
Abstract
The purpose of this meta-analysis was to investigate the association between progranulin polymorphism rs5848 and risk of the neurodegenerative diseases frontotemporal lobar degeneration (FTLD), Alzheimer’s disease (AD), Parkinson’s disease (PD), and amyotrophic lateral sclerosis (ALS). Published literature from PubMed and other databases were retrieved, and 16 case–control studies were identified as eligible: 5 on FTLD (1,439 cases, 4,461 controls), 5 on AD (2,502 cases, 2,162 controls), 3 on PD (1,605 cases, 1,591 controls), and 3 on ALS (663 cases, 811 controls). The pooled odds ratio (OR) and 95 % confidence interval (CI) were calculated. We found that rs5848 was associated with an increased risk of neurodegenerative diseases in the homozygous (TT vs. CC: OR, 1.24; 95 % CI, 1.10–1.39; P < 0.001) and recessive models (TT vs. CC + CT: OR, 1.23; 95 % CI, 1.10–1.37; P < 0.001). Stratified analyses showed associations of rs5848 with increased risk of AD and PD in the homozygous and recessive models. Our data indicate that rs5848 is associated with risk of AD and PD, suggesting important roles of progranulin in neurodegenerative processes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Neurodegenerative diseases are progressive disorders with selective neuronal loss in particular regions of the brain, including frontotemporal lobar degeneration (FTLD), Alzheimer’s disease (AD), amyotrophic lateral sclerosis (ALS), Parkinson’s disease (PD), and many others. In addition to aging, brain injury, and lifestyle, it has been acknowledged that the etiology of neurodegenerative diseases is often multifactorial (particularly gene–environment interactions). However, epidemiological evidence for an association between environmental agents and neurodegenerative disease is limited [1].
Gene defects are prominent factors in the etiology and pathogenesis of neurodegenerative diseases. To date, hundreds of genetic variants located in dozens of genes have been associated with susceptibility to various such diseases (reviewed in [2]). Although the majority of susceptibility genes do not overlap across diseases, some mutations in certain genes have been linked to diverse neurodegenerative diseases, e.g., TAR DNA-binding protein 43 and fused in sarcoma/translated in liposarcoma [3, 4]. Among them, mutations in progranulin (PGRN) have been reported in FTLD, AD, ALS, and PD [5–8]. It has been reported that PGRN mutations are a major genetic cause of FTLD, and most pathogenic PGRN mutations are associated with FTLD [9]. On the other hand, several missense mutations and deletions in PGRN have been reported in AD, ALS, and PD [10–12]. These studies suggest that PGRN mutations play important roles in neurodegenerative processes in general.
Progranulin is the precursor of granulins, and its downregulation may lead to neurodegeneration. PGRN is located 1.7 Mb centromeric of the MAPT gene (encoding tau protein) on chromosome 17q21.31, a region linked to FTLD [5]. Since the identification of mutations in PGRN, >60 different pathogenic PGRN mutations and some deletions have been identified in patients with neurodegeneration (AD&FTD mutation database) [5, 6]. Most PGRN mutations including heterozygous deletions identified to date cause null alleles that result in loss-of-function of PGRN or haploinsufficiency through nonsense-mediated decay [5, 6, 13, 14]. Notably, the single nucleotide polymorphism (SNP) rs5848, which is located in the 3′-untranslated region (3′-UTR) of PGRN and predicted to be a binding site for the microRNA miR-659, is associated with frontotemporal dementia [15]. Similarly, the PGRN genetic polymorphism rs5848 has also been demonstrated to increase the risk of AD [6, 7] and the development of PD [16]. However, other studies showed no association of rs5848 with FTLD [17], AD [18], or PD [19]. In addition, a recent study reported no major contribution of progranulin genetic variability to the etiopathogenesis of ALS [20]. These inconsistent results might be due to the limited numbers of participants included in each study, so a single study may be underpowered to estimate the effects of loci conferring small changes in disease risk.
In this study, we performed a meta-analysis by pooling all 16 case–control studies to derive a more precise estimate of the relationship between rs5848 and the risk of neurodegenerative disease.
