
Abstract
Osteoclasts are multinucleated cells of the myeloid lineage that are highly specialized for bone resorption through acidification and exocytosis of bone-degrading enzymes into the space between the osteoclast and underlying bone, termed the resorption lacuna. Physiologic osteoclast activity is essential for normal skeletal development and remodeling to repair skeletal microdamage. Osteopetrosis, a collection of monogenic diseases caused by nonfunctional or absent osteoclasts, underscores the importance of adequate osteoclast function for skeletal health. Excessive osteoclastic bone resorption also results in bone disease, including osteoporosis. In this chapter, we provide information on the cellular origin of osteoclasts, discuss the receptors and signaling pathways that drive osteoclast differentiation, and detail the transcriptional machinery required to express the osteoclast program. We detail the cytoskeletal reorganization and formation of a specialized resorption apparatus required for mineral and matrix resorption. Lastly, we discuss the consequences of perturbation of osteoclast formation or function, both in the context of genetic diseases that map to the osteoclast and in the context of pathologic conditions. The goal of this chapter is to provide a detailed overview of osteoclast formation and function that can serve as a reference for clinician’s interested in the role of the osteoclast in osteoporosis.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
FormalPara Key Points-
Osteoclasts are the only cell type known to resorb bone, and their activity is essential for normal skeletal development and remodeling to repair skeletal microdamage.
-
Osteoclast differentiation from myeloid precursors requires two key cytokines, MCSF and RANKL, as well as a second signal that is initiated by activation of an ITAM-associated receptor. During differentiation, osteoclast precursors fuse through a poorly understood mechanism to form mature multinucleated osteoclasts.
-
Osteoclasts form a tight connection to bone, termed the sealing zone, and secrete acid and degradative enzymes through a specialized membrane-rich ruffled border into the resorption lacunae.
-
Increased osteoclast activity in states of estrogen deficiency or inflammation contribute to osteoporosis. In contrast, genetic mutations, which impair osteoclast formation or activity, result in diseases such as osteopetrosis and pycnodysostosis.
Osteoclasts in the Bone Landscape
Osteoclasts are highly specialized hematopoietic cells that reside on and resorb the bone surface. Osteoclasts have some similarities to macrophages in their shared myeloid lineage and in that they are also functionally specialized for “digestion.” In contrast to macrophages, osteoclasts do not phagocytose but rather exert their digestive function outside the cell through a process called lysosomal exocytosis, in which the lysosome fuses with the plasma membrane and releases its content in the extracellular space. A further distinguishing characteristic of osteoclasts is that they are multinuclear, forming from fusion of mononuclear precursors. Normal skeletal development and remodeling require the action of osteoclasts, which are the only cells definitively shown to resorb bone. Balance between osteoclast and osteoblast activity is critical to maintain the skeleton and either over- or underactive osteoclast function can result in skeletal disease. The classic and most common disease of excess osteoclast activity is osteoporosis. As such, it is helpful for clinicians treating osteoporosis or other bone diseases to understand where osteoclasts come from, the stimuli that drive their differentiation, and the mechanism by which they resorb bone.
Osteoclasts differentiate from myeloid precursors in the presence of the key osteoclastogenic cytokine, RANKL (receptor activator of NF-κB ligand) and survival factor MCSF (macrophage colony-stimulating factor). As with many hematopoietic cells, differentiation requires a second signal, in this case provided by any one of several immunoglobulin receptors that signal through an associated immunoreceptor-based activation motif, or ITAM-containing adapter. A number of signaling pathways are activated downstream of stimulation of RANK, the receptor for RANKL, and ITAM-associated receptors, which converge to drive expression of the transcription factor NFATc1 (nuclear factor of activated T cells), the master regulator of osteoclastogenesis. NFATc1, in conjunction with the transcription factor AP-1, drives expression of a number of molecules that are required for osteoclast resorptive function, such as the protease cathepsin K and tartrate-resistant acid phosphatase (TRAP). The history of the discovery of osteoclasts as cells of the myeloid lineage and the identification of specific osteoclast precursors is discussed in detail in section “Cellular Origins of Osteoclasts.” The events of osteoclast differentiation and fusion, including an extensive discussion of the receptors, ligands, signaling pathways, and transcription factors involved, are covered in section “Osteoclast Differentiation”.
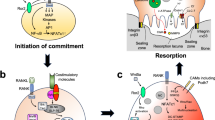
Osteoclasts do not function in isolation, but rather work in proximity with osteoblasts and osteocytes in what is termed the bone multicellular unit (BMU), diagrammed in Fig. 2.1. Within the BMU, osteoclasts and osteoblasts work in series to remodel bone. In the activation phase, remodeling can be stimulated by mechanical stress, microfractures, microischemic, or other events which release factors “trapped” in the bone microenvironment including TGFβ and IGF-1 [1]. These factors activate lining osteoblasts which can then recruit migratory mature osteoclasts as well as drive maturation of osteoclast precursors through the expression of RANK. Mature osteoclasts undergo cytoskeletal rearrangement, becoming highly polarized and form a specialized structure called the sealing zone which isolates the space between the osteoclast and underlying bone from the surrounding environment. Acidification and exocytosis of hydrolases, including the protease cathepsin K, into this space results in dissolution of the mineral and digestion of the organic matrix of bone. The process of bone resorption is reviewed in section “Functions of Osteoclasts”.

Osteoclasts in the bone multicellular unit. Osteoclasts and osteoblasts work in series to remodel the bone in the bone multicellular unit (BMU). The process of remodeling consists of four sequential and distinct phases of cellular events depicted above: activation, resorption, reversal, and formation. The coupling of osteoclasts to osteoblasts is mediated by the liberation of growth factors by process of resorption, cell contact-mediated pathways, and secreted factors produced by osteoclasts
In the reversal phase, the BMU switches from resorption to formation in what is termed the reversal phase. During this phase, digestion of bone by osteoclasts releases other factors trapped in the bone matrix, including BMPs, TGFβ, and IGF-1, which are thought to stimulate osteoblasts to form bone [2, 3]. Osteoclasts also modulate osteoblast function both through cell contact-mediated interactions and secreted factors, a regulatory function of osteoclasts discussed in section “Functions of Osteoclasts”. In the formation phase, mature osteoblasts deposit osteoid, demineralized bone matrix, followed by deposition of hydroxyapatite to generate mineralized bone (see Chap. 5).
The importance of osteoclast function for bone health is underscored by the variety of genetic diseases that map to the osteoclast, covered in section “Genetic Diseases of Osteoclast Dysfunction”. Excessive osteoclast activity also contributes to bone pathology in post-menopausal osteoporosis and inflammatory arthritis, among other conditions. In these settings, osteoclast differentiation and activity are modulated both directly by a variety of cytokines and indirectly by enhanced RANKL expression. The influence of microenvironment on osteoclasts is explored in section “Regulation of Osteoclasts By Their Environment”. Overall, this chapter attempts to provide a broad review of the cellular and molecular aspects of osteoclast differentiation and function.
Cellular Origins of Osteoclasts
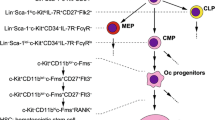
A distinct multinucleated cell type associated with bone was reported as early as 1849, though the first use of the term osteoclast was not until 1873 [4, 5]. It was not until the 1960s, however, that it was conclusively demonstrated that osteoclasts resorb bone [6,7,8]. Osteoclasts were proposed to be derived from leukocytes as early as 1911, based on their morphologic similarity to foreign body giant cells [9]. A series of elegant parabiosis and chimera experiments performed by Walker in the 1970s conclusively demonstrated the hematopoietic origin of osteoclasts [10,11,12,13]. A myeloid origin for osteoclasts was proposed early on because of the morphologic and functional similarities with macrophages and giant cells and confirmed by experiments in which labeled peripheral blood monocytes injected into mice resulted in generation of labeled osteoclasts [14].
Osteoclast progenitors have subsequently been more precisely defined though the in vitro assessment of the ability of various subsets of bone marrow or peripheral blood cells to differentiate into osteoclasts in the presence of RANKL. Each of these studies have used a variety of myeloid cell surface markers to define the osteoclast progenitor. Arai and colleagues performed the seminal studies in this area, demonstrating that the bone marrow CD11blo CD117+ (c-Kit) population contained precursors that could differentiate into osteoclasts in the presence of MCSF and RANKL [15]. Several groups have identified early myeloid progenitor populations in the bone marrow that are highly enriched for osteoclast progenitors and are distinct from the progenitors for monocytes and dendritic cells [16,17,18,19]. Peripheral blood monocytes from both mice and humans can differentiate into osteoclasts in the presence of MCSF and RANKL. Using purification based on cell surface markers in conjunction with in vitro osteoclast differentiation assays, a population of peripheral osteoclast progenitors sharing many features of classical circulating monocytes was identified in mice [17]. Circulating osteoclast progenitor populations in humans have similarly been identified as having markers overlapping with classical monocytes [20, 21]. Although both bone marrow and circulating progenitor populations efficiently differentiate into osteoclasts, the relationship between these progenitor pools and relative contribution of these progenitors to maintaining osteoclast formation is unknown.
Osteoclast Differentiation
Overview
Osteoclasts represent a terminally differentiated cell in the myeloid lineage. Similar to other differentiated myeloid cells, key cytokine stimuli are required to activate specific intracellular signaling pathways to initiate specific transcriptional programs. The master transcriptional regulator of osteoclasts, the transcription factor NFATc1, is essential for osteoclast differentiation and function [22]. The process of osteoclast differentiation from immature myeloid precursor cells is highly regulated by both positive and negative stimuli emanating from surrounding bone and immune cells. These signals orchestrate a coordinated signaling cascade that initiates precursor proliferation, fusion to multinucleated cells, cellular polarization, adherence to bone, and activation of functional resorption (Fig. 2.2).

Osteoclast differentiation. Osteoclasts develop from immature myeloid precursors. When stimulated by MCSF, they upregulate RANK, and then under the stimulation of MCSF and RANKL, they initially form mononuclear osteoclasts that fuse into multinucleated cells. The multinucleated osteoclasts polarize and adhere to bone and become functionally bone resorbing through secretion of metalloproteinases, acid, and cathepsin K (white box lower right shows osteoclast-specific genes). The figure shows in blue: Stimuli required to progress in osteoclast differentiation, in green: key transcription factors upregulated at each stage during osteoclast differentiation, in boxes within the cell: key osteoclast genes upregulated at each stage. Abbreviations: MCSF macrophage colony-stimulating factor, RANK receptor-activating NFkB, MITF microphthalmia-associated transcription factor, DC-STAMP dendritic cell-specific transmembrane protein, ECM extracellular matrix, GM-CSF, CTSK cathepsin K, TRAP tartrate resistant acid phosphatase, ITAM immunoreceptor tyrosine-based activating motif, CLC7 voltage-gated chloride channel 7, CTR calcitonin receptor, CTSK cathepsin K, CAII carbonic anhydrase II
Receptors
RANK/RANKL Signaling Is Essential for Osteoclast Differentiation
The key cytokine required to stimulate osteoclast differentiation is RANKL[23, 24] which was originally described under several names including OPGL (osteoprotegerin ligand) [25], ODF (osteoclast differentiation factor) [26] and TRANCE (TNF-related activation-induced cytokine) [27]. RANKL is in the TNF (tumor necrosis factor) cytokine family (TNFSF11) and is produced by osteoblasts, stromal cells, osteocytes, and activated immune cells as both a type II transmembrane protein and a secreted cytokine. RANKL binds to RANK (receptor activator of NF-κB) on myeloid cell precursors and serves as the key stimulus for osteoclast differentiation and activation. Studies of mice genetically deficient in RANK or RANKL demonstrated that in the absence of RANK signals, no osteoclasts are generated. Mice genetically deficient in RANK or RANKL have bones that are severely osteopetrotic, and the animals are toothless due to their inability to erupt teeth in the absence of osteoclastic degradation of the mandible [28,29,30,31]. RANKL also binds to a soluble decoy receptor OPG (osteoprotegerin or “bone protector”) which serves to prevent RANKL from interacting with RANK. Mice deficient in OPG are osteoporotic, and transgenic mice that overexpress OPG have few osteoclasts and are severely osteopetrotic [32,33,34,35,36]. The ratio of RANKL and OPG expression in the vicinity of osteoclast precursors is therefore important in determining the osteoclast differentiation response. Expression of RANKL and OPG is both highly regulated, and their production by osteoblasts/stromal cells is regulated by endocrine factors such as PTH and 1,25(OH)2D3 and inflammatory cytokines such as TNF and IL-1 [37]. Many cytokines, hormones, and growth factors regulate osteoclastogenesis indirectly, through regulation of RANKL and/or OPG expression on other cell types (Table 2.1). RANKL stimulation is required for osteoclastogenesis but is also required to activate functional resorption by mature osteoclasts, while lack of RANKL stimulation impairs osteoclast survival [37]. Given this critical role in osteoclast generation and function, RANKL was identified as an ideal therapeutic target. Denosumab (see Chap. 17) is an anti-RANKL antibody currently FDA approved for a number of indications, including treatment of postmenopausal women and men with osteoporosis at high risk for fracture, bone loss during cancer hormone ablation therapy, glucocorticoid -induced osteoporosis, and skeletal lesions in patients with bone metastases from solid tumors and giant cell tumors of the bone [38,39,40].
