
Abstract
DNA double-strand breaks arise accidentally upon exposure of DNA to radiation and chemicals or result from faulty DNA metabolic processes. DNA breaks can also be introduced in a programmed manner, such as during the maturation of the immune system, meiosis, or cancer chemo- or radiotherapy. Cells have developed a variety of repair pathways, which are fine-tuned to the specific needs of a cell. Accordingly, vegetative cells employ mechanisms that restore the integrity of broken DNA with the highest efficiency at the lowest cost of mutagenesis. In contrast, meiotic cells or developing lymphocytes exploit DNA breakage to generate diversity. Here, we review the main pathways of eukaryotic DNA double-strand break repair with the focus on homologous recombination and its various subpathways. We highlight the differences between homologous recombination and end-joining mechanisms including non-homologous end-joining and microhomology-mediated end-joining and offer insights into how these pathways are regulated. Finally, we introduce noncanonical functions of the recombination proteins, in particular during DNA replication stress.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Overview of DNA double-strand break repair pathways
Formation and types of DNA breaks
Our DNA is under a constant threat of damage from radiation, chemicals, or aberrant DNA metabolic processes (Jackson and Bartek 2009; Ciccia and Elledge 2010; Tubbs and Nussenzweig 2017). A large fraction of DNA lesions involve modifications of DNA bases, such as oxidation, ultraviolet light-induced pyrimidine dimers, methylation, or creation of abasic sites, which leave the phosphodiester backbone intact. Other abnormalities result in the disruption of the phosphodiester backbone. The most common of these are DNA single-strand breaks (SSBs), where only one DNA strand is interrupted (Fig. 1a). SSBs normally do not compromise the integrity of double-stranded DNA (dsDNA). However, if an SSB is left unrepaired and the lesion is encountered by DNA machinery that separates the DNA duplex into two-component single strands (ssDNA), such as during DNA replication, an SSB can be converted into a one-ended DNA double-strand break (DSB) (see Fig. 1b). Both SSBs and DSBs can arise as a result of ionizing radiation (IR), which may occur either directly or indirectly via generation of reactive oxygen species (Ward 1988). As a result, radiation-induced DNA damage results in complex lesions, where both SSBs and DSBs are accompanied by oxidative DNA damage (Olive et al. 1990; Olive 1998). The most common source of accidental IR exposure is the radioactive radon gas that accumulates in certain locations in the basements of old homes (Jackson and Bartek 2009). IR remains one of the most effective treatments during anticancer therapy, as it preferentially affects rapidly dividing cancer cells (Jackson and Bartek 2009; Baskar et al. 2014). SSBs and DSBs also arise during aberrant DNA topoisomerase reactions, which can occur spontaneously or upon exposure to specific inhibitors that are often used as anticancer chemotherapeutics (Holm et al. 1989; Canela et al. 2017). Finally, DNA breaks under certain conditions result from a nuclease attacking diverse DNA intermediates, including stalled DNA replication forks. As will be introduced in the “Role of recombination proteins in promoting the stability of DNA replication forks” section, DSB-like structure may also arise during a process termed replication fork reversal without template DNA breakage.

An overview of DNA breaks. a A single-stranded DNA (ssDNA) break arises when only one strand of double-stranded DNA is interrupted. b If an ssDNA break is encountered by DNA replication, it gives rise to a one-ended double-stranded DNA (dsDNA) break. c A two-ended dsDNA break forms when dsDNA is broken into two pieces
Depending on the mechanism of break formation, a DSB can either be one-ended or two-ended (Fig. 1b, c). Most one-ended DSBs arise when DNA replication encounters an SSB and the replication fork falls apart, or when a DNA replication fork stalls and one of the arms is cleaved by a nuclease. Two-ended DSBs typically form when both strands of linear dsDNA are broken simultaneously, or when two ssDNA breaks form in an immediate proximity; in this case, the broken end would contain a short stretch of overhanging ssDNA. Additionally, depending on the mechanism of the DSB formation, DNA ends may be either chemically “clean” or “dirty.” The so-called clean DNA breaks, apart from the broken phosphodiester backbone, bear normal DNA chemistry. Dirty DNA ends instead contain additional adducts that may include anything from small chemical groups to covalently attached proteins. DNA breaks induced by DNA nucleases are generally clean, but many DSBs induced by ionizing radiation are dirty (Olive et al. 1990). Likewise, DSBs induced by aberrant DNA topoisomerase reactions result in a covalently attached DNA topoisomerase at the break end (Tse et al. 1980). As will be described below, the mechanism of DSB repair depends largely on whether the break is one- or two-ended, chemically clean or dirty, as well as the cell cycle stage.
Our cells have developed numerous mechanisms for repairing DSBs with a minimal loss of genetic information. However, if a DSB is repaired incorrectly, it can lead to mutations and chromosomal rearrangements, resulting in aberrant regulation of cellular growth and cancer development or even cell death (Jackson and Bartek 2009). Therefore, accurate recognition and repair of DSBs is essential to maintain genomic integrity and prevent tumorigenesis.
End-joining and homologous recombination mechanisms repair DNA double-strand breaks
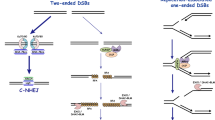
Eukaryotic cells use two main processes for DSB repair: end-joining and homologous recombination (HR) (see Fig. 2a). The end-joining pathways can be further divided into canonical non-homologous end-joining (NHEJ) and alternative non-homologous end-joining (alt-NHEJ), also termed microhomology-mediated end-joining (MMEJ). The MMEJ abbreviation will be used hereafter in this review. As the name indicates, NHEJ and MMEJ involve the direct ligation of two DSB ends with little or no sequence homology required (see below for details) (Chang et al. 2017). Therefore, a key feature of end-joining is that a repair template, such as the sister chromatid, is not required, so it can occur during any phase of the cell cycle. Both NHEJ and MMEJ processes typically lead to a limited loss of genetic information resulting in short deletions at the DSB site. Additionally, because NHEJ and MMEJ mechanisms are template independent, ligation of the incorrect ends, if multiple DSBs are present, can generate large deletions or chromosomal rearrangements (Chang et al. 2017). The end-joining pathways can only repair two-ended DSBs, and abnormal structures at the break sites (“dirty” ends, especially protein blocks) may inhibit this type of repair. In summary, end-joining pathways represent fast but potentially mutagenic DSB repair processes (Fig. 2a).

An overview of the two main pathways for DNA double-strand break repair in human cells. a Main differences between end-joining and homologous recombination pathways. b DNA double-strand break repair pathway usage gives rise to different outcomes during genome editing with CRISPR-Cas9. Whereas end-joining often results in random mutations in the vicinity of the break site that may disrupt the reading frame of the targeted gene, homologous recombination may mediate the precise replacement of genetic information
In contrast, HR requires a homologous sequence as a template for repair (Kowalczykowski 2015). This allows the recombination machinery to restore any missing genetic information in the vicinity of the break site, and as a result, HR is largely accurate. In most cases in vegetatively growing cells, the sister chromatid is used as the repair template. This restricts recombination to cell cycle stages when the sister chromatid is available, which includes the S and G2 phases, and thus necessitates a strict control mechanism (Orthwein et al. 2015). HR is capable of repairing both one- and two-ended DSBs and can also repair dirty DNA breaks, in particular those with covalently attached proteins. In contrast to end-joining, HR is mechanistically more complicated, involves a larger number of enzymes, and is thus comparatively slower but more accurate (Kowalczykowski 2015; Chang et al. 2017).
Recent years brought breakthroughs in genome editing technologies, which were spearheaded by the development of engineered nucleases such as zinc finger nucleases (ZFNs) or transcription-activator like effector nucleases (TALENs) (Lombardo et al. 2007; Bedell et al. 2012). The majority of genome editing applications now exploit the bacterial clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 system (Jinek et al. 2012; Mali et al. 2013). The common denominator of these approaches is the capacity to induce a site-specific DSB. The choice of the DSB repair pathway then dictates the result of editing (Fig. 2b). Imprecise repair by NHEJ or MMEJ gives rise to “indel” mutations (insertion or deletions, although deletions are much more common) at the break site, which may disrupt the reading frame of the targeted gene and thus result in a loss of function. Conversely, if a DNA template is provided, the recombination machinery may get involved, which can mediate precise alteration of the DNA sequence, including introduction of DNA segments or correction of a pathogenic mutation (Fig. 2b). The advance of these genome editing technologies brought renewed interest in understanding the balance between the DSB repair pathways, as the inhibition of MMEJ and NHEJ repair promotes HR-based precise genome editing (Chu et al. 2015; Mateos-Gomez et al. 2017; Schimmel et al. 2017; Zelensky et al. 2017).