Methods
Identification and eligibility of relevant studies
To identify all articles that examined the association of progranulin polymorphism with neurodegenerative disease, we conducted a literature search in the PubMed databases up to August 2013 using the MeSH terms and keywords “progranulin”, “polymorphism”, and “neurodegenerative disease”. Additional studies were identified by a manual search of other sources (e.g., Web of Knowledge), and references in original studies or review articles on these topics. Eligible studies had to meet the following criteria: (a) evaluation of an association between rs5848 and neurodegenerative disease; (b) an unrelated case–control study; if studies had partly overlapping participants, only the one with a larger sample size was selected; (c) available genotype frequency and sufficient data for estimating an odds ratio (OR) with 95 % confidence interval (CI); and (d) genotype frequencies in the control group consistent with Hardy–Weinberg equilibrium (HWE).
Data extraction
Two investigators independently assessed the articles for inclusion/exclusion, reached a consensus on all items, and extracted data. For each study, the following information was extracted: name of the first author; publication year; ethnicity (country); sample size (numbers of cases and controls); types of neurodegenerative disease; minor allele frequency; P value for the Chi-square (χ 2) HWE test in each control group.
Statistical analysis
The association between the progranulin polymorphism rs5848 and neurodegenerative disease was estimated by calculating pooled ORs and 95 % CIs. The significance of the pooled OR was determined by the Z test (P < 0.05 was considered statistically significant). The risk of rs5848 in neurodegenerative disease was evaluated by comparison with the reference wild-type homozygote. We first estimated the risks of the CT and TT genotypes in neurodegenerative disease, compared with the reference CC homozygote, and then evaluated the risks of CT + TT vs. CC and TT vs. CC + CT in neurodegenerative disease, assuming dominant and recessive effects of the variant TT allele, respectively. The I 2-based Q statistic test was performed to evaluate variations due to heterogeneity rather than chance. A random-effects (DerSimonian–Laird method) or fixed-effects (Mantel–Haenszel method) model was used to calculate pooled-effect estimates in the presence (P ≤ 0.10) or absence (P > 0.10) of heterogeneity. Publication bias was detected by Egger’s test [21] and Begg’s [22] test for the overall pooled analysis of different models of rs5848. In addition, Begg’s funnel plots were drawn. Asymmetry of the funnel plot means a potential publication bias. For one-way sensitivity analysis, a single study was excluded each time, and the new pooled results could reflect the influence of the deleted study on the overall summary OR. To obtain a measure of the degree to which the findings reported here might be false-positives, corrections for multiple comparisons were considered using the Benjamini–Hochberg false-discovery rate (FDR) adjustment [23]. FDR-adjusted P < 0.05 was considered to be potentially significant. FDR-adjustment analysis was carried out with the q value package in R software (version 3.1.0; R Foundation for Statistical Computing, Vienna, Austria) and other analyses were conducted with Stata software (version 11.0; StataCorp LP, College Station, TX), using two-sided P values.
Results
Characteristics of studies
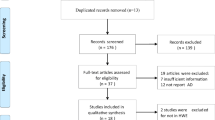
Twenty-six abstracts were retrieved through the search for “progranulin”, “polymorphism” and “neurodegenerative disease”, and 7 studies meeting the inclusion criteria were identified [17–19, 24–27]. We also included 9 studies found by manual searching [15, 16, 20, 28–33]. As a result, a total of 16 studies met the inclusion criteria and were identified as eligible articles (Fig. 1).

Flow-diagram of study identification
Five studies were included in the meta-analysis of rs5848 genotype in FTLD (1,439 cases, 4,461 controls), 5 in AD (2,502 cases, 2,162 controls), 3 in PD (1,605 cases, 1,591 controls), and 3 in ALS (663 cases, 811 controls). In terms of ethnicity, 14 studies of Caucasians and 2 of Asians were included. The detailed characteristics of each study in the meta-analysis are presented in Table 1.