RANKL and OPG are expressed by osteoblasts, stromal cells, and osteocytes; however, the relative importance of each source has only recently been redefined. Osteoblasts lining the bone surface were previously thought to be the primary source of RANKL during osteoclastogenesis. However, osteocytes, the cells residing deep within the bone, were found to express high levels of RANKL, and osteocyte-derived RANKL can reach the bone surface through osteocyte canaliculi to interact with precursor cells and stimulate differentiation (Fig. 2.3) [41]. An osteocyte-specific deletion of RANKL leads to a significant osteopetrotic bone phenotype in mice, demonstrating the importance of osteocyte-derived RANKL for basal bone remodeling [42,43,44]. Osteocytes also express OPG, which can diffuse through the lacuno-canalicular system to downregulate osteoclastogenesis. Under pathologic conditions such as mechanical unloading or “weightlessness,” osteocytes increase production of sclerostin, a Wnt inhibitor, which leads to decreased OPG and increased RANKL production to stimulate osteoclastogenesis [45, 46]. Osteocyte-derived RANKL has also been shown to be critical for the increased osteoclast formation and bone loss due to a low-calcium diet [47] and estrogen deficiency [48]; thus osteocytes are a critical source of RANKL in a variety of homeostatic and pathologic states [41]. RANKL is produced as a membrane-bound protein on the cell surface that is cleaved at the surface by enzymes (such as matrix metalloproteinase 14) to generate a soluble form. The relative importance of membrane and soluble RANKL was examined using genetically modified mice that produced a form of RANKL that could not be cleaved. The lack of soluble RANKL in adult mice led to increased cancellous bone mass and decreased osteoclast numbers, suggesting that soluble RANKL is in important ongoing osteoclast formation. However, lack of soluble RANKL did not affect bone mass in developing mice or bone loss due to estrogen deficiency suggesting that membrane RANKL is sufficient for osteoclastogenesis under other conditions [49].

Osteocytes secrete key regulator of osteoclasts. Osteoclasts differentiate under the stimulation of MCSF and RANKL. While a number of cell types produce these cytokines, including osteoblasts, stromal cells, and T cells, the cell type responsible for RANKL production important in maintaining bone homeostasis is the osteocyte. Osteocytes are highly differentiated osteoblasts imbedded in the bony matrix. Shown in the bone remodeling unit in which cells are connected to each other and the cell surface through a canalicular network that allows osteocyte cells to interact with cells at the surface of bone. Using mice deficient in RANKL only in osteocytes, it was shown that osteocytes supply RANK ligand for osteoclastogensis in both homeostatic and pathologic conditions such as low-calcium diet and estrogen deficiency
Second Signals: Co-stimulatory Receptors in Osteoclast Differentiation
Similar to other immune cells, osteoclasts require simultaneous stimulation through multiple receptor signals to initiate the cellular differentiation program (Fig. 2.4). While signaling through the RANK receptor is the key specific osteoclastogenic signal, a critical co-stimulatory signal is directed by innate receptors that utilize ITAM (immunoreceptor tyrosine-based activation motif) signaling adapters, DAP12 (DNAX-associated protein 12kD size), and FcRγ (FcεR1γ chain) [50, 51]. The ITAM motif was initially recognized as a common sequence in the cytoplasmic tails of the signaling chains associated with the T cell receptor and B cell receptor but has since been identified in a number of receptor-associated cytoplasmic domains, where it is used to link receptor activation to downstream signaling cascades. The ITAM adapter chains transduce signals from a variety of ligand-binding immunoreceptors on osteoclasts. Signaling through ITAM adapter chains in osteoclast precursors initiates the calcium flux that leads to the activation of NFATc1, the master transcriptional regulator required for osteoclastogenesis [52]. Innate immunoreceptors generally function to activate cells in response to local microenvironmental change, and it is likely that the combined input of a number of coreceptors on osteoclast precursors fine-tunes osteoclast differentiation and functional response. Each of the ITAM signaling chains pairs with specific immunoreceptors, with the best known pairs being TREM2-DAP12 and OSCAR-FcRγ [53]. Ligands that stimulate these receptors in the bone microenvironment are not well defined, though potential ligands include collagen fragments for OSCAR and apoptotic cells for TREM2 53].

RANK signaling interactions. RANK stimulation leads to binding of TRAF6 which forms a central scaffold with Gab2/TAK1/TAB2 and subsequent activation of a number of pathways including NFκB and several MAPK intracellular signaling cascades (JNK1, p38, ERK1, PI3K) and interaction with the immunoreceptor ITAM signaling pathway. In the figure receptors are shown in black, adapter proteins in blue, enzyme intermediates in signaling cascade in blue boxes, and activation of transcription factors in orange boxes, with the master regulator of osteoclastogenesis NFATc1 in the orange box outlined in red. Cooperation with the ITAM signaling pathway is shown on the right, where the interaction provides the intracellular calcium flux needed for NFATc1 translocation. Osteoclast-specific genes downstream from NFATc1 are shown in the white box lower right
Mice deficient in both of the ITAM adapter chains, DAP12, and FcRγ are severely osteopetrotic with no osteoclasts in the long bones [50, 51]. However, these mice are distinct from RANK- or RANKL-deficient mice, in that mice deficient in both DAP12 and FcRγ have teeth, because they can develop osteoclasts in the jaw needed for tooth eruption [51]. Surprisingly, despite the lack of osteoclasts in the long bones under basal conditions, following a bone-remodeling stimulus such as estrogen deficiency, DAP12−/−/ FcRγ−/− mice lose significant amounts of bone and are able to generate osteoclasts in vivo [54]. These studies suggest that the requirement for specific coreceptors can be bypassed under specific microenvironmental conditions, either due to the usage of additional coreceptors or alterations in other regulatory signals.
One additional signal comes through the MCSF receptor (CSF-1R or cFms), a tyrosine kinase-based growth factor receptor that is required for osteoclastogenesis. The identification of a mutation in the coding region of MCSF (also known as CSF-1) in the osteopetrotic op/op mice demonstrated the essential nature of MCSF receptor signals for osteoclast development [55, 56]. MCSF stimulation promotes the proliferation, survival, and differentiation of a number of myeloid cells and is similarly important during osteoclastogenesis. MCSF is produced by osteoblasts, stromal cells, and osteocytes, similar to RANKL. In osteoclasts, MCSF also stimulates cytoskeletal organization, cellular spreading, and migration [56].
Osteoclasts also interact with their surroundings through cell surface receptors, which is important for differentiation of osteoclasts to a polarized, bone degrading cell. Osteoclast-expressed integrins interact with bone matrix through αvβ3 binding to RGD peptides in the extracellular matrix. This interaction polarizes the osteoclast cell and initiates actin ring formation, creating the characteristic ruffled border [57,58,59]. The osteoclast forms an external phagolysosome adherent to the bone at the actin ring which organizes the sealing zone underneath the osteoclast where enzymatic and acidic bone degradation can take place [58,59,60]. Mice deficient in the β3 integrin subunit cannot efficiently organize their cytoskeleton for resorption and have an osteopetrotic phenotype with hypocalcemia [61]. Matrix interaction with the αvβ3 integrin induces phosphorylation of DAP12 and formation of an ITAM/Syk/Src/αvβ3 signaling complex [57, 62]. The importance of these interactions for osteoclast function is seen in β3/DAP12 double-deficient (DAP12−/−β3−/−) mice that are profoundly osteopetrotic, reflecting a severe degree of osteoclast dysfunction, which is not seen in mice lacking either αvβ3 or DAP12 alone [63]. These examples suggest that multiple receptor inputs are also required to fully activate osteoclast adherence and functional bone resorption, mimicking the need for the multiple co-stimulatory signals required in the early stages of osteoclastogenesis. Functional activation of osteoclasts is therefore a final step in the process of specialized cellular differentiation to form mature terminally differentiated osteoclasts.
Fusion and the Formation of Multinucleated Osteoclasts
One of the most unique and distinctive properties of osteoclasts is multinucleation. Multinucleation has typically thought to be a requirement for resorptive activity in higher vertebrates, although some fish species have mononuclear osteoclasts. As early as the 1980s, it was appreciated that this multinucleation occurred through fusion of mononuclear cells rather than by endoreplication [14, 64]. The precise mechanism by which homotypic membrane fusion of osteoclast precursors occurs is not known, though a number of proteins important for osteoclast fusion have been identified.
Three cell surface molecules induced by RANKL are strongly implicated in osteoclast fusion. These molecules are the multi-pass transmembrane proteins known as DC-STAMP and OC-STAMP (dendritic cell- and osteoclast- specific transmembrane protein) and ATP6V0d2 (ATPase, H+ transporting, lysosomal 38 kDa, V0 subunit d2). DC-STAMP and OC-STAMP are required for multinucleation of osteoclasts [65,66,67,68,69]. Only one cell in a cell-cell fusion needs to express STAMPs, as wild-type monocytes can fuse with STAMP-deficient monocytes. Loss of DC-STAMP results in mononuclear osteoclasts and also defective resorptive function, leading to increased trabecular bone [68]. Loss of OC-STAMP also results in mononuclear osteoclasts with diminished resorptive activity in vitro, but OC-STAMP-deficient mononuclear osteoclasts appear to function adequately in vivo as the mice have no bone phenotype [67, 70]. ATP6V0d2, a subunit of the V-ATPase complex essential for extracellular acidification, is also essential for osteoclast fusion. As with DC-STAMP, Atp6v0d2−/− mice have increased bone mass [71].
A number of additional molecules have been implicated as regulators of osteoclast fusion, though none are essential for fusion, and mice lacking these molecules have modest or no bone phenotype. These molecules include CD47 and SIRPα (signal regulatory protein alpha), tetraspanins, CD44, and ADAM8 (a disintegrin and metalloprotease 8) [72]. Although identification of molecules involved in fusion of mononuclear precursors to a mature, multinucleated osteoclast has provided insight into the requirements for fusion, we have little mechanistic insight into the fusion process, and much remains to be learned.