The key process that stands at the crossroads between end-joining and HR is the initial processing of the DNA break (Cejka 2015). NHEJ and MMEJ require little DNA end processing (see “Processing of DNA breaks for repair” section for more details). In contrast, HR (including all its subpathways) is initiated by DNA resection at the break site that exposes long tracts of ssDNA. This ssDNA is then used in search for a homologous dsDNA sequence (such as the sister chromatid) that serves as a template for the largely accurate repair of the DSB by the recombination pathway. At the same time, extended DNA end resection makes the DNA break generally nonligatable and inhibits end-joining. Therefore, extensive DNA end resection commits DSB repair to the HR-mediated pathway, and accordingly, the initiation of DNA end resection is strictly controlled (Symington and Gautier 2011; Chapman et al. 2012; Shibata 2017). This control mechanism allows HR to be initiated only when a repair template is available (S/G2 phase), and thus limits the potential for illegitimate recombination (i.e., recombination between not fully homologous DNA sequences). This is elegantly achieved through the activation of key resection factors by cyclin-dependent kinase (CDK)-catalyzed phosphorylation (Ira et al. 2004; Huertas et al. 2008). It should be pointed out that this simple model has been challenged, and there is evidence that limited or even extended DNA end resection, occurring in the G1 phase, is involved in the canonical NHEJ pathway (Biehs et al. 2017). Elucidating the details and the regulation of these processes will be an exciting direction of future research. Since misregulation of these DSB repair pathways is believed to result in genome rearrangements that are typical in many cancer types, understanding these processes is highly relevant for human health.
End-joining and recombination processes involve several subpathways
Both HR and end-joining processes are not simple linear pathways. Both can be divided into several subpathways, which significantly differ in terms of repair mechanisms and enzyme requirements. Here, we will introduce the basic principles of these repair processes; a more detailed description that includes the key enzymatic players will be provided in subsequent sections.
With regard to the end-joining mechanisms, canonical NHEJ significantly differs from MMEJ (Fig. 3). Whereas canonical NHEJ requires no or very limited homology (less than 4 nt) between the broken DNA molecules, MMEJ was found as a DNA end-joining event that occurs independently of the key NHEJ factors and usually involves short stretches of microhomology (2–20 nt) between the two broken DNA ends to mediate repair (Seol et al. 2017). Therefore, MMEJ is sometimes considered a separate process that stands between the NHEJ and HR pathways.

An overview of DNA end-joining repair mechanisms. a Overview and main factors of non-homologous end-joining. b Overview and main factors of microhomology-mediated end-joining
Recombination processes, in a broad sense, can be divided into single-strand annealing (SSA), synthesis-dependent strand annealing (SDSA), break-induced replication (BIR), and canonical HR (also called canonical DNA double-strand break repair, DSBR) (Kowalczykowski 2015). The main conceptual differences between the mechanisms of these subpathways are schematically illustrated in Fig. 4. Depending on whether the flanking sequences of the recombining DNA molecules are exchanged or not, recombination leads to crossover or noncrossover recombination products. A crossover is defined as an event where the distal arm of the broken DNA is swapped with the distal arm of the template DNA molecule. As schematically depicted in Fig. 4d, a crossover results in the “blue” DNA molecule ultimately joined with the “red” one. Crossovers that occur between two homologous loci of sister chromatids give rise to “equal” sister chromatid exchanges, which are mutagenically silent (Fig. 4). In contrast, crossovers that occur between two ectopic loci (non-homologous loci, such as in repetitive sequences) of sister chromatids give rise to “unequal” sister chromatid exchanges. Likewise, crossovers resulting from recombination between nonsister (homologous) chromosomes also lead to gross genome rearrangements. Furthermore, events when genetic information from one DNA molecule gets unidirectionally transferred into another DNA molecule are referred to as gene conversions. This occurs when a DNA sequence is copied from a donor template to the broken DNA molecule during BIR, SDSA, or canonical HR. As above, gene conversion can be mutagenic when an ectopic site of a sister chromatid or nonsister chromosome is used as a template. Mutations arising in the course of DSB repair by either NHEJ or HR may give rise to a loss of heterozygosity, which is a key driver of tumorigenesis (Fearon and Vogelstein 1990).

An overview of homologous recombination pathways. A schematic representation of a single-strand annealing, b synthesis-dependent strand annealing, c break-induced replication, and d canonical DNA double-strand break repair pathway that involves generation of a double Holliday junction, which can be processed by either topologic dissolution (d1) or nucleolytic resolution (d2). The various pathways differ in terms of mutagenic potential and whether they lead to crossover or noncrossover products, as indicated. The green triangles indicate DNA replication sites. Newly synthesized DNA is illustrated using dashed lines
SSA involves DNA end resection to reveal repetitive DNA sequences, which are subsequently annealed. The resulting DNA flaps are cleaved and the strands are ligated. SSA leads to the deletion of the DNA sequence between the two repeats and is thus a very mutagenic repair process (Fig. 4a). SSA is restricted to situations when two repeats flank the break site, and can successfully restore the integrity of DSBs within repetitive DNA sequences. Although SSA is conceptually similar to MMEJ, it is usually grouped together with recombination mechanisms because of enzyme requirements (see below). Additionally, unlike MMEJ, SSA requires extensive DNA end resection, which is shared among all recombination subpathways (Bhargava et al. 2016).
In most cases, DNA end resection is followed by the invasion of the resected DNA into the dsDNA template, forming a joint molecule intermediate (Kowalczykowski 2015). This initiates DNA synthesis, which restores missing genetic information at the break site. In SDSA, the joint molecule intermediate is destabilized and the nascent DNA is annealed to the other end of the broken DNA molecule (Fig. 4b). SDSA is one of the least mutagenic recombination subpathways, resulting in a noncrossover repair product. In BIR, DNA synthesis proceeds all the way to the end of the template DNA, copying the sequence of the entire chromosome arm (Fig. 4c). BIR is thus a unique pathway for repair of one-ended DSBs resulting from collapsed DNA replication forks (Sakofsky and Malkova 2017). Additionally, BIR allows telomere lengthening in the absence of telomerase (Sakofsky and Malkova 2017). The genetic result of BIR is a nonreciprocal crossover.
In the canonical recombinational DSBR pathway, the joint molecule (D-loop) intermediate is processed into double Holliday junctions (dHJs), which may be processed into noncrossovers or crossovers, depending on the dHJ processing pathway utilized (see section “Processing recombination intermediates”) (Fig. 4d) (Szostak et al. 1983). Although the various repair events described above share some processing steps, they have different degrees of mutagenic potential. To date, many of the key repair factors for each pathway have been identified; however, how a cell determines which pathway to use for DSB repair is still poorly understood. The initial processing of broken DNA ends seems to be the key step that determines which pathway is used to repair a DSB, and will be described in the next section.
Processing of DNA breaks for repair
DNA end resection involves the degradation of the 5′-terminated DNA strand in the 5′ to 3′ direction from the break site to generate a 3′ ssDNA overhang. Generation of this 3′-terminated ssDNA is essential to allow for the usage of homologous DNA sequences for repair. The homology may be either between the two resected ends of the broken DNA molecule, such as in the case of MMEJ or SSA, or between the resected broken DNA molecule and an intact dsDNA template, such as in the case of BIR, SDSA, and canonical recombinational DSBR. Nucleolytic resection of DNA ends typically inhibits canonical NHEJ.
Processing of DNA ends for canonical NHEJ
Many canonical NHEJ events involve little or no processing of the broken DNA ends. The initial step of NHEJ involves the binding of the DNA ends by the Ku70-80 heterodimer, which forms a ring that encircles the duplex DNA (Gottlieb and Jackson 1993; Ramsden and Gellert 1998). This protects DNA ends from degradation and recruits additional NHEJ components (Fig. 3a). Next, the Ku70-80-bound ends are tethered by DNA-dependent protein kinase catalytic subunit (DNA-PKcs), followed by ligation of the broken DNA ends by the XRCC4-XLF complex and DNA ligase IV. The yeast Mre11-Rad50-Xrs2 (MRX) complex has a structural role to promote ligation, while the function of the human MRE11-RAD50-NBS1 (MRN) complex in NHEJ is less apparent (Chen et al. 2001; Huang and Dynan 2002; Zhang et al. 2007; Rass et al. 2009; Xie et al. 2009).
In the case of DNA ends that are not directly ligatable, which may include those with DNA overhangs, gaps, or blocking chemical groups, limited DNA end processing may be required. This involves a nucleolytic removal of overhangs or chemical groups by the human Artemis nuclease, which cleaves at the junctions of single- and double-stranded DNA, and is activated by DNA-PKcs (Ma et al. 2002; Chang et al. 2017; Lobrich and Jeggo 2017). Artemis may not be the only NHEJ nuclease; other proteins, including APLF, Werner’s syndrome helicase (WRN), the MRN complex, FEN1, and EXO1 may also play a role in some cases (Chang et al. 2017). Alternatively, the filling of DNA gaps at breaks may facilitate ligation, which is carried out by DNA polymerases μ and λ (Bebenek et al. 2014; Moon et al. 2014). Additionally, polynucleotide kinase (PNK) may remove 3′ phosphate groups and phosphorylate 5′ OH groups, which may be necessary for ligation (Chappell et al. 2002). Finally, one recent study found that extended DNA end resection can occur prior to NHEJ in G1 (Biehs et al. 2017), but further mechanistic analysis is required to fully understand this process.
