na+/H+ exchange in primary rat astrocytes: role of myosin light-chain kinase. Am J Physiol 269:C257–C266
Smith JB, Smith L (1987) Na+/K+/Cl− cotransport in cultured vascular smooth muscle cells: stimulation by angiotensin II and calcium ionophores, inhibition by cyclic AMP and calmodulin antagonists. J Membr Biol 99:51–63
Takeda M, Homma T, Breyer MD, Horiba N, Hoover RL, Kawamoto S, Ichikawa I, Kon V (1993) Volume and agonist-induced regulation of myosin light-chain phosphorylation in glomerular mesangial cells. Am J Physiol 264:F421–F426
Tseng H, Berk BC (1992) The Na/K/2Cl cotransporter is increased in hypertrophied vascular smooth muscle cells. J Biol Chem 267:8161–8167
Wagner CA, Huber SM, Warntges S, Zempel G, Kaba NK, Fux R, Orth N, Busch GL, Waldegger S, Lambert I, Nilius B, Heinle H, Lang F (2000) Effect of urea and osmotic cell shrinkage on Ca2+ entry and contraction of vascular smooth muscle cells. Pflugers Arch 440:295–301
Acknowledgements
This work was supported by grants from the Heart and Stroke Foundation of Canada, and the INTAS Young Scientist Fellowship (YSF 2001/2-0168). The editorial assistance help of Ovid Da Silva, Editor, Research Support Office, Research Centre, CHUM, is appreciated.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Anfinogenova, Y.J., Baskakov, M.B., Kovalev, I.V. et al. Cell-volume-dependent vascular smooth muscle contraction: role of Na+, K+, 2Cl− cotransport, intracellular Cl− and L-type Ca2+ channels. Pflugers Arch - Eur J Physiol 449, 42–55 (2004). https://doi.org/10.1007/s00424-004-1316-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-004-1316-z