Downstream Events: Signaling Cascades and Transcriptional Activation
Osteoclastogenesis requires the activation of a number of transcription factors to induce the transcriptional program that defines the osteoclast, including expression of TRAP, integrin β3, cathepsin K, matrix metalloprotease 9, and calcitonin receptor [73]. RANKL stimulation leads to the upregulation and activation of NFATc1, the master regulator of osteoclast differentiation [22], through activation of a number of signaling pathways, including the canonical NF-κB and AP-1 pathways and facilitation of calcium signaling by ITAM-associated receptors. The complexity of RANK-induced signaling is outlined in Fig. 2.4. While significant advances have been detailed by numerous studies, these complex interactions remain incompletely understood, and new key signaling factors are still being described [73]. The delineation of intracellular signaling during osteoclastogenesis has been a topic of considerable interest given that identification of critical signaling intermediates may suggest new therapeutic targets to block bone loss and will also further our understanding of how these pathways are dysregulated by medications or pathologic or inflammatory disease states.
Signaling Cascades in Osteoclast Differentiation
RANKL interaction with the RANK receptor initiates a signaling cascade beginning with the binding of the adapter molecule TRAF6, which forms scaffolds that lead to activation of JNK, p38, and NF-κB [73, 74] (Fig. 2.4). While there are multiple TRAF adapters, the key role for TRAF6 in osteoclastogenesis was shown when the TRAF6-deficient mouse was found to develop severe osteopetrosis with impaired osteoclast differentiation and bone resorption [75].
RANK/TRAF6 signaling recruits IKK-α (IκB kinase alpha) and IKKβ- (IκB kinase beta), also known as IKK1 and 2, an upstream enzyme complex in the NF-κB signaling cascade. The α- and β-subunits together are catalytically active as a serine-threonine kinase and are modified by IKK3/IKK-β or NEMO (NF-κB essential modifier of NF-κB kinase), a subunit of the IKK complex that serves a regulatory function. Activation of the IKK complex leads to binding of NEMO to IKK-α and IKK-β with subsequent serine phosphorylation of IκB, which binds NF-κB and retains it in the cytoplasm [76]. Phosphorylation of IκB leads to its ubiquitination and degradation by the proteasome, releasing NF-κB and allowing its translocation to the nucleus where it initiates gene transcription. Mice lacking NF-κB subunits develop osteopetrosis due to a severe defect in osteoclast differentiation [77]. In the NF-κB-null mice, development of macrophages and osteoclast precursors is preserved, suggesting that NF-κB is not essential during early osteoclast differentiation [78, 79]. Gene targeting studies have demonstrated that different transcription factors are required at different stages of osteoclast differentiation and therefore differentially affect other myeloid lineages (Fig. 2.2) [73].
TRAF6 also links RANK to multiple MAPK (mitogen-activated protein kinase) pathways: ERK, JNK and p38, through formation of complexes with TAK1 (TGF-β-activated kinase), TAB1 and TAB2 (TAK-1-binding proteins 1 and 2) [73]. Ablation of TAK1 in myeloid cells results in defective osteoclastogenesis and development of osteopetrosis in mice [80]. Interestingly, TAK1 deficiency alters signaling through NF-κB, p38 MAPK, and Smad1/5/8 and has been shown to alter expression of multiple transcription factors, including PU.1, MITF, c-Fos, and NFATc1, suggesting that TAK1 acts as a regulator at multiple points during osteoclast differentiation [80].
RANK stimulation of MAPK activation leads to activation of downstream targets of ERK, JNK, and p38 in osteoclast precursors, which include c-Fos, AP-1 transcription factors, and MITF, respectively [73]. AP-1 (activator protein-1), which is composed of a protein complex of Fos (c-Fos, FosB, Fra-1 and Fra-2) and Jun (c-Jun, JunB, and JunD) proteins, is critical during osteoclastogenesis, because genetic deletion of c-Fos also abrogates osteoclastogenesis resulting in osteopetrosis [81]. Interestingly, cFos-deficient animals have increased macrophages; thus AP-1 regulation of osteoclast and macrophage differentiation is in opposing directions [81]. Transgenic mice expressing dominant negative c-Jun in the osteoclast lineage also demonstrate severe osteopetrosis with defective osteoclastogenesis [81]. The role of p38 MAPK is more complex as, although p38-deficient cells have defective osteoclastogenesis and p38 MAPK inhibitors can inhibit in vitro osteoclastogenesis, p38 MAPK deficiency in monocytes led to only a minor increase in bone mass in young animals, while older animals developed osteoporosis and an increase in osteoclastogenesis. The absence of p38 led to increased monocyte proliferation and increased size of the osteoclast progenitor pool in the aged mice, demonstrating a complex role for p38 that varies with age [82, 83]. ERK1 positively regulates osteoclast development and bone resorption, and genetic deletion of ERK1 in hematopoietic cells resulted in reduced osteoclast progenitor cell number, decreased osteoclast function with defective pit formation, and diminished MCSF-mediated adhesion and migration [84].
RANK also activates the PI3K (phosphoinositide 3-kinase)/AKT pathway. PI3K activation leads to the production of phosphatidylinositol-(3,4,5)-phosphate (PIP3) at the plasma membrane, where it recruits AKT. The critical nature of PI3K/AKT for osteoclasts was demonstrated by deletion of the p85 regulatory subunit of the Class IA PI3K, which results in an osteopetrotic phenotype caused by a defect in osteoclast resorption of bone. Class IA PI3K was found to be required to initiate ruffled border formation and vesicular transport, but not for the formation of the sealing zone [85]. p85α/β doubly deficient osteoclasts showed defective AKT activation and loss of resorption, which could be recovered by expression of activated AKT. Simultaneous blockage of both AKT and MEK1/2 causes rapid apoptosis of nearly all osteoclasts, which suggests a role for PI3K in osteoclast survival. In keeping with this finding, PI3K inhibitors can also lead to rapid osteoclast apoptosis [86].
Activation of PI3K/AKT by RANK is modulated by Src kinase activity, thus integrating RANK and ITAM-associated receptor signaling. This collaborative activation of PI3K/AKT is demonstrated by the loss of RANKL-mediated AKT activation in cells genetically deficient for c-Src. PI3K is also activated downstream of αvβ3 integrin and CSF-1 receptor, which may be of importance in regulation of osteoclast function [73]. AKT activation requires PIP3 production, which is negatively regulated by PTEN (phosphatase and tensin homolog) and SHIP1 (SH2-containing inositol phosphatase 1). As would be predicted, both PTEN and SHIP1 negatively regulate osteoclast differentiation, with deficiency of either SHIP1 or PTEN, leading to increased osteoclastogenesis and severe osteoporosis in mice [87, 88].
Transcription Factors in Osteoclast Differentiation
The transcription factor PU.1, an ETS-domain transcription factor, is expressed at all stages of osteoclast differentiation but plays a critical role early in osteoclastogenesis and is essential for development of all myeloid lineage cells. In osteoclast precursors, PU.1 regulates expression of the CSF-1 receptor and RANK which are required for osteoclastogenesis. Consistent with this, PU.1 deletion in mice causes osteopetrosis and lack of both osteoclasts and macrophages [89, 90]. PU.1 also cooperatively regulates gene transcription with other key osteoclastogenic transcription factors MITF and NFATc1 and thus plays a role in later osteoclast differentiation as well [89]. MITF plays a later role in osteoclast differentiation, around the time of precursor cell fusion to multinucleated cells. Mutations in MITF lead to osteopetrosis with formation of only mononuclear osteoclasts that are defective in bone resorption with a lack of ruffled border formation on bone [91,92,93,94].
NFATc1 was termed the master switch for regulating the terminal differentiation of osteoclasts because ectopic expression of NFATc1 in precursor cells led to efficient differentiation to osteoclasts in the absence of RANKL signaling [22]. NFATc1-deficient embryonic stem cells also failed to differentiate into OCs in response to RANKL stimulation; thus the expression of NFATc1 was both necessary and sufficient to drive osteoclastogenesis [22]. NFAT transcription factors are regulated primarily by intracellular calcium signaling. Signals through the ITAM adapters initiate calcium signaling that is required in the basal state to drive osteoclastogenesis and NFATc1 activation [50, 51]. In osteoclast precursors, stimulation of ITAM-associated receptors leads to phosphorylation of the tyrosine residues in the ITAM motif through the action of Src family kinases. The activated ITAM motif then recruits the tyrosine kinase Syk which initiates a signaling cascade involving the intermediates BTK/Tec, BLNK (B cell linker)/SLP76 and phospholipase C-γ2 [51, 52, 95. PLCγ2 is activated through phosphorylation which increases its catalytic function to hydrolyze phosphatidylinositol-4,5 bisphosphate into inositol-1,4,5-triphosphate (IP3) and diacylglycerol. IP3 then activates receptors on the endoplasmic reticulum to stimulate Ca2+ release from the endoplasmic reticulum to the cytoplasm [52, 96]. The increase in cytoplasmic Ca2+ activates calcineurin, a cytoplasmic phosphatase that dephosphorylates NFATc1, allowing it to translocate to the nucleus to initiate and regulate gene transcription. Consistent with this, calcineurin inhibitors such as FK506 and cyclosporin A strongly inhibit osteoclastogenesis [52]. NFATc1 also autoamplifies its own gene, possibly by binding to its own promoter, and associates with AP-1 to initiate gene transcription of essential osteoclast genes such as TRAP, calcitonin receptor, cathepsin K, and β3 integrin [97].
The transcription factor c-MYC is strongly upregulated by RANKL stimulation and promotes osteoclastogenesis in vitro. Recent studies examining the role of MYC have highlighted the role of cellular metabolism in osteoclastogenesis [98]. MYC has been shown to function to drive metabolic reprogramming during osteoclast differentiation, and switching cellular metabolism to an oxidative state enhances both osteoclastogenesis and function [98]. Osteoclasts contain abundant mitochondria and undergo metabolic adaptation during the course of differentiation to meet the bioenergetic demands required for functional resorption of bone. PGC-1β (PPARγ coactivator-1β) is induced during osteoclast differentiation by CREB via reactive oxygen species (ROS) and also stimulates mitochondrial biogenesis [99]. During this switch MYC induces estrogen receptor-related receptor α (ERRα), a nuclear receptor that cooperates NFATc1 to drive osteoclastogenesis [98]. While a complex array of transcriptional activators must be engaged through RANK/RANKL stimulation to drive osteoclast differentiation, an important additional function of RANK stimulation on osteoclast precursors is to downregulate expression of transcriptional repressors to enable osteoclastogenesis to take place [100].
Negative Regulators of Osteoclast Differentiation
A host of negative regulatory mechanisms exist to ensure that osteoclasts are generated only in the correct time and place. Downregulation of transcriptional repressors during RANK stimulation is required for osteoclastogenesis to proceed. Repressors of gene transcription that are downregulated during osteoclastogenesis include Ids (inhibitors of differentiation/DNA binding), Eos, MafB (v-maf musculoaponeurotic fibrosarcoma oncogene family protein B), C/EBPβ (CCAAT-enhancer-binding protein β), IRF-8 (interferon regulatory factor 8), and Bcl-6 (B cell lymphoma 6) [100, 101]. The negative regulatory transcription factors also inhibit osteoclastogenesis at specific points during differentatiation; Ids, IRF-8, and MafB are inhbitiory during early osteoclastogenesis (within 24 h after RANKL stimulation), while Eos and Bcl6 expression are inhibitory at later time points during osteoclast development.
MafB expression is downregulated following RANKL stimulation during osteoclastogenesis and MafB has since been shown to negatively regulate osteoclast formation. MafB is a basic leucine zipper transcription factor that plays an important role in the regulation of lineage-specific hematopoiesis, and overexpression of MafB inhibits the formation of TRAP+ multinuclear osteoclasts. In osteoclasts, MafB abrogates NFATc1 expression and interferes with the DNA binding of cFos, Mitf, and NFATc1 transcription factors [54].