Processing of DNA end for homologous recombination
In contrast to NHEJ, extended DNA end resection is an obligate step that initiates all recombination pathways. DNA end resection of the 5′-terminated DNA strand occurs in two main steps (Mimitou and Symington 2008; Zhu et al. 2008). The first step is catalyzed by the MRN complex and CtIP in human cells, and MRX and Sae2 in S. cerevisiae (Johzuka and Ogawa 1995; Keeney and Kleckner 1995; Paull and Gellert 1998; Sartori et al. 2007). The nucleolytic processing by these proteins is limited to the vicinity of the DNA end (generally up to 300 nt in yeast) and is thus referred to as short-range DNA end resection (Zhu et al. 2008). The most likely mechanism for the short-range resection by MRX/N is illustrated in Fig. 5. Resection is initiated by the endonucleolytic cleavage of the 5′-terminated DNA strand away from the DNA end, followed by 3′→5′ exonuclease that proceeds back toward the DNA end.

An overview of DNA end resection in human cells. The first (short-range) resection step involves the MRE11 nuclease and the second (long-range) step either EXO1 or DNA2 nuclease. DNA end resection leads to the generation of 3′ overhanged DNA at DNA double-strand break sites. The dashed blue lines indicate degraded DNA
Both exonuclease and endonuclease activities during the first resection step are likely catalyzed by MRE11/Mre11 in human and yeast cells (Neale et al. 2005; Garcia et al. 2011; Cannavo and Cejka 2014; Shibata et al. 2014). MRE11/Mre11 first endonucleolytically cleaves 5′-terminated DNA in the vicinity of the DNA end. This endonucleolytic cleavage requires the ATPase activity of RAD50/Rad50 as well as CtIP/Sae2 and NBS1 (but not strictly Xrs2 in yeast) as co-factors (Cannavo and Cejka 2014; Anand et al. 2016; Deshpande et al. 2016; Oh et al. 2016; Kim et al. 2017). Importantly, the capacity of Sae2 and CtIP to promote MRE11/Mre11 depends on phosphorylation of key residues in CtIP/Sae2, at least some of which are under cyclin-dependent kinase CDK control (Huertas et al. 2008; Huertas and Jackson 2009; Cannavo and Cejka 2014; Anand et al. 2016; Deshpande et al. 2016). Cell-cycle-dependent phosphorylation of CtIP/Sae2 represents one of the key control mechanisms that allow resection (and hence recombination) to initiate only in S and G2 phases of the cell cycle when a sister chromatid is available as a template for repair (Orthwein et al. 2015). Downstream of the endonuclease cut, MRE11/Mre11 subsequently uses its 3′→5′ exonuclease activity to proceed back toward the DNA end, generating a 3′ ssDNA overhang. Additionally, another nuclease, EXD2, may function alongside MRE11 exonuclease in human cells (Broderick et al. 2016). It has been also proposed that CtIP/Sae2 are nucleases (Lengsfeld et al. 2007; Makharashvili et al. 2014; Wang et al. 2014), but their potential catalytic functions in resection remain undefined.
The initial endonucleolytic cleavage away from the DNA end allows the resection machinery to bypass end-binding factors or noncanonical structures that may be present at the break end. This includes protein blocks, such as Spo11 in meiosis, stalled topoisomerases, or Ku (Keeney and Kleckner 1995; Keeney et al. 1997; Neale et al. 2005; Bonetti et al. 2010; Mimitou and Symington 2010; Langerak et al. 2011; Chanut et al. 2016). Indeed, the efficiency of 5′ DNA end cleavage in vitro by MRN-CtIP or MRX-Sae2 is stimulated by the presence of protein blocks at DNA ends (Cannavo and Cejka 2014; Anand et al. 2016; Deshpande et al. 2016). The short-range DNA end resection pathway is absolutely required for the processing of protein-blocked DNA ends but may be dispensable for the resection of clean DNA ends in yeast (Neale et al. 2005; Zhu et al. 2008; Mimitou and Symington 2010). Instead, both MRE11 and CtIP are required for all resection events in human cells, although it remains to be defined whether resection always depends on the nuclease of MRE11 (Sartori et al. 2007).
The initial endonucleolytic cleavage by MRE11/Mre11 creates entry sites for the long-range resection enzymes. These subsequently catalyze resection in the 5′→3′ direction away from the DNA end to generate extended ssDNA overhangs (up to several kilobases in length) and represent the second step of the resection process. The 3′→5′ exonucleolytic DNA degradation by MRE11/Mre11 and the 5′→3′ degradation by the long-range enzymes downstream of the endonucleolytic cut have been termed “bidirectional” resection. In addition to the nuclease function, the MRN/MRX complex has noncatalytic (i.e., structural) roles to recruit the long-range resection enzymes (Cejka et al. 2010a; Nicolette et al. 2010; Niu et al. 2010; Nimonkar et al. 2011). The long-range resection factors include either of two nucleases, EXO1/Exo1 or DNA2/Dna2, which are well conserved between human and yeast cells (Gravel et al. 2008; Mimitou and Symington 2008; Zhu et al. 2008). EXO1/Exo1 is a dsDNA-specific exonuclease (Tran et al. 2002), which specifically degrades the 5′-terminated DNA strand within dsDNA, generating 3′ ssDNA overhangs. In contrast, DNA2/Dna2 is an ssDNA-specific 5′→3′ nuclease that cannot process dsDNA on its own, and requires a cognate RecQ family helicase partner (Bae et al. 1998; Zhu et al. 2008; Levikova et al. 2013). This includes Sgs1 in yeast and either Bloom syndrome helicase (BLM) or WRN in human cells (Sturzenegger et al. 2014; Pinto et al. 2016; Levikova et al. 2017). Sgs1/BLM/WRN unwinds dsDNA to generate ssDNA, which becomes rapidly coated by replication protein A (RPA). RPA-coated ssDNA is subject to degradation by DNA2/Dna2 (Cejka et al. 2010a; Niu et al. 2010). RPA was found to promote 5′ DNA end degradation by DNA2/Dna2, while at the same time inhibiting 3′ end degradation. RPA, which physically interacts with the RecQ family helicase and DNA2/Dna2, is thus a critical factor that enforces the correct DNA polarity of DNA end resection by DNA2/Dna2 (Cejka et al. 2010a; Niu et al. 2010). Both human DNA2 and yeast Dna2, in addition to their essential nuclease activity, contain a helicase domain, which likely functions as an ssDNA translocase to facilitate the degradation of 5′-overhanged DNA by the DNA2/Dna2 nuclease (Levikova et al. 2017; Miller et al. 2017). The individual subunits of the Sgs1-Dna2-RPA, BLM-DNA2-RPA, and WRN-DNA2-RPA complexes stimulate the activities of their partners to form integrated molecular resection machines (Cejka et al. 2010a; Niu et al. 2010; Pinto et al. 2016).
Multiple DNA end resection mechanisms described above lead to the formation of 3′-tailed ssDNA coated by RPA. The key function of RPA is to protect ssDNA from the action of nucleases and prevent the formation of secondary structures that might arise by self-annealing of ssDNA (Wold 1997). As will be described in the section below, in SDSA, BIR, and canonical recombinational DSBR pathways, RPA must be replaced with the strand exchange protein RAD51. In contrast, SSA shares the initial DNA end resection with other recombination subpathways but is RAD51-independent. Instead of using intact dsDNA as a template, SSA functions by annealing the two resected strands of DNA, using large stretches of sequence homology to form a stable complex between the two resected broken DNA molecules (Fig. 4a). This is dependent on RAD52/Rad52, which has a capacity to anneal RPA-coated ssDNA. After annealing, the noncomplementary sequences are cleaved by XPF-ERCC1 (Rad1-Rad10 in yeast), and any remaining gaps are filled and ligated to complete the repair of the DSB (Bardwell et al. 1994; Ivanov et al. 1996; Mortensen et al. 1996; Shinohara et al. 1998).
Broken DNA molecules signal the presence of DNA damage to the cellular checkpoint machinery. Generally, DSBs activate the ATM (Tel1 in yeast) kinase. In both systems, MRN/MRX plays a structural role to activate ATM/Tel1 (Carson et al. 2003; Uziel et al. 2003; Lee and Paull 2004; Lee and Paull 2005). Upon resection, RPA-coated ssDNA then activates the ATR-ATRIP (Mec1-Ddc2 in yeast) pathway (Zou and Elledge 2003). ATM/Tel1 and ATR/Mec1 sensors phosphorylate hundreds of protein targets at SQ/TQ motifs, which activate the proper response to DNA damage. This includes regulation of DNA repair components and checkpoint proteins, leading to cell cycle arrest and thus providing time for repair (Jackson and Bartek 2009). Additionally, unsuccessful repair and prolonged cell cycle arrest lead to the activation of apoptosis in higher eukaryotes (Jackson and Bartek 2009).