Quantitative synthesis
The results of the meta-analysis on the association between rs5848 and risk of neurodegenerative disease are shown in Table 2. By pooling all the studies, the results showed that rs5848 was associated with an increased risk of all neurodegenerative diseases in the homozygous (TT vs. CC: OR, 1.24; 95 % CI, 1.10–1.39; P < 0.001) but not the heterozygous models (CT vs. CC: OR, 1.00; 95 % CI, 0.93–1.08; P = 0.983). Furthermore, we found that rs5848 was significantly associated with an increased risk of all neurodegenerative diseases in a recessive model (TT vs. CC + CT: OR, 1.23; 95 % CI, 1.10–1.37; P < 0.001), but not in a dominant model (CT + TT vs. CC: OR, 1.04; 95 % CI, 0.98–1.11; P = 0.263). We also performed subgroup analyses and found that rs5848 polymorphism was associated with increased risk of all neurodegenerative diseases in Caucasians in the homozygous (TT vs. CC: OR, 1.18; 95 % CI, 1.04–1.34; P = 0.012) and recessive models (TT vs. CC + CT: OR, 1.18; 95 % CI, 1.04–1.33; P = 0.008). Similar associations were also found in Asians.
We next performed subgroup analysis on the association of the rs5848 polymorphism with each neurodegenerative disease. The results showed that this polymorphism was not associated with FTLD in different models (Table 2; Fig. 2). For AD, we found that rs5848 was associated with an increased risk in the homozygous (TT vs. CC: OR, 1.36; 95 % CI, 1.11–1.66; P = 0.003) and recessive models (TT vs. CC + CT: OR, 1.31; 95 % CI, 1.08–1.58; P = 0.006). As for PD, rs5848 was associated with an increased risk in the homozygous (TT vs. CC: OR, 1.34; 95 % CI, 1.05–1.69; P = 0.017) and recessive models (TT vs. CC + CT: OR, 1.28; 95 % CI, 1.02–1.60; P = 0.034). On the contrary, we found that rs5848 was associated with a decreased risk of ALS in the heterozygous (TC vs. CC: OR, 0.80; 95 % CI, 0.64–1.00; P = 0.047) and dominant models (TT + CT vs. CC: OR, 0.79; 95 % CI, 0.64–0.97; P = 0.026).

Forest plots of the association between rs5848 and risk of neurodegenerative diseases. The association between rs5848 and risk of neurodegenerative diseases was examined in heterozygous (a), homozygous (b), dominant (c), and recessive models (d). The squares and horizontal lines correspond to the OR and 95 % CI of a specific study, and the area of squares reflects the study weight (inverse of the variance). The diamond represents the pooled OR and its 95 % CI
Potential publication bias and sensitivity analysis
Publication bias was first assessed by Begg’s test for the overall pooled analysis of different models of rs5848. This test showed that the P values of rs5848 were 0.096, 0.096, 0.065, and 0.260 for the heterozygous, homozygous, dominant, and recessive models, respectively, and the corresponding funnel plots showed a symmetrical distribution (Fig. 3). Egger’s test also showed that the P values of rs5848 were 0.023, 0.053, 0.026, and 0.141, respectively, suggesting a slight publication bias. Sensitivity analysis showed that exclusion of each study did not influence the result (Fig. 4).

Funnel plots showed symmetric distribution. Log OR was plotted against the standard error of log OR for the association of rs5848 with risk of neurodegenerative diseases in heterozygote (a), homozygote (b), dominant (c), and recessive models (d). The dots represent specific studies for the indicated association

Sensitivity analysis of the summary OR on the association between rs5848 and risk of neurodegenerative diseases. The association of rs5848 with risk of neurodegenerative diseases was computed by omitting each study in turn in heterozygous (a), homozygous (b), dominant (c) or recessive models (d). The two ends of the dotted lines represent the 95 % CI of the OR
Discussion
In the present meta-analysis, we found that rs5848 was associated with increased risk of neurodegenerative diseases in homozygous and recessive models. In the subgroup analysis, however, rs5848 was associated with a decreased risk of ALS in the heterozygous and dominant models. It should be noted that only 663 ALS cases and 811 controls were included. Thus, the protective effect of rs5848 on ALS development awaits further investigation. As for PD, an early study by Jasinska–Myga et al. [19] reported a lack of association between rs5848 and PD risk in the US and Poland, while a recent study showed that PGRN rs5848 affects the risk of developing PD in a Taiwanese population [16]. This discrepancy may be due to a differential effect of rs5848 on PD risk between Eastern and Western populations. By pooling all studies, our data showed that rs5848 was associated with increased risk of PD in the homozygous and recessive models. Future studies are required to verify this association since our data were only based on three studies. In addition, we found significant associations between rs5848 and increased risk of neurodegenerative diseases in both Caucasians and Asians; however, only two studies of Asians were included. The explanation for the limited evidence on Asians should be treated with caution.