Similarly, RANKL stimulation downregulates the Ids helix-loop-helix (HLH) transcription factors encoded by the Id1, Id2, and Id3 genes. Overexpression of the three Id genes negatively affects osteoclast differentiation [102] Overexpression of Eos also leads to defective osteoclast differentiation, with selective repression of transcription of MITF/PU.1 targets such as Ctsk (encoding cathepsin K) and Acp5 (encoding TRAP) [103] Eos forms a complex with MITF and PU.1 at their target gene promoters and suppresses transcription through recruitment of corepressors. In myeloid progenitors prior to the initiation of osteoclast differentiation, Eos directly interacts with MITF and PU.1 to suppress transcription. Later in osteoclast differentiation, Eos association, for example, at Ctsk and Acp5 promoters, decreases significantly allowing transcription to proceed.
IRF-8 is a transcription factor critical for lineage commitment in the maturation of myeloid precursors [104]. IRF-8 is expressed in macrophages derived from bone marrow and spleen, and downregulation of IRF8 is required for these cells to initiate osteoclastogenesis. IRF-8 suppresses osteoclastogenesis by inhibiting NFATc1 expression and physically interacts with NFATc1 to inhibit its function [105].
The downregulation of these negative regulators of osteoclastogenesis is in fact controlled by RANK stimulation. RANKL induces expression of Blimp1 (B lymphocyte-induced maturation protein-1) via NFATc1 during osteoclastogenesis. Blimp1 functions as a transcriptional repressor of anti-osteoclastogenic regulators such as IRF-8, MafB, and Bcl6. Overexpression of Blimp1 leads to an increase in osteoclast formation, while deficiency of Blimp1 leads to defective osteoclast differentiation. Thus, while Blimp1 is a positive regulator of osteoclastogenesis in itself, its primary function is to suppress the transcription of negative regulators. Mice with an osteoclast-specific deficiency of Blimp1 exhibit a high bone mass phenotype caused by a decreased number of osteoclasts [101]. In the absence of Blimpl1, osteoclastogenesis is impaired through increase of Irf8 and MafB and by upregulating Bcl6. Bcl6 suppresses expression of osteoclastic genes downstream of NFATc1 which includes cathepsin K, dendritic cell-specific transmembrane protein (DC-STAMP), and NFATc1 itself [106]. RANKL also induces the IFN-β (interferon-beta) gene in osteoclast precursor cells. In a negative regulatory feedback loop, IFN-β then functions to limit osteoclastogenesis by interfering with the RANKL-induced expression of c-Fos [107].
Signaling during osteoclastogenesis is also regulated by ubiquitination of specific substrates. RANK regulates the de-ubiquitinase CYLD, which inactivates TRAF6 by removal of polyubiquitin chains, resulting in inhibition of osteoclast formation. CYLD deficiency leads to severe osteoporosis and osteoclasts that are hyper-responsive to RANK stimulation [108]. NUMB/NUMB-like (NUMBL) is an intracellular adapter protein that directly interacts with TRAF6 and NEMO and induces their ubiquitination and proteasomal degradation. NUMBL has been shown to be downregulated by RANKL stimulation, and its presence inhibits osteoclast differentiation and function [109]. Downstream of RANKL, TAK1 is also important in inhibiting expression of NUMBL because the TAK1-deficient mouse showed increased NUMBL expression. The TAK1/TAB2 complex mediates the polyubiquitination of NUMBL which marks it for proteasomal degradation [80]. NUMBL has also been shown to regulate NOTCH signaling and with increased NUMBL expression in myeloid cells, there is increased degradation of NICD and subsequent accumulation of RBPJ. In other studies RBPJ has been shown to be a significant inhibitor of osteoclast differentiation [110]. Thus, NUMBL acts as an endogenous negative regulator of NF-κB signaling in osteoclasts by targeting the TAK1/TRAF6/NEMO complex which leads to indirect negative regulation of RBPJ [109]. RBPJ negatively regulates osteoclastogenesis induced by both RANKL and TNF and may be of particular importance in inflammatory bone loss. RBPJ inhibits activation of PLCγ2 downstream from the ITAM-associated receptors and has been demonstrated to function as a negative regulator of osteoclastogenesis by suppressing induction of NFATc1, BLIMP1, and c-Fos [110].
As evident from the discussion above, osteoclast formation is a highly regulated process, requiring MCSF stimulation of CSF-1R for precursor survival, RANKL-RANK pathway stimulation and a second signal through and an ITAM-associated immunoreceptor, with further modulation by negative regulatory pathways. The ability to differentially regulate the combination and balance of these signals, as well as the potential for site and/or condition-specific ligand expression for ITAM-associated receptors, results in a highly tunable program of osteoclastogenesis. This likely allows for location- and environment-specific regulation of osteoclast formation and function.
Functions of Osteoclasts
Bone resorption is the canonical function of osteoclasts and they are the only cells capable of resorbing bone. Their resorptive function is essential for the formation of the bone marrow cavity during skeletogenesis, and they actively remodel bone throughout life, resorbing approximately 10% of skeletal bone annually by some estimates. However, it has increasingly been appreciated that osteoclasts are more than just bone resorbing cells. Osteoclasts are able to regulate other biological processes through the production of cytokines and heterocellular signaling [111]. Moreover, several lines of evidence support the idea that communication between osteoclasts and osteoblasts, referred to as coupling, is bidirectional, with osteoclasts actively promoting osteoblast function [112]. Thus, one can divide osteoclast functions into canonical bone resorptive/remodeling functions and what might be termed “regulatory functions,” consisting of regulation of bone formation through coupling, autocrine regulatory pathways, and angiogenesis [113].
Bone Resorbing Function
Within bone, osteoclasts reside on the periosteal and trabecular surfaces and in Haversian canals. Osteoclasts are highly motile cells, migrating along the bone surface to resorb bone at multiple sites. The mature differentiated osteoclast, after reabsorbing bone in a specific area, is able to adopt a migratory state to move to a new site of resorption. The migratory osteoclast has a lamellipodic front to back “horizontal” migratory polarity with the majority of the cytoplasm at the leading edge. When it reaches a new resorption site, attracted by cytokines released by osteoblasts, the osteoclast changes its morphology to a static conformation that facilitates bone reabsorption [114, 115]. The hallmark of a resorbing osteoclast is the reorganization of the cytoskeleton to form a “vertical” polarized cell. The cytosol is reorganized, with the new position of the organelles inside the cells reflecting the different activity of the opposing surfaces of the osteoclast. The nuclei, Golgi apparatus, and the rough endoplasmic reticulum are on the basolateral side of the cell, in contact with the microvasculature. The lysosomes together with mitochondria and components of the endocytic compartment move close to the apical side of the cell, juxtaposed to bone. This polarization reflects the different activities that occur at the two cell surfaces, with apical surface producing degradative enzymes to deliver into the reabsorption lacunae, whereas the basolateral surface is in charge of “packing” the products of reabsorption and delivering them into the main circulation (Fig. 2.5) [113, 116, 117].

Functional polarization in the osteoclast. Resorbing osteoclasts are highly polarized, with the resorption machinery located on the apical surface (highlighted in green) adjacent to the bone surface, while the basolateral membrane (highlighted in blue) transports resorbed molecules to the adjacent circulation. The cytoskeleton is reorganized to form a specialized actin structure, the podosome. The acid and hydrolases required for resorption are isolated from surrounding bone and cells by the formation of a tightly bone adherent sealing zone. The apical surface of the osteoclast has a highly invaginated membrane or ruffled border that greatly expands the membrane surface available for transport. The V-H-ATPase required for acidification and ion channels required to maintain intracellular electroneutrality are located in the ruffled border. Degradative enzymes, including cathepsin K (CTS K), TRAP, and matrix metalloproteinases (MMPs), are exocytosed into the resorption lacuna via fusion of lysosomal membrane (highlighed in red) with the ruffled border
The functional domains of the active reabsorbing osteoclast can be divided into the sealing zone, the membrane-rich ruffled border, the functional secretory domain, and the basolateral domain. The sealing zone has the key function of mediating the attachment of the osteoclasts to the underlying bone matrix, forming a distinctive and isolated “pouch” called the Howship or resorption lacuna, into which the osteoclast pumps protons and degradative enzymes to digest the bone matrix. The extremely tight connection between the apical surface and bone is mediated by a structure called the podosome ring. Podosomes are highly specialized adhesions that consist of actin microfilaments and integrins, together with several other regulatory proteins. Individual podosomes cluster into groups and then migrate to encircle the outside circumference of the sealing zone forming the so-called podosome belt. The formation of the podosome belt is the hallmark of a sealed attachment of the osteoclast to the bone [113, 115, 118, 119].
The membrane-rich ruffled border is centrally positioned relative to the sealing zone and is composed of an irregular array of membrane expansions. The ruffled border is divided into functional subdomains: the outer “secretive zone” and the inner “reuptake zone.” The secretive zone is characterized by secretion of lysosomal enzymes into the resorption lacuna and the presence of ion transporters to discharge protons resulting in acidification of the lacuna. The reuptake zone is specialized for the reuptake of the calcium, phosphorus, and other bone components digested by the released enzymes. The functional secretory domain of the basolateral surface of the cell is connected with the microvasculature and is important for the passage of the reabsorption products into the general circulation [113, 115]. The functional secretory domain is anatomically connected with the sealing zone by what is referred to as the basolateral domain [115].
This complex reorganization creates a lacunar “pouch” between the osteoclast and the underlying bone that is isolated from the surrounding environment. Now the osteoclast can safely secrete protons, driven by V-H-ATPases, to acidify the lacuna and dissolve the inorganic hydroxyapatite component of the bone. Removal of mineral unmasks the organic component of the bone, mostly composed of type 1 collagen. The organic matrix is then digested by a number of hydrolases secreted into lacuna via lysosomal exocytosis. Acidification of the lacuna not only unmasks the organic component of the bone but also creates the acidic environment needed for optimal hydrolase activity, as lysosomal enzymes perform best between pH 4.0 and 5.0 [113].
The critical nature of the highly specialized resorptive apparatus described above is revealed by the causal loss-of-function mutations described for several human bone diseases. Many genetic diseases involving the osteoclasts are characterized by abnormal function of the ruffled border, as detailed in the section “Genetic Diseases of Osteoclast Dysfunction.”
Regulatory Functions
Osteoclasts are reported to secrete cytokines that act in an autocrine fashion to either promote or inhibit osteoclastogenesis. The IL-6 family member cardiotrophin-1 (CT-1) is produced by and stimulates osteoclast formation and has also been hypothesized to have paracrine effects promoting osteoblast formation [120]. Stimulation of osteoclasts with autoantibodies to citrullinated peptides that are found in rheumatoid arthritis was reported to induce an autocrine loop of IL-8-stimulated osteoclastogenesis [121]. On the other hand, osteoclast progenitors have been reported to express OPG, which would inhibit RANKL-stimulated osteoclast formation [122] Paracrine functions of osteoclasts include secretion of platelet-derived growth factor-BB (PDGF-BB) by osteoclast progenitors. PDGF-BB induces Type H capillary formation in bone; thus one paracrine action of osteoclasts appears to be promoting the coupling of angiogenesis and osteogenesis mediated by Type H capillaries [123].