Processing of DNA ends for alt-EJ (MMEJ)
In the absence of the key canonical NHEJ factors, including Ku70-80, DNA-PKcs, XRCC4, XLF, and DNA ligase IV, cells can still repair DSBs through an end-joining event referred to as MMEJ (Simsek and Jasin 2010; Chang et al. 2017) (Fig. 3b). The frequency of MMEJ in DSB repair is not very clear yet (Sfeir and Symington 2015), but it has been established that the process is more common in human cells compared to yeast. One of the hallmarks of MMEJ is the use of shared sequence microhomology between the break points, which is generally limited to ~ 2–20 nt in length (Chang et al. 2017). Overlapping homologies were observed in junction points upon translocations in human cancers (Stephens et al. 2009), suggesting that they may arise due to MMEJ. Currently, only a few factors have been implicated in mediating human MMEJ, including FANCA, PARP1, DNA ligase III, CtIP, DNA2, and Pol θ (Pol theta, also known as PolQ), but none of these appear to be absolutely essential for MMEJ (Audebert et al. 2004; Bennardo et al. 2008; Simsek et al. 2011; Howard et al. 2015; Mateos-Gomez et al. 2015, 2017). The use of microhomology for repair indicates that MMEJ and recombination-based mechanisms may share elements of DNA end resection. This is also supported by observations that Ku70-80 inhibits MMEJ, and Ku is known to be inhibitory for DNA end resection (Mimitou and Symington 2010). The end resection factors CtIP and DNA2 in human cells have been identified as being important to promote MMEJ events, but it is still unclear if this is due to a direct role in DNA end resection (Zhang and Jasin 2011; Howard et al. 2015). Likewise, the genetic requirements for MMEJ remain to be defined in yeast. Interestingly, MMEJ was increased in mre11, sae2, and sgs1 exo1 mutants, indicating that the absence of the canonical DNA end resection factors is not limiting for MMEJ (Wang et al. 2006; Deng et al. 2014). Whether another yet unidentified nuclease is essential for MMEJ is not clear. RPA is clearly a strong inhibitor of MMEJ in both yeast and human cells (Deng et al. 2014; Mateos-Gomez et al. 2017). This shows that the more extensive resection creating ssDNA of lengths that strongly associate with RPA channels repairs toward HR. In accord, the motor activity of human Pol θ was found to displace RPA to promote MMEJ, showing that the balance between HR and MMEJ is controlled by the opposing activities of Pol θ and RPA (Mateos-Gomez et al. 2017).
Formation of RAD51-ssDNA filament on resected DNA
Resection of the 5′-terminated DNA strand at DSBs leads to the formation of 3′-overhanged ssDNA, which is initially coated by RPA. In a subsequent step, RPA is replaced by the key recombination protein RAD51 (Rad51 in yeast) (Fig. 6). RAD51 and ssDNA form a nucleoprotein filament, also called a presynaptic filament. This catalyzes the signature step of the recombination pathway, which includes homology search, pairing with the intact donor (also called template) dsDNA and strand invasion (Benson et al. 1994; Sugiyama et al. 1997). The transient interaction of the RAD51 nucleoprotein filament with the template dsDNA is referred to as a synaptic complex. This ultimately leads to the displacement of one of the template DNA strands forming a displacement loop (D-loop, also called joint molecule) intermediate (Fig. 6), which represents the postsynaptic stage. All these steps are controlled (both positively and negatively) by a number of recombination regulators, which often physically interact with RAD51 and allow recombination to occur only in the proper context.

An overview of the RAD51 filament formation (presynaptic phase) and the invasion of template dsDNA (postsynaptic phase) in human cells. Both positive and negative regulators of the process are indicated
To understand the mechanisms underlying the regulation of RAD51, it is important to understand that RAD51 has a capacity to bind both single- and double-stranded DNA, but only ssDNA binding is thought to promote recombination, while dsDNA binding by RAD51 is generally inhibitory (Zaitseva et al. 1999). Furthermore, RAD51 binds and hydrolyzes ATP. ATP binding, but not ATP hydrolysis, by RAD51 is required for stable RAD51-DNA binding; in contrast, ATP hydrolysis by RAD51 is required for the steps downstream of DNA invasion (Sung 1994; Baumann et al. 1996; van Mameren et al. 2009). RAD51 can also hydrolyze ATP nonproductively (i.e., without catalyzing strand invasion); also in this case, ATP hydrolysis leads to a reduction in its capacity to bind DNA. Recombination can be stimulated on several levels by affecting these RAD51 activities. This includes RAD51 loading on RPA-coated ssDNA (i.e., exchange of RPA with RAD51), reduction of RAD51’s capacity to bind dsDNA, stabilization of the nascent nucleoprotein filament by inhibiting its disassembly, as well as remodeling of the RAD51 nucleoprotein filament into a conformation that is optimally permissive for DNA strand exchange. These control mechanisms are in place to prevent aberrant recombination (Heyer 2015). This includes recombination between non-homologous sequences, which may lead to DNA translocations and genome rearrangements, or recombination during physiological processes of DNA metabolism when ssDNA is present, such as during unperturbed DNA replication or transcription. The proper interplay of both positive and negative recombination regulators ascertains that recombination occurs only when it is needed to optimally maintain genome stability (Heyer 2015).
The first of the control mechanisms is the replacement of RPA with RAD51 on resected ssDNA. Due to the higher affinity toward ssDNA, RPA initially outcompetes RAD51 for ssDNA binding. To overcome this apparent inhibitory effect of RPA, cells have various recombination mediator proteins that help load RAD51 onto ssDNA, displacing RPA in the process. In yeast, the key recombination mediator that displaces RPA from ssDNA is Rad52. The interaction of Rad51 with Rad52 is required for this process (Benson et al. 1998; Song and Sung 2000). Human RAD52, in contrast, possesses no recombination mediator activity, and its contribution to recombination in human cells is much more subtle than in yeast, while Caenorhabditis elegans and Drosophila melanogaster lack RAD52. The main mediator protein in higher eukaryotes, including worms, flies, and humans, is the product of the breast cancer susceptibility gene 2 (BRCA2) and its homologs (Yang et al. 2005; Petalcorin et al. 2007; Jensen et al. 2010). Depletion of BRCA2 in cells treated with IR leads to persistent RPA and decreased RAD51 foci formation on broken DNA, clearly indicating its role in RAD51 loading (Yuan et al. 1999). BRCA2 contains eight BRC repeats (conserved motifs of about 35 amino acids), which can all independently interact with RAD51, although only four to five repeats associate with RAD51 at a given time (Carreira et al. 2009; Jensen et al. 2010). The repeats 1–4 bind RAD51 with a high affinity and promote RAD51 nucleation on ssDNA; repeats 5–8 show a lower affinity to free RAD51 and rather stimulate the growth of the nascent RAD51 filament (Jensen et al. 2010; Liu et al. 2010; Thorslund et al. 2010). Mechanistically, the BRC repeats 1–4 of BRCA2 promote ssDNA binding of RAD51 by inhibiting its ATPase activity, which stabilizes ssDNA binding of RAD51. Additionally, BRCA2 promotes recombination by inhibiting RAD51’s dsDNA binding activity (Jensen et al. 2010). The displacement of RPA from ssDNA by BRCA2 is further facilitated by DSS1, a direct interaction partner of BRCA2 (Yang et al. 2002; Zhao et al. 2015). BRCA2 also interacts with PALB2, which was shown to promote RAD51-mediated DNA strand exchange on its own (Buisson et al. 2010). In cells, PALB2 mediates BRCA2’s recruitment to DNA damage and bridges BRCA2’s interaction with BRCA1 (Sy et al. 2009). Cellular assays established that these interactions are critical for recombination, although the mechanisms on how BRCA1 and PALB2 proteins affect BRCA2’s recombination mediator activity remain to be defined. RAD51 nucleoprotein filament assembly is also stimulated by RAD54, independently of RAD54’s ATPase activity (Wolner and Peterson 2005). Additional proteins including the MMS22L-TONSL complex may promote the RAD51 nucleoprotein filament assembly during perturbed DNA replication (Piwko et al. 2016).