AD is associated with impaired clearance of β-amyloid from the brain, a process normally facilitated by apolipoprotein E (APOE). The ε4 variant of APOE is a major risk for AD [34]. A recent study by Lee et al. [24] has demonstrated that rs5848 TT of the PGRN genotype increases the risk of AD in a Taiwanese population; interestingly, this association is independent of the APOE ε4 allele. Experimental studies have shown that the PGRN level is significantly correlated with amyloid load in mouse models of AD [35]. Furthermore, the T allele of rs5848 has been shown to lead to decreased levels of PGRN and might be a risk factor for hippocampal sclerosis in patients with AD [36]. Therefore, the APOE and PGRN proteins may modulate the pathogenesis of AD via different pathways.
Previously, Rademakers and colleagues reported an association between rs5848 and frontotemporal dementia in a homogeneous cohort of an autopsy-confirmed FTD-U series (FTD with cortical ubiquitin-only neuropathology) [15]. However, the study was not confirmed in a larger population or in other cohorts [17, 25]. By pooling 5 studies, we found no association of rs5848 with FTLD. Actually, there are discrepancies concerning the role of this SNP in sporadic FTLD; for example, the diagnosis was based on a less heterogeneous autopsy-confirmed FTD-U series in the study by Rademakers [15], while the later studies lacked an autopsy-proven diagnosis [17, 25].
Progranulin is a secreted growth factor and regulates multiple physiological and pathological processes, including tissue repair, tumorigenesis, inflammation, and embryonic brain development [37]. Dysregulation of the PGRN level can lead to neurodegeneration or cancer [38]. Although the role of PGRN in the development of neurodegenerative diseases has not been fully characterized, the precise regulation of PGRN level plays a crucial role in maintaining proper neuronal morphology and the connections between neurons [39]. rs5848 is one of the PGRN variants that can regulate PGRN levels by shifting the miR-659 binding site [15]. Carriers homozygous for the T allele of rs5848 have a 3.2-fold increased risk of developing FTLD compared with homozygous C allele carriers [15]. With regard to the mechanism, miR-659 can regulate PGRN expression by binding more efficiently to the high-risk T allele, resulting in augmented translational inhibition of PGRN [15]. The present meta-analysis demonstrated that the rs5848 polymorphism was associated with increased risks of AD and PD, suggesting that this polymorphism is a promising predictor for the diagnosis of neurodegenerative diseases as well as a drug target of miR-659 in these diseases.
The major limitation of this meta-analysis is that we only pooled studies on the association of rs5848 with neurodegenerative diseases. Previous studies have examined the associations between other PGRN polymorphisms (e.g., rs9897526, rs850713, and rs25646) and the risks of different neurodegenerative diseases, and the data are inconsistent [18, 20, 25, 26, 29, 31, 32]. Due to the limited number of studies, we did not evaluate the effects of other PGRN polymorphisms on the risks of different neurodegenerative diseases (see Supplementary Data Tables). Another limitation is that our meta-analysis did not include genome-wide association studies due to lack of sufficient data on the genotype frequency of rs5848 for cases and controls. However, the publication bias analyses with the Egger’s and Begg’s tests showed no evident bias, suggesting that we collected sufficient published data. On the other hand, previous reports have suggested a female gender bias in the risk association of rs5848 with PD [16], but a male-oriented bias in the risk association for AD [29]. We did not discern a gender effect in the present meta-analysis due to limited data.
In summary, we for the first time performed meta-analysis by pooling all studies, and found that rs5848 is associated with an increased risk of AD and PD in homozygous and recessive models, suggesting an important role of PGRN in neurodegenerative processes.