The term “clastokine” has been coined and is often used to describe factors secreted by osteoclasts with putative paracrine actions on osteoblasts. While osteoblasts are widely accepted to modulate osteoclast formation and function through expression of RANKL and OPG, osteoclasts traditionally have been thought to contribute to osteoblast regulation through liberation of previously trapped cytokines from the bone matrix. More recently the concept of osteoclast-secreted “clastokines” has emerged. Clastokines are hypothesized to attract and facilitate the maturation of pre-osteoblast cells into mature bone-forming osteoblasts. A number of putative clastokines, including collagen triple repeat containing 1 (CTHCR1), sphingosine-1-phosphate (S1P), and complement factor 3a (C3a), have been described in vitro though relative in vivo significance of these putative clastokines is not entirely clear (reviewed in [111, 112]), and the osteoclast-specific expression of CTHRC1 in bone has been challenged [124]. The axon guidance molecule SLIT3 was recently proposed to act as a clastokine [125], though similar to CTHRC1, the source and cellular target of SLIT3 in bone are a source of debate. Loss of SLIT3 results in decreased bone mass, though whether this is via osteoclast-derived SLIT3 actions to promote osteoblast proliferation and migration via activation of the beta-catenin pathway [125] or via osteoblast production of SLIT3 promoting the development of the Type H vascular endothelium in bone [126] is not settled. The concept, however, is particularly exciting as it suggests the possible existence of novel mechanisms to stimulate bone anabolic activity which might prove attractive therapeutic targets.
Osteoclast regulation of osteoblast lineage cells can also occur via cell contact-mediated mechanisms. Semaphorin 4D expressed by osteoclasts inhibits osteoblast migration by binding to Plexin-B1, its cognate receptor on osteoblasts. In contrast, Ephrin B2 expressed on osteoclasts stimulates bone formation through binding EphB4, its receptor on osteoblasts [127, 128] Recently, osteoclast RANK stimulation of reverse signaling through RANKL on osteoblasts was proposed as a key mechanism coupling bone resorption and formation. Although reverse signaling could be stimulated through a cell contact-mediated mechanism, RANK was shown to be released from osteoclasts in small extracellular vesicles and thus likely acts in a paracrine fashion [129]. In summary, osteoclast regulatory functions have broad impact on the local bone microenvironment, influencing bone resorption, formation, and angiogenesis. However, future research is needed to clarify the roles of many of the aforementioned coupling factors in human bone remodeling.
Genetic Diseases of Osteoclast Dysfunction
The identification of causative mutations underlying monogenic traits responsible for bone syndromes has added greatly to our understanding of osteoclast biology. Recognizing the gene/protein impaired in a specific rare bone disease had revealed the role of several proteins involved in the maturation and/or reabsorption machinery of the osteoclasts. Table 2.2
provides a list of genes and corresponding diseases. The genetic diseases primarily involving the osteoclast can be divided into two broad categories: those with normal or increased osteoclast function and those with decreased osteoclast function.
The spectrum of mutations in TNFRSF11A (RANK) are emblematic of these categories. Loss-of-function mutations in TNFRSF11A are responsible for osteopetrosis type VII, in which osteoclast differentiation and thus bone resorption are impaired and therefore active [130]. In contrast, gain-of-function mutations in TNFRSF11A are responsible for two diseases, expansile skeletal hyperphosphatasia and familial expansile osteolysis; these conditions are characterized by increased osteoclast activity which in turn leads to an excessive immature and disorganized bone formation [131]. A disease characterized by focal lesions with increased osteoclast activity is Paget’s disease of bone (PDB), hereditary forms of which have been linked to heterozygous loss-of-function mutations of genes important for osteoclast maturation, SQSTM1 and VCP [132, 133]. SQSTM1 encodes sequestosome 1, a scaffolding protein important for RANK signaling. A SQSTM1 mutation commonly associated with PDB has been shown to impair association with the TRAF6 deubiquitnase CYLD described above, resulting in increased poly-ubiquitinated TRAF6, RANK signaling, and osteoclastogenesis [134].
Monogenic diseases with decreased osteoclast function present with a phenotype of osteopetrosis, or “stone bone,” with dramatically increased bone density and loss of bone marrow cavity. Osteopetroses are divided in three categories based on the mechanism of transmission: autosomal-dominant osteopetrosis (ADO), autosomal-recessive osteopetrosis (ARO), and X-linked osteopetrosis. ADOs are usually more benign and occur in adulthood or in some cases represent an incidental finding in radiographic exams, whereas AROs result in severe skeletal involvement, are diagnosed in early childhood, and result in more morbidity. ADOs and AROs can develop from a heterozygous or homozygous mutation of the same gene; it is the involvement of one or both alleles that determine the severity of the disease [135, 136]. This is the case for mutations in the chloride channel CLCN7; heterozygous mutations in CLCN7 cause ADO type II or Albers-Schonberg disease, whereas a homozygous mutation or a composite heterozygous mutation is responsible for ARO type IV [137, 138]. The only known X-linked osteopetrotic syndrome involves the gene IKBKG necessary for translocation of the transcription factor NF-κB into the nucleus [139].
A more biologically based approach to classifying osteopetrosis is considering whether osteoclasts do not form (osteoclast-poor osteopetrosis) or form but do not function (osteoclast-rich osteopetrosis). In the category of osteoclast-poor osteopetrosis are mutations involving TNFRSF11A (RANK) causing ARO type VII [130] and X-linked IKBKG mutations (anhidrotic ectodermal dysplasia, lymphedema, and immunodeficiency), underscoring the importance of the NF-κB pathway (described in the section “Osteoclast Differentiation”) downstream of RANKL-RANK signaling in the development of the mature osteoclast [139]. In this category, we can also include mutations in genes expressed primarily by osteoblasts which indirectly alter osteoclast maturation and differentiation. These include mutations in TNFSF11 encoding RANKL and TNFRSF11B encoding OPG, resulting in ARO type II and juvenile Paget’s disease, respectively [140, 141].
Osteoclast-rich osteopetrosis can be subdivided into diseases with defects in cytoplasmic proteins, podosome formation, or lysosomal defects caused either by mutations in lysosomal proteins or defects in ruffled border maturation. The only identified defect in a cytoplasmic protein described to date is mutation in the gene encoding carbonic anhydrase type II, the enzyme necessary to maintain intracellular neutrality during acidification of the lacuna. Patients with this mutation not only manifest osteopetrosis but also renal tubular acidosis since the same isoform is present in tubular cells [142]. Mutation of KIND3, which encodes KINDLIN-3, impairs podosome assembly into the sealing zone and results in leucocyte adhesion deficiency with osteopetrosis [143].
Mutations in lysosomal proteases can cause bone disease even if the ruffled border formation is normal. Mutations of the gene encoding the abundant osteoclast protease cathepsin k results in pycnodysostosis, characterized by short stature with increased bone density [144]. Mutation of other lysosomal proteases cause bone disease, though not osteopetrosis. Patients with mutations in metalloproteinases 9 and 13 and tartrate-resistant acid phosphatase (TRAP) have been described and cause recessive and dominant metaphyseal dysplasia and spondyloenchondrodysplasia, respectively [145]. Mutations that impair fusion of lysosomes with the ruffled border comprise the largest subtype of osteopetrosis and are characterized by the inability of the abnormal ruffled border to acidify and secrete enzymes into the resorption lacuna. Mutations in TCIRG1 cause ARO type I, the most common of the ARO types with more than 50% of ARO patients carrying a mutation in TCIRG1. TCIRG1 encodes the subunit a3 of the V-ATPase complex, which is necessary to localize the V-ATPase complex in the ruffled border. Absence of this subunit impairs acidification of the resorption lacuna. ARO IV, also relatively frequent, is characterized by mutations in CLCN7 , which encodes a Chloride-hydrogen antiporter in the lysosomal membrane. Mutations in OSTM1, encoding for a protein that binds CLCN7, is responsible for ARO type V. Mutations in PLEKHM1, which encodes a protein that interacts with the vesicular trafficking protein RAB-7, and mutations in SNX10, which encodes a protein involved in intracellular endosomal trafficking, cause much rarer types of ARO [146, 147] These monogenic diseases affecting osteoclast function not only illuminate aspects of osteoclast biology, they provide indisputable evidence for the essential function of osteoclasts in skeletal biology.
Regulation of Osteoclasts by their Environment
A variety of perturbations in the physiologic state can promote osteoclastogenesis and bone loss, with perhaps the best characterized being inflammatory states such as rheumatoid arthritis. Inflammation promotes osteoclast formation at many levels, including through increasing the abundance of osteoclast progenitors. In mice, an increase in bone marrow osteoclast progenitors and in differentiation of circulating monocytes to osteoclasts is enhanced in inflammatory arthritis [16, 148,149,150] Circulating monocytes from patients with inflammatory arthritis, including rheumatoid arthritis, psoriatic arthritis, and ankylosing spondylitis, generate more osteoclasts in vitro cultures, suggesting an increase in circulating osteoclast progenitors within the monocyte pool [151,152,153,154]. Thus, the enhanced osteoclast activity seen in inflammatory arthritis may result in part from an increase in or skewing of progenitors toward an osteoclast fate.
The local microenvironment further regulates osteoclast differentiation through the relative expression of RANKL and OPG. The degree of osteoclast differentiation is likely further tuned by differential expression of the ligands that activate the ITAM-associated receptors essential for osteoclast differentiation. Superimposed on this is an additional layer of regulation by cytokines, which can affect osteoclast differentiation and activity both directly and indirectly by enhanced RANKL expression.
A number of inflammatory cytokines promote osteoclastogenesis, with TNF being the canonical example. TNF-induced bone resorption is implicated in both post-menopausal bone loss and the formation of bone erosions and generalized osteopenia of inflammatory arthritis. TNF acts directly to promote osteoclastogenesis by inducing expression of RANK on progenitors and through TNF receptor-mediated NF-κB activation [155, 156]. A large body of evidence has demonstrated that TNF has a critical role in pathologic osteoclastogenesis through promoting RANKL expression by osteoblasts, osteocytes, and synovial cells in inflammatory arthritis [157]. In mouse models, TNF contributes significantly to estrogen deficiency-induced bone loss, as demonstrated by the effectiveness of TNF inhibitor treatment or genetic deficiency in TNF or TNF receptor p55-deficient mice in preventing ovariectomy-induced bone loss [158, 159].
Other inflammatory cytokines that promote osteoclastogenesis include IL-1 and IL-6. Similar to TNF, IL-1 both directly stimulates osteoclast progenitors and increases expression of RANKL by the environment [160]. IL-6 and IL-6 family members promote osteoclastogenesis indirectly via enhancing RANKL expression, and also appear to have direct effects on progenitors, though whether IL-6 stimulates or inhibits differentiation is controversial [161]. IL-6 is thought to be increased by estrogen deficiency and is a putative mediator of post-menopausal bone loss. However, blocking IL-6 did not prevent bone loss in a mouse ovariectomy model, and there are conflicting reports on the effect of IL-6 deficiency on ovariectomy-induced bone loss [158, 162, 163]. Other cytokines, particularly Th2 cytokines, inhibit osteoclast differentiation either by promoting OPG expression or by direct actions on osteoclasts. These cytokines include IL-4, IL-13, IL-33, and IL-10 [164,165,166,167]. See also Table 2.1 for list of the effect of various cytokines on RANKL and OPG.
Estrogen deficiency may also promote bone loss through expansion of a T cell subset termed Th17 for their production of IL-17. Two mechanisms explain the pro-osteoclastogenic effect of T cells: IL-17 induces RANKL expression and Th17 cells themselves express RANKL. IL-17 also induces TNF and IL-1, further promoting a pro-osteoclastogenic environment [168]. Although Th17 cells function as an osteoclastogenic helper cell T cell subset, other T cell subsets are inhibitory. Activated Th1 CD4+ T cells produce IFNγ, a potent inhibitor of osteoclastogenesis [169]. Regulatory T cells directly inhibit osteoclast differentiation through their expression of CTLA-4. CTLA-4 on regulatory T cells interacting with CD80 and CD86 on osteoclast precursors induces reverse signaling in the myeloid cells to induce IDO (indolamine oxidase) which inhibits osteoclastogenesis [170,171,172]
Many stimuli discussed here play multiple roles in the immune system and on myeloid cell development. Therefore, the impact of any of the individual regulatory pathways on osteoclastogenesis likely depends mostly on the homeostatic or pathologic state in which they are deployed. Similar to other immune cells, osteoclasts are differentiated and activated in response to their environment and in pathologic or inflammatory disease, and the effect of specific positive and negative regulatory stimuli differs depending on the situation. While the focus of our discussion of regulation of osteoclasts has centered on differentiation, osteoclast survival and function are also of importance and likely have additional regulatory elements that center on the fine-tuning of the bone degradation response.