The activity of RAD51 is also promoted by a group of proteins termed RAD51 paralogs. The respective genes likely arose during evolution through a duplication of the RAD51 gene and share around 20 to 30% of sequence homology with RAD51. The paralogs are represented by five polypeptides in human cells: RAD51B, RAD51C, RAD51D, XRCC2, and XRCC3, which form two major complexes: RAD51B-RAD51C-RAD51D-XRCC2 (BCDX2) and RAD51C-XRCC3 (CX3) (Masson et al. 2001). Neither of the paralog proteins nor complexes exhibits DNA strand exchange activity on their own, and all likely function via regulating RAD51. Phenotypically, depletion of these complexes generally results in fewer RAD51 foci in response to ionizing radiation. This resembles, although to a lesser extent, the depletion of BRCA2. The paralogs likely function in the same pathway as BRCA2 as indicated by epistatic interactions (Qing et al. 2011). The first mechanistic report indicated that the RAD51B-RAD51C proteins, similar to BRCA2, have a recombination mediator activity to facilitate loading of RAD51 on RPA-coated ssDNA, displacing RPA in the process (Sigurdsson et al. 2001). In yeast, the RAD51 paralogs include Rad55 and Rad57 (Sung 1997; Liu et al. 2011a). Rad55-Rad57 physically interact with the Shu complex, comprising of Csm2, Psy3, Shu1 and Shu2 polypeptides, and Rad52. The subunits of the supercomplex synergize in their capacity to displace RPA and facilitate Rad51-ssDNA nucleoprotein filament formation (Gaines et al. 2015). The human ortholog of Shu2 is SWS1, which forms a complex with SWSAP1 (Martin et al. 2006; Liu et al. 2011b). The heterodimer binds DNA and interacts with RAD51, RAD51D, and XRCC2. Depletion of SWS1 results in a reduction of spontaneous and radiation-induced RAD51 foci. This suggests that SWS1 in humans also likely functions in conjunction with the RAD51 paralogs, but their interplay, as well as their relationship to BRCA2’s function, remains undefined. Evidence from the C. elegans model system instead suggests that the RAD51 paralogs, represented by RFS-1 and RIP-1, function downstream of BRCA2, specifically in the stabilization of the RAD51 nucleoprotein filament and its remodeling into a species that is optimally capable to invade template dsDNA (Taylor et al. 2015, 2016). RAD51 filaments are also stabilized by the SWI5-SFR1 complex (Akamatsu and Jasin 2010; Tsai et al. 2012).
It is well established that there is a balance between factors that promote the formation and disrupt RAD51/Rad51 nucleoprotein filaments (Heyer 2015). In yeast, Srs2 can dismantle Rad51 filaments due to its ATP-powered DNA translocase activity and additionally via stimulating the ATP hydrolysis of Rad51 through a direct physical interaction, which destabilizes the nucleoprotein filament (Krejci et al. 2003; Veaute et al. 2003; Liu et al. 2011a). In humans, no direct homolog of Srs2 has been identified, yet the most probable candidates for this function are RECQ5, FBH1, PARI, or BLM helicases (Bugreev et al. 2007b; Hu et al. 2007; Fugger et al. 2009; Schwendener et al. 2010; Moldovan et al. 2012; Patel et al. 2017). To elucidate the functional interplay of the various RAD51 regulators represents a challenge for future research. The proper balance between pro- and antirecombination factors is required to execute recombination only when it is needed and thus prevent illegitimate recombination and genome rearrangements.
Homology search and DNA strand exchange
Once the presynaptic filament is formed and stabilized, it begins the search for a homologous sequence. In most cases in vegetative cells, the sister chromatid is used as a repair template. A recent report demonstrated that the human BRCA1 protein, which is similar as BRCA2, a tumor suppressor that is frequently mutated in familiar breast and ovarian cancers, directly promotes DNA invasion. This likely occurs through promoting the assembly of the synaptic complex or promoting the homology search (Zhao et al. 2017). Despite the complex nuclear environment, the course of the homology search—the process that occurs immediately before the synaptic phase—is relatively fast. In yeast, it has been demonstrated that the mobility of a cut chromosome is increased, which allows the Rad51 filaments to explore a larger nuclear volume (Dion et al. 2012; Mine-Hattab and Rothstein 2012). Additionally, broken DNA relocalizes to the nuclear periphery (Oza et al. 2009; Chiolo et al. 2011; Horigome et al. 2014; Ryu et al. 2015). Although the exact mechanism of homology search is still undefined, it is suggested that the presynaptic filament randomly probes the genome by making multiple temporary contacts with different DNA duplexes (Forget and Kowalczykowski 2012; Renkawitz et al. 2014; Qi et al. 2015). Contacts with very short microhomologies are unstable; instead, contacts with more than 7 nt of homology are more stable, which allows the presynaptic complex to probe flanking sequences for additional homology. Once homology is identified, the presynaptic filament invades the duplex DNA, displaces the original strand, and binds its complementary sequence by Watson-Crick pairing. It has been estimated that the efficiency of repair in yeast was decreased to ~ 14% when template sequence diverged by about one in every eight nucleotides (Anand et al. 2017). Following strand invasion, the displaced ssDNA within the D-loop structure is stabilized by RPA, which prevents reversal of the D-loop formation (Lavery and Kowalczykowski 1992; Eggler et al. 2002). Additionally, RAD51 is removed from the heteroduplex dsDNA of the D-loop structure. This function is catalyzed by RAD54, a dsDNA translocase that uses its ATPase-powered motor activity to displace RAD51 from dsDNA (Solinger et al. 2002; Mason et al. 2015) (Fig. 6). RAD54 thus promotes D-loop stability, which allows DNA synthesis and facilitates canonical HR (Ceballos and Heyer 2011; Wright and Heyer 2014).
The 3′-terminated strands within D-loop structures that resist disassembly can prime DNA synthesis. Although translesion polymerases have been implicated in HR (Kawamoto et al. 2005; McIlwraith et al. 2005), most of DNA synthesis during recombination is likely catalyzed by either polymerase δ or polymerase ε (Li et al. 2009; Hicks et al. 2010; Wilson et al. 2013). However, the DNA synthesis during recombination is about three orders of magnitude more error-prone than in DNA replication, most likely due to limited activity of DNA mismatch repair during recombination-dependent repair DNA synthesis (Hicks et al. 2010). DNA synthesis extends the length of the paired duplex, which further stabilizes the joint molecule, leading to the recovery of the missing genetic information in the broken DNA molecule by using the invaded molecule as a template. Downstream of strand invasion and DNA synthesis, recombination proceeds into either of three recombination subpathways: BIR, SDSA, and canonical HR (see section “End-joining and recombination processes involve several sub pathways” for a general introduction to these pathways and Fig. 4). In BIR, the invaded DNA molecule is stabilized, and DNA synthesis proceeds along the whole length of the template DNA via a bubble-like structure to the chromosome end (Llorente et al. 2008; Sakofsky and Malkova 2017). The extended DNA synthesis is likely catalyzed by polymerase δ, in conjunction with the Pif1 helicase, which promotes the strand displacement activity of polymerase δ (Wilson et al. 2013). Interestingly, DNA synthesis during BIR occurs conservatively and thus dramatically differs from canonical DNA replication (Donnianni and Symington 2013). How exactly the synthesis of the complementary strand is achieved remains undefined. The BIR pathway occurs at the cost of elevated mutagenesis and is thus primarily utilized when no alternative is available, such as in the absence of the second DNA end.
In SDSA, the extended invaded DNA strand separates from the template DNA and anneals to the second end of the broken DNA molecule. DNA synthesis and ligation then complete the repair process. In canonical HR instead, through a process termed second DNA end capture, the second broken DNA end anneals to the displaced ssDNA strand of the D-loop structure (Fig. 4). The strand annealing employs the Rad52 protein in yeast (Nimonkar et al. 2009). Although human RAD52 also possesses the strand annealing activity (McIlwraith and West 2008; Jensen et al. 2010), the effects of RAD52 deficiency are modest and are particularly revealed only in the absence of BRCA2 (Feng et al. 2011). This raises questions whether the second end capture in human cells is mediated by annealing or a second DNA strand invasion event (Kowalczykowski 2015).
The balance between SDSA and canonical recombinational DSBR is regulated by activities that either disrupt or promote stability of the D-loop structure. In most cases, the stability of D-loops is dictated by motor proteins such as DNA helicases that act by moving the junctions. Mph1 in yeast cells and BLM, RECQ1, and RTEL1 in human cells have been implicated in D-loop dissociation and promotion of SDSA (Bugreev et al. 2007b, 2008; Barber et al. 2008; Prakash et al. 2009; Daley et al. 2013; Mitchel et al. 2013). In yeast, D-loops can also be disrupted or “dissolved” by an alternative mechanism that involves topoisomerase III (Top3), which is also thought to promote SDSA (Fasching et al. 2015). Whether this process also functions in human cells is not yet clear. Additionally, RAD54’s branch migration activity has been implicated to disrupt D-loops downstream of RAD51-mediated strand invasion in vitro (Bugreev et al. 2007a), but the biological significance of this function may be restricted to limiting recombination with non-homologous DNA (Ceballos and Heyer 2011; Wright and Heyer 2014). Regulating the balance between SDSA and HR is important, as it affects the genetic outcome of recombination. Whereas SDSA only leads to noncrossover products, canonical HR is a pathway that can potentially produce crossovers, as will be described in the next section.
Processing recombination intermediates
D-loop stability determines the pathway choice between SDSA and canonical recombinational DSBR. In SDSA, the D-loop is disrupted, whereas in recombinational DSBR, the D-loop is stabilized and becomes a substrate for annealing with the second resected dsDNA end (Fig. 4). This gives rise to a “double” or “complement-stabilized” D-loop. This structure forms as a result of annealing activity catalyzed by Rad52 in yeast and does not represent a second strand invasion step (Nimonkar et al. 2009). Rad52 was shown to promote annealing of ssDNA to D-loops generated by cognate Rad51 and Rad54 in the presence of RPA, indicating that the process likely requires direct protein-protein interactions. This function of Rad52 is apparent in unicellular eukaryotes, while RAD52 in human cells has a much less defined function (Feng et al. 2011). It is possible that in high eukaryotes, other proteins such as BRCA2 might be involved in second end capture in addition to RAD52. It remains to be established whether second end capture in human cells employs annealing and/or strand invasion mechanisms.