References
Brown RC, Lockwood AH, Sonawane BR (2005) Neurodegenerative diseases: an overview of environmental risk factors. Environ Health Perspect 113:1250–1256
Lill CM, Bertram L (2011) Towards unveiling the genetics of neurodegenerative diseases. Semin Neurol 31:531–541
Lagier-Tourenne C, Cleveland DW (2009) Rethinking ALS: the FUS about TDP-43. Cell 136:1001–1004
Rohrer JD, Guerreiro R, Vandrovcova J, Uphill J, Reiman D et al (2009) The heritability and genetics of frontotemporal lobar degeneration. Neurology 73:1451–1456
Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R et al (2006) Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 442:916–919
Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H et al (2006) Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature 442:920–924
Schymick JC, Yang Y, Andersen PM, Vonsattel JP, Greenway M et al (2007) Progranulin mutations and amyotrophic lateral sclerosis or amyotrophic lateral sclerosis-frontotemporal dementia phenotypes. J Neurol Neurosurg Psychiatry 78:754–756
Brouwers N, Nuytemans K, van der Zee J, Gijselinck I, Engelborghs S et al (2007) Alzheimer and Parkinson diagnoses in progranulin null mutation carriers in an extended founder family. Arch Neurol 64:1436–1446
Ward ME, Miller BL (2011) Potential mechanisms of progranulin-deficient FTLD. J Mol Neurosci 45:574–582
Perry DC, Lehmann M, Yokoyama JS, Karydas A, Lee JJ et al (2013) Progranulin mutations as risk factors for Alzheimer disease. JAMA Neurol 70:774–778
Cannon A, Fujioka S, Rutherford NJ, Ferman TJ, Broderick DF et al (2013) Clinicopathologic variability of the GRN A9D mutation, including amyotrophic lateral sclerosis. Neurology 80:1771–1777
Rovelet-Lecrux A, Deramecourt V, Legallic S, Maurage CA, Le Ber I et al (2008) Deletion of the progranulin gene in patients with frontotemporal lobar degeneration or Parkinson disease. Neurobiol Dis 31:41–45
Gass J, Cannon A, Mackenzie IR, Boeve B, Baker M et al (2006) Mutations in progranulin are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Hum Mol Genet 15:2988–3001
Cruts M, Van Broeckhoven C (2008) Loss of progranulin function in frontotemporal lobar degeneration. Trends Genet 24:186–194
Rademakers R, Eriksen JL, Baker M, Robinson T, Ahmed Z et al (2008) Common variation in the miR-659 binding-site of GRN is a major risk factor for TDP43-positive frontotemporal dementia. Hum Mol Genet 17:3631–3642
Chang KH, Chen CM, Chen YC, Hsiao YC, Huang CC et al (2013) Association between GRN rs5848 polymorphism and Parkinson’s disease in Taiwanese population. PLoS One 8:e54448
Rollinson S, Rohrer JD, van der Zee J, Sleegers K, Mead S et al (2011) No association of PGRN 3′UTR rs5848 in frontotemporal lobar degeneration. Neurobiol Aging 32:754–755
Fenoglio C, Galimberti D, Cortini F, Kauwe JS, Cruchaga C et al (2009) Rs5848 variant influences GRN mRNA levels in brain and peripheral mononuclear cells in patients with Alzheimer’s disease. J Alzheimers Dis 18:603–612
Jasinska-Myga B, Wider C, Opala G, Krygowska-Wajs A, Barcikowska M et al (2009) GRN 3′UTR+ 78 C>T is not associated with risk for Parkinson’s disease. Eur J Neurol 16:909–911
Del Bo R, Corti S, Santoro D, Ghione I, Fenoglio C et al (2011) No major progranulin genetic variability contribution to disease etiopathogenesis in an ALS Italian cohort. Neurobiol Aging 32:1157–1158
Hayashino Y, Noguchi Y, Fukui T (2005) Systematic evaluation and comparison of statistical tests for publication bias. J Epidemiol 15:235–243
Begg CB, Mazumdar M (1994) Operating characteristics of a rank correlation test for publication bias. Biometrics 50:1088–1101
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B57:289–300