References
Amarasekara DS, et al. Regulation of osteoclast differentiation by cytokine networks. Immune Netw. 2018;18:e8.
Raggatt LJ, Partridge NC. Cellular and molecular mechanisms of bone remodeling. J Biol Chem. 2010;285:25103–8.
Hauschka PV, Mavrakos AE, Iafrati MD, Doleman SE, Klagsbrun M. Growth factors in bone matrix. Isolation of multiple types by affinity chromatography on heparin-Sepharose. J Biol Chem. 1986;261:12665–74.
Robin C. Sur l’existence de deux especes nouvelles d’elements anatomiques qui se trouvent dans le canal medullaire des os. C R Seanc Soc Biol. 1849;1:149–50.
Kölliker A. Die normale Resorption des Knochengewebes und ihre Bedeutung für die Entstehung der typischen Knochenformen. Leipzig; Vogel. 1873.
Arnold JS, Jee WS. Bone growth and osteoclastic activity as indicated by radioautographic distribution of plutonium. Am J Anat. 1957;101:367–417.
Luben RA, Wong GL, Cohn DV. Parathormone-stimulated resorption of devitalised bone by cultured osteoclast-type bone cells. Nature. 1977;265:629–30.
Scott BL, Pease DC. Electron microscopy of the epiphyseal apparatus. Anat Rec. 1956;126:465–95.
Mallory FB. Giant Cell Sarcoma. J Med Res. 1911;24:463–468.3.
Walker DG. Congenital osteopetrosis in mice cured by parabiotic union with normal siblings. Endocrinology. 1972;91:916–20.
Walker DG. Osteopetrosis cured by temporary parabiosis. Science. 1973;180:875.
Walker DG. Control of bone resorption by hematopoietic tissue. The induction and reversal of congenital osteopetrosis in mice through use of bone marrow and splenic transplants. J Exp Med. 1975;142:651–63.
Walker DG. Bone resorption restored in osteopetrotic mice by transplants of normal bone marrow and spleen cells. Science. 1975;190:784–5.
Tinkler SM, Linder JE, Williams DM, Johnson NW. Formation of osteoclasts from blood monocytes during 1 alpha-OH Vit D-stimulated bone resorption in mice. J Anat. 1981;133:389–96.
Arai F, et al. Commitment and differentiation of osteoclast precursor cells by the sequential expression of c-Fms and receptor activator of nuclear factor kappaB (RANK) receptors. J Exp Med. 1999;190:1741–54.
Charles JF, et al. Inflammatory arthritis increases mouse osteoclast precursors with myeloid suppressor function. J Clin Invest. 2012;122:4592–605.
Jacome-Galarza CE, Lee S-K, Lorenzo JA, Aguila HL. Identification, characterization, and isolation of a common progenitor for osteoclasts, macrophages, and dendritic cells from murine bone marrow and periphery. J Bone Miner Res Off J Am Soc Bone Miner Res. 2013;28:1203–13.
Jacquin C, Gran DE, Lee SK, Lorenzo JA, Aguila HL. Identification of multiple osteoclast precursor populations in murine bone marrow. J Bone Miner Res Off J Am Soc Bone Miner Res. 2006;21:67–77.
Muto A, et al. Lineage-committed osteoclast precursors circulate in blood and settle down into bone. J Bone Miner Res Off J Am Soc Bone Miner Res. 2011;26:2978–90.
Komano Y, Nanki T, Hayashida K, Taniguchi K, Miyasaka N. Identification of a human peripheral blood monocyte subset that differentiates into osteoclasts. Arthritis Res Ther. 2006;8:R152.
Lari R, Kitchener PD, Hamilton JA. The proliferative human monocyte subpopulation contains osteoclast precursors. Arthritis Res Ther. 2009;11:R23.
Takayanagi H, et al. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell. 2002;3:889–901.
American Society for Bone and Mineral Research President’s Committee on Nomenclature. Proposed standard nomenclature for new tumor necrosis factor family members involved in the regulation of bone resorption. The American Society for Bone and Mineral Research President’s committee on nomenclature. J Bone Miner Res Off J Am Soc Bone Miner Res. 2000;15:2293–6.
Anderson DM, et al. A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature. 1997;390:175–9.
Burgess TL, et al. The ligand for osteoprotegerin (OPGL) directly activates mature osteoclasts. J Cell Biol. 1999;145:527–38.
Yasuda H, et al. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci U S A. 1998;95:3597–602.
Wong BR, et al. TRANCE is a novel ligand of the tumor necrosis factor receptor family that activates c-Jun N-terminal kinase in T cells. J Biol Chem. 1997;272:25190–4.
Nakagawa N, et al. RANK is the essential signaling receptor for osteoclast differentiation factor in osteoclastogenesis. Biochem Biophys Res Commun. 1998;253:395–400.
Kong YY, et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature. 1999;397:315–23.
Li J, et al. RANK is the intrinsic hematopoietic cell surface receptor that controls osteoclastogenesis and regulation of bone mass and calcium metabolism. Proc Natl Acad Sci U S A. 2000;97:1566–71.
Hsu H, et al. Tumor necrosis factor receptor family member RANK mediates osteoclast differentiation and activation induced by osteoprotegerin ligand. Proc Natl Acad Sci U S A. 1999;96:3540–5.
Simonet WS, et al. Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell. 1997;89:309–19.
Tsuda E, et al. Isolation of a novel cytokine from human fibroblasts that specifically inhibits osteoclastogenesis. Biochem Biophys Res Commun. 1997;234:137–42.
Kwon BS, et al. TR1, a new member of the tumor necrosis factor receptor superfamily, induces fibroblast proliferation and inhibits osteoclastogenesis and bone resorption. FASEB J Off Publ Fed Am Soc Exp Biol. 1998;12:845–54.
Yun TJ, et al. OPG/FDCR-1, a TNF receptor family member, is expressed in lymphoid cells and is up-regulated by ligating CD40. J Immunol Baltim Md. 1998;1950(161):6113–21.
Bucay N, et al. Osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev. 1998;12:1260–8.
Walsh MC, Choi Y. Biology of the RANKL-RANK-OPG system in immunity, bone, and beyond. Front Immunol. 2014;5:511.
McClung MR. Inhibition of RANKL as a treatment for osteoporosis: preclinical and early clinical studies. Curr Osteoporos Rep. 2006;4:28–33.
Prolia (denosumab) [package insert]. 2018.
Xgeva (denosumab) [package insert]. 2013.
Bellido T. Osteocyte-driven bone remodeling. Calcif Tissue Int. 2014;94:25–34.
Xiong J, et al. Osteocytes, not osteoblasts or lining cells, are the Main source of the RANKL required for osteoclast formation in remodeling bone. PLoS One. 2015;10:e0138189.
Nakashima T, et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat Med. 2011;17:1231–4.
Xiong J, et al. Matrix-embedded cells control osteoclast formation. Nat Med. 2011;17:1235–41.
Cabahug-Zuckerman P, et al. Osteocyte apoptosis caused by Hindlimb unloading is required to trigger osteocyte RANKL production and subsequent resorption of cortical and trabecular bone in mice femurs. J Bone Miner Res Off J Am Soc Bone Miner Res. 2016;31:1356–65.
Sapir-Koren R, Livshits G. Osteocyte control of bone remodeling: is sclerostin a key molecular coordinator of the balanced bone resorption-formation cycles? Osteoporos Int J Establ Result Coop Eur Found Osteoporos Natl Osteoporos Found USA. 2014;25:2685–700.
Xiong J, et al. Osteocyte-derived RANKL is a critical mediator of the increased bone resorption caused by dietary calcium deficiency. Bone. 2014;66:146–54.
Fujiwara Y, et al. RANKL (receptor activator of NFκB ligand) produced by osteocytes is required for the increase in B cells and bone loss caused by estrogen deficiency in mice. J Biol Chem. 2016;291:24838–50.
Xiong J, et al. Soluble RANKL contributes to osteoclast formation in adult mice but not ovariectomy-induced bone loss. Nat Commun. 2018;9:2909.
Koga T, et al. Costimulatory signals mediated by the ITAM motif cooperate with RANKL for bone homeostasis. Nature. 2004;428:758–63.
Mócsai A, et al. The immunomodulatory adapter proteins DAP12 and fc receptor gamma-chain (FcRgamma) regulate development of functional osteoclasts through the Syk tyrosine kinase. Proc Natl Acad Sci U S A. 2004;101:6158–63.
Humphrey MB, Lanier LL, Nakamura MC. Role of ITAM-containing adapter proteins and their receptors in the immune system and bone. Immunol Rev. 2005;208:50–65.
Humphrey MB, Nakamura MC. A comprehensive review of Immunoreceptor regulation of osteoclasts. Clin Rev Allergy Immunol. 2016;51:48–58.
Kim K, et al. MafB negatively regulates RANKL-mediated osteoclast differentiation. Blood. 2007;109:3253–9.
Kodama H, et al. Congenital osteoclast deficiency in osteopetrotic (op/op) mice is cured by injections of macrophage colony-stimulating factor. J Exp Med. 1991;173:269–72.
Cecchini MG, Hofstetter W, Halasy J, Wetterwald A, Felix R. Role of CSF-1 in bone and bone marrow development. Mol Reprod Dev. 1997;46:75–83.; discussion 83-84.
Zou W, et al. Syk, c-Src, the alphavbeta3 integrin, and ITAM immunoreceptors, in concert, regulate osteoclastic bone resorption. J Cell Biol. 2007;176:877–88.
Duong LT, Lakkakorpi P, Nakamura I, Rodan GA. Integrins and signaling in osteoclast function. Matrix Biol J Int Soc Matrix Biol. 2000;19:97–105.
Nakamura I, Duong LT, Rodan SB, Rodan GA. Involvement of alpha(v)beta3 integrins in osteoclast function. J Bone Miner Metab. 2007;25:337–44.
McHugh KP, et al. Role of cell-matrix interactions in osteoclast differentiation. Adv Exp Med Biol. 2007;602:107–11.
McHugh KP, et al. Mice lacking beta3 integrins are osteosclerotic because of dysfunctional osteoclasts. J Clin Invest. 2000;105:433–40.
Elsegood CL, et al. M-CSF induces the stable interaction of cFms with alphaVbeta3 integrin in osteoclasts. Int J Biochem Cell Biol. 2006;38:1518–29.
Zou W, Teitelbaum SL. Absence of Dap12 and the αvβ3 integrin causes severe osteopetrosis. J Cell Biol. 2015;208:125–36.
Zallone AZ, Teti A, Primavera MV. Monocytes from circulating blood fuse in vitro with purified osteoclasts in primary culture. J Cell Sci. 1984;66:335–42.
Khan UA, Hashimi SM, Bakr MM, Forwood MR, Morrison NA. Foreign body giant cells and osteoclasts are TRAP positive, have podosome-belts and both require OC-STAMP for cell fusion. J Cell Biochem. 2013;114:1772–8.
Kukita T, et al. RANKL-induced DC-STAMP is essential for osteoclastogenesis. J Exp Med. 2004;200:941–6.
Miyamoto H, et al. Osteoclast stimulatory transmembrane protein and dendritic cell–specific transmembrane protein cooperatively modulate cell–cell fusion to form osteoclasts and foreign body giant cells. J Bone Miner Res Off J Am Soc Bone Miner Res. 2012;27:1289–97.
Yagi M, et al. DC-STAMP is essential for cell-cell fusion in osteoclasts and foreign body giant cells. J Exp Med. 2005;202:345–51.