Following second end capture, DNA synthesis and ligation gives rise to a central intermediate of canonical HR, termed a dHJ (Duckett et al. 1988). As both DNA molecules are physically linked at the junction points, HJs need to be processed prior to separation of both DNA molecules (Fig. 7). A failure to process HJs leads to chromosome segregation defects and may be one of the mechanisms responsible for genome instability (Wechsler et al. 2011). Due to the homology between the recombining DNA molecules, a key feature of endogenous HJs is their mobility: the junction points can move in either direction to a limited extent spontaneously, or more extensively in ATP-hydrolysis-driven reactions catalyzed by molecular machines, such as DNA helicases or translocases. Double HJs can be processed by resolution or dissolution-based mechanisms. As will be described below, these processes are enzymatically distinct and lead to diverse genetic outcomes. Whereas dissolution leads to noncrossover products, resolution gives rise to both crossovers and noncrossovers (Figs. 4 and 7).

An overview of double Holliday junction processing mechanisms in the canonical recombinational DSBR pathway. In vegetative cells, double Holliday junctions can be processed by either dissolution (a) that involves helicase-coupled topoisomerase or by resolution (b) that involves nucleolytic cleavage of the junctions. Meiotic cells in most organisms preferentially use a dedicated crossover pathway (c), and to a smaller degree nucleases that are common to both meiotic and mitotic cells (b). For simplicity, human nomenclature is used. The involvement of nucleases listed in panels b and c in human (or mouse) meiotic cells generally remains to be defined. Yeast S. cerevisiae cells use a crossover-specific pathway that involves the Exo1-Mlh1-Mlh3 complex and the structure-specific nucleases Mus81-Mms4, Slx1-Slx4, and Yen1. See text for details
Dissolution of double Holliday junctions
Dissolution separates the recombining DNA molecules without exchanging the flanking sequences. As somatic cells employ mechanisms to maximally preserve genome integrity, dissolution is the default mode of dHJ processing. In yeast, dissolution is carried out by the Sgs1-Top3-Rmi1 (STR) complex (Cejka et al. 2010b), while in human cells, the “dissolvasome” consists of BLM, topoisomerase IIIα, and RMI1-RMI2, forming the BTRR complex (Wu and Hickson 2003; Singh et al. 2008; Xue et al. 2013). As Sgs1, BLM can unwind various DNA structures and branch migrate HJs in vitro (Wu and Hickson 2003). The dissolution reaction involves migration of the two Holliday junctions toward each other (i.e., convergent branch migration) by the combined activity of the RecQ family BLM/Sgs1 helicase and the strand passage activity of the TOPOIIIα/Top3 type IA topoisomerase (Fig. 7a). As both Holliday junctions link two DNA molecules of long lengths, the ends are not free to rotate, and the dHJ structure is topologically constrained. Therefore, a branch migration activity of Sgs1/BLM is not sufficient to migrate endogenous HJs (Cejka et al. 2010b; Chen et al. 2014). It has been demonstrated that the strand passage activity of TOPOIIIα/Top3 is required to relieve positive supercoiling that would otherwise form ahead of (between) the convergently migrating junctions and prevent further movement (Chen et al. 2014). Mechanistically, Top3/TOPOIIIα creates a transient nick in ssDNA that allows the other ssDNA strand to pass through it, thereby allowing the relaxation of the torsional stress forming between the junctions during convergent branch migration. RMI1/Rmi1 does not significantly affect the initial DNA branch migration step, but seems to specifically promote a late step just prior to dissolution (Cejka et al. 2010b; Bocquet et al. 2014). The last predicted intermediate of convergent branch migration is a hemicatenane: this structure represents a junction between two dsDNA molecules, where one strand of one duplex is wrapped around another strand from the second DNA duplex (Fig. 7a). It has been proposed that RMI1/Rmi1 specifically promotes the processing of a hemicatenane (Cejka et al. 2012). RMI2, which is only present in high eukaryotes, likely has only a minor function in dHJ dissolution and may have other roles such as to target BLM to blocked DNA replication forks (Singh et al. 2008). Finally, RPA, which physically interacts with BLM/Sgs1 and RMI1/Rmi1 subunits of the STR/BTRR complexes, also stimulates dissolution (Cejka et al. 2010b; Xue et al. 2013). This likely stems from RPA’s capacity to promote the helicase of BLM/Sgs1 and strand passage of TOPOIIIα-RMI1/Top3-Rmi1, likely through its ssDNA binding activity to prevent reannealing, as well as because of the direct physical interaction of RPA with RMI1/Rmi1 (Brosh et al. 2000; Plank et al. 2006; Cejka et al. 2012; Xue et al. 2013). The unique mechanism of dHJ processing by dissolution exclusively results in noncrossover events, which maintains genome stability in vegetative cells (Fig. 7a). BLM-deficient cells are characterized by elevated levels of sister chromatid exchanges, which are a hallmark of genome rearrangements and may contribute to cancer predisposition of Bloom syndrome patients (Chaganti et al. 1974). These rearrangements result from elevated usage of pathways that are alternative to dissolution and that are dependent on the structure-specific nucleases described in the next section.
Resolution of double Holliday junctions
Double HJs that evade dissolution are resolved by structure-specific nucleases later in the cell cycle to give rise to both crossover and noncrossover recombination products. Nucleolytic cleavage also represents the only option for the processing of single HJs. These may form during DSB repair where only one strand invades template DNA, or when one of the D-loop arms has been cleaved prior to second end capture (Wechsler et al. 2011; Shah Punatar et al. 2017). To date, three structure-specific nucleases capable of processing HJs or similar structures have been identified in eukaryotic cells, including human MUS81-EME1 and yeast Mus81-Mms4, human SLX1-SLX4 and yeast Slx1-Slx4 as well as human GEN1 and yeast Yen1 (Dehe and Gaillard 2017). The Mus81-Mms4 complex in yeast was found to be essential for viability in cells lacking Sgs1-Top3, indicating its role in the processing of recombination intermediates that would have otherwise been processed by the Sgs1-Top3 complex (Hickson and Mankouri 2011). Mus81 belongs to the XPF family of nucleases and represents the catalytic subunit of the heterodimer. Both yeast and human Mus81-Mms4/MUS81-EME1 complexes prefer to cleave replication forks, 3′ flaps, and nicked HJs (Gaillard et al. 2003; Ehmsen and Heyer 2008). MUS81-EME1/Mus81-Mms4 cleave intact HJs inefficiently in an asymmetric manner by introducing a nick a few nucleotides away from the junction, producing gapped and flapped DNA products, which are unsuitable for ligation and necessitate further processing (Wyatt et al. 2013). In human cells, MUS81 forms another complex with EME2, which has a broader substrate specificity than MUS81-EME1, and has roles in replication fork restart (Pepe and West 2014b). SLX1/Slx1 belongs to the GYI-YIG family of nucleases and is the catalytic subunit of the SLX1-SLX4 heterodimer. The SLX1-SLX4/Slx1-Slx4 complex can cleave branched structures with a preference toward Y-structures, 5′ flaps, and replication forks. While it can cleave intact HJs, the cleavage is inefficient and asymmetrical and creates poorly ligatable products (Fricke and Brill 2003; Fekairi et al. 2009; Svendsen et al. 2009; Wyatt et al. 2013). Both MUS81 and SLX1 complexes are thus noncanonical HJ resolvases. The only bona fide eukaryotic HJ resolvase is GEN1/Yen1, which belongs to the RAD2/XPG family of nucleases. As the Escherichia coli RuvC, GEN1/Yen1 dimerizes on HJs and cleaves HJs by a dual incision mechanism that produces two ligatable products (Dunderdale et al. 1991; Ip et al. 2008; Rass et al. 2010). However, both GEN1 and Yen1 are capable of cleaving other branched DNA structures as well. The random nature of HJ cleavage by the various nuclease complexes gives rise to both crossover and noncrossover recombination products (Shah Punatar et al. 2017) (Fig. 7).