Lee MJ, Chen TF, Cheng TW, Chiu MJ (2011) rs5848 variant of progranulin gene is a risk of Alzheimer’s disease in the Taiwanese population. Neurodegener Dis 8:216–220
Galimberti D, Fenoglio C, Cortini F, Serpente M, Venturelli E et al (2010) GRN variability contributes to sporadic frontotemporal lobar degeneration. J Alzheimers Dis 19:171–177
Sleegers K, Brouwers N, Maurer-Stroh S, van Es MA, Van Damme P et al (2008) Progranulin genetic variability contributes to amyotrophic lateral sclerosis. Neurology 71:253–259
Nuytemans K, Pals P, Sleegers K, Engelborghs S, Corsmit E et al (2008) Progranulin variability has no major role in Parkinson disease genetic etiology. Neurology 71:1147–1151
Kamalainen A, Viswanathan J, Natunen T, Helisalmi S, Kauppinen T et al (2013) GRN variant rs5848 reduces plasma and brain levels of granulin in Alzheimer’s disease patients. J Alzheimers Dis 33:23–27
Viswanathan J, Makinen P, Helisalmi S, Haapasalo A, Soininen H et al (2009) An association study between granulin gene polymorphisms and Alzheimer’s disease in Finnish population. Am J Med Genet B Neuropsychiatr Genet 150B:747–750
Simon-Sanchez J, Seelaar H, Bochdanovits Z, Deeg DJ, van Swieten JC et al (2009) Variation at GRN 3′-UTR rs5848 is not associated with a risk of frontotemporal lobar degeneration in Dutch population. PLoS One 4:e7494
Xiao S, Sato C, Kawarai T, Goodall EF, Pall HS et al (2008) Genetic studies of GRN and IFT74 in amyotrophic lateral sclerosis. Neurobiol Aging 29:1279–1282
Brouwers N, Sleegers K, Engelborghs S, Maurer-Stroh S, Gijselinck I et al (2008) Genetic variability in progranulin contributes to risk for clinically diagnosed Alzheimer disease. Neurology 71:656–664
van der Zee J, Le Ber I, Maurer-Stroh S, Engelborghs S, Gijselinck I et al (2007) Mutations other than null mutations producing a pathogenic loss of progranulin in frontotemporal dementia. Hum Mutat 28:416
Slooter AJ, Cruts M, Kalmijn S, Hofman A, Breteler MM et al (1998) Risk estimates of dementia by apolipoprotein E genotypes from a population-based incidence study: the Rotterdam Study. Arch Neurol 55:964–968
Pereson S, Wils H, Kleinberger G, McGowan E, Vandewoestyne M et al (2009) Progranulin expression correlates with dense-core amyloid plaque burden in Alzheimer disease mouse models. J Pathol 219:173–181
Dickson DW, Baker M, Rademakers R (2010) Common variant in GRN is a genetic risk factor for hippocampal sclerosis in the elderly. Neurodegener Dis 7:170–174
He Z, Bateman A (2003) Progranulin (granulin-epithelin precursor, PC-cell-derived growth factor, acrogranin) mediates tissue repair and tumorigenesis. J Mol Med (Berl) 81:600–612
Bateman A, Bennett HP (2009) The granulin gene family: from cancer to dementia. Bioessays 31:1245–1254
Gass J, Prudencio M, Stetler C, Petrucelli L (2012) Progranulin: an emerging target for FTLD therapies. Brain Res 1462:118–128
Acknowledgments
This study was supported by the Qianjiang Talents Program of Zhejiang Province (2013R10041) and the Program for the Cultivation of Younger Innovative Health Talents, Zhejiang Province Health Bureau (2013-137). The authors are grateful to Drs. Christine Van Broeckhoven and Kristel Sleegers, University of Antwerp, Belgium, for providing the frequencies of rs5848 of subjects.
Conflicts of interest
The authors declare that they have no conflict of interest.
Ethical standard
The manuscript does not contain clinical studies or patient data.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Chen, Y., Li, S., Su, L. et al. Association of progranulin polymorphism rs5848 with neurodegenerative diseases: a meta-analysis. J Neurol 262, 814–822 (2015). https://doi.org/10.1007/s00415-014-7630-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-014-7630-2