Yang M, et al. Osteoclast stimulatory transmembrane protein (OC-STAMP), a novel protein induced by RANKL that promotes osteoclast differentiation. J Cell Physiol. 2008;215:497–505.
Witwicka H, et al. Studies of OC-STAMP in osteoclast fusion: a new knockout mouse model, Rescue of Cell Fusion, and transmembrane topology. PLoS One. 2015;10(6):e0128275.
Lee S-H, et al. V-ATPase V0 subunit d2-deficient mice exhibit impaired osteoclast fusion and increased bone formation. Nat Med. 2006;12:1403–9.
Xing L, Xiu Y, Boyce BF. Osteoclast fusion and regulation by RANKL-dependent and independent factors. World J Orthop. 2012;3:212–22.
Asagiri M, Takayanagi H. The molecular understanding of osteoclast differentiation. Bone. 2007;40:251–64.
Ikeda K, Takeshita S. The role of osteoclast differentiation and function in skeletal homeostasis. J Biochem (Tokyo). 2016;159(1):1–8.
Lomaga MA, et al. TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and LPS signaling. Genes Dev. 1999;13:1015–24.
Israël A. The IKK complex, a central regulator of NF-kappaB activation. Cold Spring Harb Perspect Biol. 2010;2:a000158.
Soysa NS, et al. The pivotal role of the alternative NF-kappaB pathway in maintenance of basal bone homeostasis and osteoclastogenesis. J Bone Miner Res Off J Am Soc Bone Miner Res. 2010;25:809–18.
Xing L, et al. NF-kappaB p50 and p52 expression is not required for RANK-expressing osteoclast progenitor formation but is essential for RANK- and cytokine-mediated osteoclastogenesis. J Bone Miner Res Off J Am Soc Bone Miner Res. 2002;17:1200–10.
Iotsova V, et al. Osteopetrosis in mice lacking NF-kappaB1 and NF-kappaB2. Nat Med. 1997;3:1285–9.
Swarnkar G, Karuppaiah K, Mbalaviele G, Chen TH-P, Abu-Amer Y. Osteopetrosis in TAK1-deficient mice owing to defective NF-κB and NOTCH signaling. Proc Natl Acad Sci U S A. 2015;112:154–9.
Grigoriadis AE, et al. C-Fos: a key regulator of osteoclast-macrophage lineage determination and bone remodeling. Science. 1994;266:443–8.
Thouverey C, Caverzasio J. Focus on the p38 MAPK signaling pathway in bone development and maintenance. BoneKEy Rep. 2015;4:711.
Cong Q, et al. p38α MAPK regulates proliferation and differentiation of osteoclast progenitors and bone remodeling in an aging-dependent manner. Sci Rep. 2017;7:45964.
He Y, et al. Erk1 positively regulates osteoclast differentiation and bone resorptive activity. PLoS One. 2011;6:e24780.
Shinohara M, et al. Class IA phosphatidylinositol 3-kinase regulates osteoclastic bone resorption through protein kinase B-mediated vesicle transport. J Bone Miner Res Off J Am Soc Bone Miner Res. 2012;27:2464–75.
Gingery A, Bradley E, Shaw A, Oursler MJ. Phosphatidylinositol 3-kinase coordinately activates the MEK/ERK and AKT/NFkappaB pathways to maintain osteoclast survival. J Cell Biochem. 2003;89:165–79.
Takeshita S, et al. SHIP-deficient mice are severely osteoporotic due to increased numbers of hyper-resorptive osteoclasts. Nat Med. 2002;8:943–9.
Jang HD, Noh JY, Shin JH, Lin JJ, Lee SY. PTEN regulation by the Akt/GSK-3β axis during RANKL signaling. Bone. 2013;55:126–31.
Kwon OH, Lee C-K, Lee YI, Paik S-G, Lee H-J. The hematopoietic transcription factor PU.1 regulates RANK gene expression in myeloid progenitors. Biochem Biophys Res Commun. 2005;335:437–46.
Tondravi MM, et al. Osteopetrosis in mice lacking haematopoietic transcription factor PU.1. Nature. 1997;386:81–4.
Lu S-Y, Li M, Lin Y-L. Mitf induction by RANKL is critical for osteoclastogenesis. Mol Biol Cell. 2010;21:1763–71.
Sharma SM, et al. MITF and PU.1 recruit p38 MAPK and NFATc1 to target genes during osteoclast differentiation. J Biol Chem. 2007;282:15921–9.
Thesingh CW, Scherft JP. Fusion disability of embryonic osteoclast precursor cells and macrophages in the microphthalmic osteopetrotic mouse. Bone. 1985;6:43–52.
Weilbaecher KN, et al. Linkage of M-CSF signaling to Mitf, TFE3, and the osteoclast defect in Mitf(mi/mi) mice. Mol Cell. 2001;8:749–58.
Shinohara M, et al. Tyrosine kinases Btk and Tec regulate osteoclast differentiation by linking RANK and ITAM signals. Cell. 2008;132:794–806.
Epple H, et al. Phospholipase Cgamma2 modulates integrin signaling in the osteoclast by affecting the localization and activation of Src kinase. Mol Cell Biol. 2008;28:3610–22.
Asagiri M, et al. Autoamplification of NFATc1 expression determines its essential role in bone homeostasis. J Exp Med. 2005;202:1261–9.
Bae S, et al. MYC-dependent oxidative metabolism regulates osteoclastogenesis via nuclear receptor ERRα. J Clin Invest. 2017;127:2555–68.
Indo Y, et al. Metabolic regulation of osteoclast differentiation and function. J Bone Miner Res Off J Am Soc Bone Miner Res. 2013;28:2392–9.
Zhao B, Ivashkiv LB. Negative regulation of osteoclastogenesis and bone resorption by cytokines and transcriptional repressors. Arthritis Res Ther. 2011;13:234.
Nishikawa K, et al. Blimp1-mediated repression of negative regulators is required for osteoclast differentiation. Proc Natl Acad Sci U S A. 2010;107:3117–22.
Lee J, et al. Id helix-loop-helix proteins negatively regulate TRANCE-mediated osteoclast differentiation. Blood. 2006;107:2686–93.
Hu R, et al. Eos, MITF, and PU.1 recruit corepressors to osteoclast-specific genes in committed myeloid progenitors. Mol Cell Biol. 2007;27:4018–27.
Tamura T, Kurotaki D, Koizumi S. Regulation of myelopoiesis by the transcription factor IRF8. Int J Hematol. 2015;101:342–51.
Zhao B, et al. Interferon regulatory factor-8 regulates bone metabolism by suppressing osteoclastogenesis. Nat Med. 2009;15:1066–71.
Miyauchi Y, et al. The Blimp1-Bcl6 axis is critical to regulate osteoclast differentiation and bone homeostasis. J Exp Med. 2010;207:751–62.
Takayanagi H, et al. RANKL maintains bone homeostasis through c-Fos-dependent induction of interferon-beta. Nature. 2002;416:744–9.
Jin W, et al. Deubiquitinating enzyme CYLD negatively regulates RANK signaling and osteoclastogenesis in mice. J Clin Invest. 2008;118:1858–66.
Swarnkar G, et al. NUMBL interacts with TAK1, TRAF6 and NEMO to negatively regulate NF-κB signaling during Osteoclastogenesis. Sci Rep. 2017;7:12600.
Li S, et al. RBP-J imposes a requirement for ITAM-mediated costimulation of osteoclastogenesis. J Clin Invest. 2014;124:5057–73.
Han Y, You X, Xing W, Zhang Z, Zou W. Paracrine and endocrine actions of bone-the functions of secretory proteins from osteoblasts, osteocytes, and osteoclasts. Bone Res. 2018;6:16.
Charles JF, Aliprantis AO. Osteoclasts: more than ‘bone eaters’. Trends Mol Med. 2014;20:449–59.
Cappariello A, Maurizi A, Veeriah V, Teti A. The great beauty of the osteoclast. Arch Biochem Biophys. 2014;558:70–8.
Novack DV, Faccio R. Osteoclast motility: putting the brakes on bone resorption. Ageing Res Rev. 2011;10:54–61.
Georgess D, Machuca-Gayet I, Blangy A, Jurdic P. Podosome organization drives osteoclast-mediated bone resorption. Cell Adhes Migr. 2014;8:191–204.
Zallone AZ, Teti A, Primavera MV, Naldini L, Marchisio PC. Osteoclasts and monocytes have similar cytoskeletal structures and adhesion property in vitro. J Anat. 1983;137(Pt 1):57–70.
Hunter SJ, Schraer H, Gay CV. Characterization of the cytoskeleton of isolated chick osteoclasts: effect of calcitonin. J Histochem Cytochem Off J Histochem Soc. 1989;37:1529–37.
Mulari MTK, Zhao H, Lakkakorpi PT, Väänänen HK. Osteoclast ruffled border has distinct subdomains for secretion and degraded matrix uptake. Traffic Cph Den. 2003;4:113–25.
Mulari M, Vääräniemi J, Väänänen HK. Intracellular membrane trafficking in bone resorbing osteoclasts. Microsc Res Tech. 2003;61:496–503.
Walker EC, et al. Cardiotrophin-1 is an osteoclast-derived stimulus of bone formation required for normal bone remodeling. J Bone Miner Res Off J Am Soc Bone Miner Res. 2008;23:2025–32.
Krishnamurthy A, et al. Identification of a novel chemokine-dependent molecular mechanism underlying rheumatoid arthritis-associated autoantibody-mediated bone loss. Ann Rheum Dis. 2016;75:721–9.
Aliprantis AO, et al. NFATc1 in mice represses osteoprotegerin during osteoclastogenesis and dissociates systemic osteopenia from inflammation in cherubism. J Clin Invest. 2008;118:3775–89.
Xie H, et al. PDGF-BB secreted by preosteoclasts induces angiogenesis during coupling with osteogenesis. Nat Med. 2014;20:1270–8.
Jin Y-R, et al. Inhibition of osteoclast differentiation and collagen antibody-induced arthritis by CTHRC1. Bone. 2017;97:153–67.
Kim B-J, et al. Osteoclast-secreted SLIT3 coordinates bone resorption and formation. J Clin Invest. 2018;128:1429–41.
Xu R, et al. Targeting skeletal endothelium to ameliorate bone loss. Nat Med. 2018;24:823–33.
Negishi-Koga T, et al. Suppression of bone formation by osteoclastic expression of semaphorin 4D. Nat Med. 2011;17:1473–80.
Zhao C, et al. Bidirectional ephrinB2-EphB4 signaling controls bone homeostasis. Cell Metab. 2006;4:111–21.
Ikebuchi Y, et al. Coupling of bone resorption and formation by RANKL reverse signalling. Nature. 2018;561:195–200.
Guerrini MM, et al. Human osteoclast-poor osteopetrosis with hypogammaglobulinemia due to TNFRSF11A (RANK) mutations. Am J Hum Genet. 2008;83:64–76.
Hughes AE, et al. Mutations in TNFRSF11A, affecting the signal peptide of RANK, cause familial expansile osteolysis. Nat Genet. 2000;24:45–8.
Laurin N, Brown JP, Morissette J, Raymond V. Recurrent mutation of the gene encoding sequestosome 1 (SQSTM1/p62) in Paget disease of bone. Am J Hum Genet. 2002;70:1582–8.
Watts GDJ, et al. Novel VCP mutations in inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia. Clin Genet. 2007;72:420–6.
Sundaram K, Shanmugarajan S, Rao DS, Reddy SV. Mutant p62P392L stimulation of osteoclast differentiation in Paget’s disease of bone. Endocrinology. 2011;152:4180–9.
Sobacchi C, Schulz A, Coxon FP, Villa A, Helfrich MH. Osteopetrosis: genetics, treatment and new insights into osteoclast function. Nat Rev Endocrinol. 2013;9:522–36.