In human cells, both the SLX1-SLX4 and MUS81-EME1 dimers associate to form the SLX1-SLX4-MUS81-EME1 tetramer (SM complex). The formation of the SM complex is mediated by the interaction between MUS81 and SLX4 polypeptides (Wyatt et al. 2013). Biochemical studies demonstrated that the SM complex exhibits higher HJ resolution activity compared to its individual subunits. Mechanistically, SLX1-SLX4 makes the first nick in the HJ creating a substrate for MUS81-EME1, which then makes a second counter-nick in the opposing strand, allowing for efficient HJ resolution (Castor et al. 2013; Wyatt et al. 2013). This activity is further enhanced by interaction with a third nuclease complex, the XPF-ERCC1 dimer, which has a structural (i.e., noncatalytic) role to promote HJ cleavage (Wyatt et al. 2017) (Fig. 7b). The catalytic subunit within the XPF-ERCC1 dimer is XPF, which has established functions in nucleotide excision repair. It should be pointed out that the functions of MUS81-EME1/Mus81-Mms4 and SLX1-SLX4/Slx1-Slx4 are not specific to processing recombination products, and both enzyme complexes may cleave structures arising during other DNA repair processes, as well as during replication or telomere maintenance. Likewise, the SLX1-SLX4-MUS81-EME1-XPF-ERCC1 (SMX) trinuclease complex has a broad substrate specificity that is not restricted to HJ cleavage and likely functions in multiple DNA metabolic pathways (Wyatt et al. 2017). The formation of the nuclease complexes appears specific to higher eukaryotes, as no such collaborative action of nucleases has been observed in yeast.
Regulation of Holliday junction processing
To maximally preserve genome stability, vegetative cells evolved mechanisms that facilitate dissolution pathway usage over resolution, which limits crossover formation. Additionally, the activity of the nucleases capable of cleaving branched DNA structures must be carefully controlled to avoid promiscuous DNA cleavage. The preferential employment of dissolution over resolution by structure-specific nucleases is governed by tight spatial and temporal control (Matos and West 2014). The STR/BTRR complex is likely active in any phase of the cell cycle and, thus, processes the majority of dHJs from S phase to mitosis in both yeast and human cells. The activity of yeast Mus81-Mms4 is instead low in S phase and becomes elevated at the onset of mitosis by phosphorylation of the Mms4 subunit by CDK and Cdc5 (Matos et al. 2011; Matos et al. 2013). In human cells, the phosphorylation of EME1 does not activate MUS81 directly but rather promotes the formation of the SLX1-SLX4-MUS81-EME1 (SM) complex with increased activity toward HJs (Wyatt et al. 2013). Yeast Yen1 is inactive in its phosphorylated state in S phase and becomes activated by Cdc14-mediated dephosphorylation late in mitosis. Interestingly, phosphorylation of Yen1 not only inhibits its catalytic activity by limiting its association with DNA but also prevents nuclear import (Kosugi et al. 2009; Blanco et al. 2014). Nuclear exclusion appears to be the primary mechanism that restricts human GEN1 activity (Chan and West 2014). The nuclear export sequence within GEN1 ascertains that GEN1 can process recombination intermediates only when the nuclear envelope breaks down during mitosis. These regulatory mechanisms collectively ascertain that the structure-specific nucleases are only activated late in the cell cycle to remove any residual junctions that were not processed by the dissolvasome complex. Although potentially mutagenic, efficient processing of all joint molecules by resolution mechanisms is essential to prevent chromosome missegregation in mitosis (Wechsler et al. 2011).
Specialized roles of DSB repair pathways
Cells evolved mechanisms to repair accidental DNA breaks to achieve maximal efficiency and accuracy in the maintenance of genome integrity. However, not all DSBs are pathological, and there are several cases when DSBs are introduced deliberately, which serves specific physiological purposes. The best examples are processes occurring during lymphocyte development and in meiosis. As will be seen below, during these events, cells make use of the DSB repair pathways to instead generate diversity. Both processes represent fascinating examples of the plasticity of the DSB repair systems.
DSB repair pathways in the immune system
V(D)J recombination is a process that occurs during B and T lymphocyte development and involves a random rearrangement of the variable (V), diversity (D), and joining (J) segments of immunoglobulin genes (Arya and Bassing 2017) (Fig. 8a). The random assembly of these VDJ segments allows producing a wide variety of antigen receptors from a limited number of gene segments. Despite its name, V(D)J recombination is facilitated by canonical NHEJ factors and does not involve homologous recombination. Disruption of key NHEJ factors results in severe immune disorders in humans and mice, indicating their essential role in V(D)J recombination. The MRN complex was found to have a function in V(D)J recombination, but this appears to be dependent on its capacity to promote checkpoint signaling via activation of the ATM kinase, and not a role in HR (Uziel et al. 2003; Helmink et al. 2009).

An overview of specialized roles of the DSB repair pathways in human lymphocytes. a Mechanism of V(D)J recombination, where DNA breaks are introduced by RAG1-RAG2. b Mechanism of class switch recombination, where DNA breaks are indirectly caused by the action of AID. In both cases, end-joining, but not homologous recombination pathways, is responsible for DNA break repair
V(D)J recombination is initiated by the recombination-activating genes 1 and 2 (RAG1 and RAG2), which are expressed in developing lymphocytes and therefore restrict this process to these cells (McBlane et al. 1995; Schatz and Swanson 2011). RAGs bind to the recombination signal sequences (RSS) flanking the V, D, and J gene segments and first create an ssDNA nick at the junction between the coding region and the RSS. There are two types of RS sequences, which differ with respect to the length of a spacer region between the two identical sequences. The spacers are of 12 or 23 bps in length, giving rise to RS12 and RS23 sequences (Fig. 8a). Next, RAGs catalyze the formation of a paired complex, where the RS12 and RS23 sequences associate with the same RAG complex. The free hydroxyl group of the nicked strand then invades the phosphodiester bond of the intact strand, generating a DSB with different DNA ends. The coding end of the DSB contains a hairpin loop, whereas the signal end is blunt. Next, the blunt signal ends are ligated together to form a circular piece of DNA that is lost during subsequent cell divisions. The hairpin coding ends are processed and joined by the NHEJ machinery to fuse the respective gene segments. First, Ku70-80 and DNA-PKcs bind to the coding ends and recruit other additional NHEJ factors, including Artemis, XRCC4-XLF, and DNA ligase IV (Fig. 8a). Next, the Artemis nuclease is activated upon autophosphorylation of DNA-PKcs and opens the hairpin loop of the coding ends, which can then be ligated to the other coding ends by the XRCC4-XLF and DNA ligase IV complex (Li et al. 1995; Casellas et al. 1998; Ma et al. 2002). The creation of DSBs with different ends and the requirement for association of the 12- and 23-RS sequences in human B and T cells are essential for regulated processing to ensure that coding sequences of V, D, and J, but not homotypic gene segments, are joined. The final product of V(D)J recombination is a DNA coding sequence consisting of randomly rearranged V, D, and J gene segments (Schatz and Swanson 2011).
Class switch recombination (CSR) and somatic hypermutation (SHM) only occur in activated germinal center B cells. CSR leads to the change of antibody isotype and thus its effector function, while SHM affects the variable regions of the immunoglobulin genes to promote diversity of antibodies. Both CSR and SHM are independent of RAGs and instead are triggered by activation-induced cytidine deaminase (AID) (Muramatsu et al. 2000; Arakawa et al. 2002; Petersen-Mahrt et al. 2002) (Fig. 8b). AID specifically deaminates cytosines into uracils, resulting in the formation of U:G mismatches. During CSR, the Cμ exon is exchanged by Cγ/ε/α exons, resulting in the change of the antibody isotype from IgM to IgG, IgE, or IgA (Methot and Di Noia 2017). Unlike RAGs that are targeted to very specific sequences, AID in CSR is active on large regions (1–3 kb) of repetitive DNA sequences upstream of CH genes (coding for constant regions of immunoglobulin heavy chains), called the switch (S) regions. The U:G mismatches are mainly processed by the base excision repair machinery (BER), which creates ssDNA breaks as intermediates of the repair process (Rada et al. 2002; Imai et al. 2003; Schrader et al. 2005). Closely spaced ssDNA breaks then give rise to staggered DSBs (DSBs with ssDNA overhangs) (Fig. 8b). Additionally, a smaller fraction of DNA breaks may form as a result of a noncanonical function of the DNA mismatch repair machinery, which involves the exonuclease activity of Exo1 (Ehrenstein and Neuberger 1999; Schrader et al. 2007; Bregenhorn et al. 2016). The exonucleolytic processing of one DNA strand then collides with a nick in the opposite DNA strand, leading to DSBs (Fig. 8b). Irrespectively of the exact mechanism, the resulting DSBs often contain ssDNA overhangs, which need to be either filled or cleaved prior to joining by the canonical NHEJ machinery. DSBs with overhangs bearing microhomologies may also be joined by the MMEJ pathway (Lee-Theilen et al. 2011). As with V(D)J recombination, CSR is also dependent on end-joining mechanisms, and it is thus not a homologous recombination process. Interestingly, MRN was found important for CSR, where it likely has a structural role to promote both canonical NHEJ and MMEJ pathways (Dinkelmann et al. 2009).