Boudin E, Fijalkowski I, Hendrickx G, Van Hul W. Genetic control of bone mass. Mol Cell Endocrinol. 2016;432:3–13.
Kornak U, et al. Loss of the ClC-7 chloride channel leads to osteopetrosis in mice and man. Cell. 2001;104:205–15.
Campos-Xavier AB, Saraiva JM, Ribeiro LM, Munnich A, Cormier-Daire V. Chloride channel 7 (CLCN7) gene mutations in intermediate autosomal recessive osteopetrosis. Hum Genet. 2003;112:186–9.
Döffinger R, et al. X-linked anhidrotic ectodermal dysplasia with immunodeficiency is caused by impaired NF-kappaB signaling. Nat Genet. 2001;27:277–85.
Sobacchi C, et al. Osteoclast-poor human osteopetrosis due to mutations in the gene encoding RANKL. Nat Genet. 2007;39:960–2.
Whyte MP, et al. Osteoprotegerin deficiency and juvenile Paget’s disease. N Engl J Med. 2002;347:175–84.
Sly WS, et al. Carbonic anhydrase II deficiency in 12 families with the autosomal recessive syndrome of osteopetrosis with renal tubular acidosis and cerebral calcification. N Engl J Med. 1985;313:139–45.
Malinin NL, et al. A point mutation in KINDLIN3 ablates activation of three integrin subfamilies in humans. Nat Med. 2009;15:313–8.
Gelb BD, Shi GP, Chapman HA, Desnick RJ. Pycnodysostosis, a lysosomal disease caused by cathepsin K deficiency. Science. 1996;273:1236–8.
Kornak U, Mundlos S. Genetic disorders of the skeleton: a developmental approach. Am J Hum Genet. 2003;73:447–74.
Van Wesenbeeck L, et al. Involvement of PLEKHM1 in osteoclastic vesicular transport and osteopetrosis in incisors absent rats and humans. J Clin Invest. 2007;117:919–30.
Aker M, et al. An SNX10 mutation causes malignant osteopetrosis of infancy. J Med Genet. 2012;49:221–6.
Li P, et al. Systemic tumor necrosis factor alpha mediates an increase in peripheral CD11bhigh osteoclast precursors in tumor necrosis factor alpha-transgenic mice. Arthritis Rheum. 2004;50:265–76.
Yao Z, et al. Tumor necrosis factor-α increases circulating osteoclast precursor numbers by promoting their proliferation and differentiation in the bone marrow through up-regulation of c-Fms expression. J Biol Chem. 2006;281:11846–55.
Chitu V, et al. PSTPIP2 deficiency in mice causes osteopenia and increased differentiation of multipotent myeloid precursors into osteoclasts. Blood. 2012;120:3126–35.
Chiu YG, et al. CD16 (FcRgammaIII) as a potential marker of osteoclast precursors in psoriatic arthritis. Arthritis Res Ther. 2010;12:R14.
Durand M, et al. The increased in vitro osteoclastogenesis in patients with rheumatoid arthritis is due to increased percentage of precursors and decreased apoptosis - the in vitro osteoclast differentiation in arthritis (IODA) study. Bone. 2011;48:588–96.
Kawanaka N, et al. CD14+,CD16+ blood monocytes and joint inflammation in rheumatoid arthritis. Arthritis Rheum. 2002;46:2578–86.
Ritchlin CT, Haas-Smith SA, Li P, Hicks DG, Schwarz EM. Mechanisms of TNF-alpha- and RANKL-mediated osteoclastogenesis and bone resorption in psoriatic arthritis. J Clin Invest. 2003;111:821–31.
Zhang YH, Heulsmann A, Tondravi MM, Mukherjee A, Abu-Amer Y. Tumor necrosis factor-alpha (TNF) stimulates RANKL-induced osteoclastogenesis via coupling of TNF type 1 receptor and RANK signaling pathways. J Biol Chem. 2001;276:563–8.
Azuma Y, Kaji K, Katogi R, Takeshita S, Kudo A. Tumor necrosis factor-alpha induces differentiation of and bone resorption by osteoclasts. J Biol Chem. 2000;275:4858–64.
Okamoto K, Takayanagi H. Osteoimmunology. Cold Spring Harb Perspect Med. 2018; https://doi.org/10.1101/cshperspect.a031245.
Kimble RB, Bain S, Pacifici R. The functional block of TNF but not of IL-6 prevents bone loss in ovariectomized mice. J Bone Miner Res Off J Am Soc Bone Miner Res. 1997;12:935–41.
Roggia C, et al. Up-regulation of TNF-producing T cells in the bone marrow: a key mechanism by which estrogen deficiency induces bone loss in vivo. Proc Natl Acad Sci U S A. 2001;98:13960–5.
Kim JH, et al. The mechanism of osteoclast differentiation induced by IL-1. J. Immunol. Baltim. Md. 2009;1950(183):1862–70.
Blanchard F, Duplomb L, Baud’huin M, Brounais B. The dual role of IL-6-type cytokines on bone remodeling and bone tumors. Cytokine Growth Factor Rev. 2009;20:19–28.
Poli V, et al. Interleukin-6 deficient mice are protected from bone loss caused by estrogen depletion. EMBO J. 1994;13:1189–96.
Zhu S, et al. Ovariectomy-induced bone loss in TNFα and IL6 gene knockout mice is regulated by different mechanisms. J Mol Endocrinol. 2018;60:185–98.
Hiasa M, et al. GM-CSF and IL-4 induce dendritic cell differentiation and disrupt osteoclastogenesis through M-CSF receptor shedding by up-regulation of TNF-alpha converting enzyme (TACE). Blood. 2009;114:4517–26.
Evans KE, Fox SW. Interleukin-10 inhibits osteoclastogenesis by reducing NFATc1 expression and preventing its translocation to the nucleus. BMC Cell Biol. 2007;8:4.
Park-Min K-H, et al. IL-10 suppresses calcium-mediated costimulation of receptor activator NF-kappa B signaling during human osteoclast differentiation by inhibiting TREM-2 expression. J. Immunol. Baltim. Md. 2009;1950(183):2444–55.
Yamada A, et al. Interleukin-4 inhibition of osteoclast differentiation is stronger than that of interleukin-13 and they are equivalent for induction of osteoprotegerin production from osteoblasts. Immunology. 2007;120:573–9.
Lubberts E, Koenders MI, van den Berg WB. The role of T-cell interleukin-17 in conducting destructive arthritis: lessons from animal models. Arthritis Res Ther. 2005;7:29–37.
Takayanagi H, et al. T-cell-mediated regulation of osteoclastogenesis by signalling cross-talk between RANKL and IFN-gamma. Nature. 2000;408:600–5.
Zaiss MM, et al. Treg cells suppress osteoclast formation: a new link between the immune system and bone. Arthritis Rheum. 2007;56:4104–12.
Axmann R, et al. CTLA-4 directly inhibits osteoclast formation. Ann Rheum Dis. 2008;67:1603–9.
Bozec A, et al. T cell costimulation molecules CD80/86 inhibit osteoclast differentiation by inducing the IDO/tryptophan pathway. Sci Transl Med. 2014;6:235ra60.
Lee SK, Lorenzo JA. Parathyroid hormone stimulates TRANCE and inhibits osteoprotegerin messenger ribonucleic acid expression in murine bone marrow cultures: correlation with osteoclast-like cell formation. Endocrinology. 1999;140:3552–61.
Huang JC, et al. PTH differentially regulates expression of RANKL and OPG. J Bone Miner Res Off J Am Soc Bone Miner Res. 2004;19:235–44.
Kumamoto H, Ooya K. Expression of parathyroid hormone-related protein (PTHrP), osteoclast differentiation factor (ODF)/receptor activator of nuclear factor-kappaB ligand (RANKL) and osteoclastogenesis inhibitory factor (OCIF)/osteoprotegerin (OPG) in ameloblastomas. J Oral Pathol Med Off Publ Int Assoc Oral Pathol Am Acad Oral Pathol. 2004;33:46–52.
Thomas RJ, et al. Breast cancer cells interact with osteoblasts to support osteoclast formation. Endocrinology. 1999;140:4451–8.
Hofbauer LC, Dunstan CR, Spelsberg TC, Riggs BL, Khosla S. Osteoprotegerin production by human osteoblast lineage cells is stimulated by vitamin D, bone morphogenetic protein-2, and cytokines. Biochem Biophys Res Commun. 1998;250:776–81.
Palmqvist P, Persson E, Conaway HH, Lerner UH. IL-6, leukemia inhibitory factor, and oncostatin M stimulate bone resorption and regulate the expression of receptor activator of NF-kappa B ligand, osteoprotegerin, and receptor activator of NF-kappa B in mouse calvariae. J. Immunol. Baltim. Md. 2002;1950(169):3353–62.
Glass DA, et al. Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev Cell. 2005;8:751–64.
Saika M, Inoue D, Kido S, Matsumoto T. 17beta-estradiol stimulates expression of osteoprotegerin by a mouse stromal cell line, ST-2, via estrogen receptor-alpha. Endocrinology. 2001;142:2205–12.
Brändström H, Björkman T, Ljunggren O. Regulation of osteoprotegerin secretion from primary cultures of human bone marrow stromal cells. Biochem Biophys Res Commun. 2001;280:831–5.
Suda K, et al. Suppression of osteoprotegerin expression by prostaglandin E2 is crucially involved in lipopolysaccharide-induced osteoclast formation. J. Immunol. Baltim. Md. 2004;1950(172):2504–10.
Tan YY, Yang Y-Q, Chai L, Wong RWK, Rabie ABM. Effects of vascular endothelial growth factor (VEGF) on MC3T3-E1. Orthod Craniofac Res. 2010;13:223–8.
Rubin J, et al. IGF-I regulates osteoprotegerin (OPG) and receptor activator of nuclear factor-kappaB ligand in vitro and OPG in vivo. J Clin Endocrinol Metab. 2002;87:4273–9.
O’Sullivan S, et al. Tyrosine kinase inhibitors regulate OPG through inhibition of PDGFRβ. PLoS One. 2016;11:e0164727.
Nakashima T, et al. Protein expression and functional difference of membrane-bound and soluble receptor activator of NF-kappaB ligand: modulation of the expression by osteotropic factors and cytokines. Biochem Biophys Res Commun. 2000;275:768–75.
Ahlen J, et al. Characterization of the bone-resorptive effect of interleukin-11 in cultured mouse calvarial bones. Bone. 2002;31:242–51.
Dai S-M, Nishioka K, Yudoh K. Interleukin (IL) 18 stimulates osteoclast formation through synovial T cells in rheumatoid arthritis: comparison with IL1 beta and tumour necrosis factor alpha. Ann Rheum Dis. 2004;63:1379–86.
Thirunavukkarasu K, et al. Stimulation of osteoprotegerin (OPG) gene expression by transforming growth factor-beta (TGF-beta). Mapping of the OPG promoter region that mediates TGF-beta effects. J Biol Chem. 2001;276:36241–50.
Quinn JM, et al. Transforming growth factor beta affects osteoclast differentiation via direct and indirect actions. J Bone Miner Res Off J Am Soc Bone Miner Res. 2001;16:1787–94.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Alesi, N., Charles, J.F., Nakamura, M.C. (2020). Basic Aspects of Osteoclast Differentiation and Function. In: Leder, B., Wein, M. (eds) Osteoporosis. Contemporary Endocrinology. Humana, Cham. https://doi.org/10.1007/978-3-319-69287-6_2
Download citation
DOI: https://doi.org/10.1007/978-3-319-69287-6_2
Published:
Publisher Name: Humana, Cham
Print ISBN: 978-3-319-69286-9
Online ISBN: 978-3-319-69287-6
eBook Packages: MedicineMedicine (R0)