Somatic hypermutation is, as CSR, dependent on AID (Arakawa et al. 2002; Pham et al. 2003). In contrast to CSR, SHM leads to antibody diversification through mutagenesis in the V regions of light and heavy chains of immunoglobulins. It has been estimated that the mutation rate in the V regions during SHM is about 6 orders of magnitude higher than in the rest of the genome. The majority of mutations are single base substitutions, with a small fraction of short insertions or deletions. This suggests that the formation of a DSB is not an obligate step in SHM, and in fact NHEJ-deficient cells do not show significant defects in SHM. However, some DSBs form during SHM (Papavasiliou and Schatz 2000). To this point, it has been demonstrated that DSBs during SHM can be repaired by homologous recombination (Papavasiliou and Schatz 2000; Zan et al. 2003); furthermore, HR has been implicated as a safeguard against off target AID activity (Hasham et al. 2010; Zahn et al. 2014). Interestingly, MRN was found to promote SHM, but the underlying mechanisms remain unclear (Yabuki et al. 2005).
Finally, the insertion of DNA fragments of various sizes has been recently reported to occur in the switch region of immunoglobulin genes (Tan et al. 2016; Pieper et al. 2017). The DNA sequences originate from another part of the genome, and the transfer occurs via a copy-paste rather than a cut-paste process. This may represent a novel mechanism for antibody diversification, although the molecular pathways required for these transactions are unknown. Interestingly, insertions of fragments from the collagen receptor LAIR1 into immunoglobulin genes lead to antibodies that are broadly reactive to malaria (Tan et al. 2016; Pieper et al. 2017). It is important to point out that during B cell maturation, clones that produce high affinity antibodies are positively selected while low affinity clones are eliminated. Due to the selection process, even infrequent events may thus become physiologically relevant (Pieper et al. 2017).
Homologous recombination in meiosis
Meiosis is a specialized cell division that is required for genome haploidization (Keeney et al. 2014). This is essential to form spores or gametes in sexually reproducing unicellular or multicellular eukaryotes, and makes sure that ploidy is maintained with each successive generation (Fig. 9). DSBs are introduced during meiosis and become substrates for the homologous recombination machinery (Sun et al. 1989, 1991). The function of meiotic recombination in most organisms including yeast and vertebrates (but not Drosophila and Caenorhabditis) is to make physical connections between homologous chromosomes to facilitate their proper alignment and subsequent segregation. Additionally, recombination creates genetic diversity in the population by exchanging DNA regions between maternal and paternal chromosomes. The key mechanistic differences between HR in vegetative cells and during meiosis are (a) the formation of DNA breaks, which are induced in a programmed manner during meiosis; (b) the preferential usage of the homologous chromosome as a template for repair; and (c) a bias for preferential resolution of recombination intermediates into crossovers that are nonrandomly spaced (Fig. 9) (Hunter 2015).

An overview of the specialized roles of the homologous recombination pathway in meiosis. The main differences between recombination in meiotic and vegetative cells are indicated
DSBs are introduced during the first meiotic division by the SPO11/Spo11 transesterase, which is evolutionarily conserved from yeast to mammals (Keeney and Kleckner 1995; Keeney et al. 1997; Mahadevaiah et al. 2001; Robert et al. 2016). SPO11 is a topoisomerase-like protein that cleaves both strands of dsDNA, but remains covalently attached to the 5′-terminated DNA strand upon cleavage. In yeast, at least nine other proteins are required for Spo11 to cleave DNA, including the MRX complex (Keeney 2008). Interestingly, the nuclease activity of MRE11 is dispensable for DNA cleavage by Spo11 in yeast, but it is instead essential to initiate the processing of Spo11-bound DNA breaks (Borde et al. 2004; Lam and Keeney 2014). As with other protein-blocked DNA ends, the first resection step requires the nuclease activity of the MRN/MRX complex with its co-factor CtIP/Sae2 to remove SPO11/Spo11 from the break ends (Keeney and Kleckner 1995; Neale et al. 2005). Resection by MRX-Sae2 can proceed up to several hundreds of nucleotides in length. This is followed by long-range resection, which appears to be exclusively carried out by the Exo1 branch in yeast meiotic cells (Zakharyevich et al. 2010; Mimitou et al. 2017). In mice, the average resection length is 0.9 kb (Lange et al. 2016), which is similar to the value obtained in yeast (Zakharyevich et al. 2010).
The resected 3′ DNA tail is bound by RPA and successively replaced by DMC1 (a meiosis-specific strand exchange protein) together with RAD51 (Bishop et al. 1992; Cloud et al. 2012). Whereas RAD51 is the only strand exchange protein in vegetative cells, meiotic cells employ both RAD51 and DMC1. In addition to the recombination mediators such as yeast Rad52 and human BRCA2 that function during recombination in both vegetative cells and in meiosis, meiotic cells make use of an additional regulator, the HOP2-MND1 complex (Leu et al. 1998; Petukhova et al. 2005). The heterodimer stabilizes the RAD51/DMC1 complex on ssDNA, reduces the affinity of RAD51 to dsDNA, and modulates the conformation of the nucleoprotein filament to promote DNA strand exchange (Chi et al. 2007; Pezza et al. 2007).
The second key dissimilarity between meiotic and mitotic HR is the template choice for repair. Meiotic cells preferentially use the homologous chromosome instead of the sister chromatid in order to fulfill the requirement to generate genetic variability (Schwacha and Kleckner 1994; Baudat et al. 2000; Peoples et al. 2002). How this is achieved is not yet fully understood. The template bias is regulated by cohesion (Kim et al. 2010), depends in part on the role of DMC1 to promote strand exchange between homologs, and requires the DNA damage response cascade involving Mec1, Tel1, and meiosis-specific components such as Mek1 (Schwacha and Kleckner 1997; Niu et al. 2005; Carballo et al. 2008).
The third key characteristic of meiotic HR is a more frequent resolution of joint molecule intermediates such as dHJs or their precursors into crossovers. Most meiotic noncrossover products appear early and result from the processing of unstable intermediates by SDSA (Allers and Lichten 2001; Bishop and Zickler 2004). The joint molecules that can be readily observed are termed single-end invasions (containing presumably D-loop structures), which later mature into dHJs (Hunter and Kleckner 2001). Most of these joint molecules are resolved synchronously later, which is triggered by the phosphorylation of yet undefined targets by the Cdc5 kinase (Allers and Lichten 2001; Sourirajan and Lichten 2008). Although the molecular mechanism remains unclear, the preferential processing of joint molecule intermediates into crossovers is achieved by the usage of a meiosis-specific procrossover pathway, which is responsible for up to ~ 80% of meiotic crossovers in S. cerevisiae (Zakharyevich et al. 2012). This pathway ensures that every chromosome receives at least one crossover (positive crossover interference), thereby assuring the exchange of genetic information between the homologous chromosomes. At the same time, the formation of one crossover suppresses the formation of additional crossovers in its vicinity (negative crossover interference), making sure that the crossovers are evenly spaced, as two crossovers in immediate vicinity limit the exchange of genetic information between the homologous chromosomes. The proper number of crossovers and their distribution is thus balanced to enable efficient chromosome segregation and genetic exchange (Hunter 2015).
The exact mechanism of crossover interference remains to be defined. It is apparent that it is regulated by the components of the synaptonemal complex that juxtaposes homologs along their entire length (Sym and Roeder 1994; Hunter 2015). The interference signaling involves Tel1 (Garcia et al. 2015). Crossover interference also requires topoisomerase II, suggesting that interference may be regulated by mechanical or topological stress along the meiotic chromosome axes (Zhang et al. 2014). The meiotic crossover-specific pathway involves the ZMM group of proteins, which includes Zip1, Zip2, Zip3, Zip4, Spo16, Mer3, and Msh4-Msh5 in S. cerevisiae (Lynn et al. 2007; Shinohara et al. 2008). These proteins form or facilitate the formation of the synaptonemal complex, or function more downstream to stabilize D-loop structures in a way that protects their disassembly by motor proteins (Mazina et al. 2004; Snowden et al. 2004). Similarly in mice, the putative SUMO-conjugating factor RNF212 was found to stabilize meiotic recombination factors including MSH4-MSH5 at designated recombination sites, which is essential for crossing over. This is likely achieved via sumoylation of selected targets along the chromosome axes, which functionally interacts with HEI10-dependent ubiquitination and proteasomal degradation (Wei et al. 2003; Reynolds et al. 2013; Rao et al. 2017).
The crossover-specific resolution in budding yeast, plants, and mammals depends on the nuclease activity of Mlh3, which is a part of the Mlh1-Mlh3 heterodimer (Lipkin et al. 2002; Nishant et al. 2008; Zakharyevich et al. 2012; Ranjha et al. 2014; Rogacheva et al. 2014). Unexpectedly, Exo1 was found to have a noncatalytic function in crossover resolution together with Mlh1-Mlh3 (Zakharyevich et al. 2010). How the ZMM proteins and Mlh1-Mlh3 achieve the crossover-specific resolution of dHJs or their precursors remains to be described (Fig. 7c). Interestingly, only crossovers from the ZMM-Mlh1-Mlh3 pathway—termed class I crossovers—display interference. Joint molecules that are processed by the structure-specific nucleases (Mus81-Mms4, Slx1-Slx4, and Yen1 in yeast) result in both crossovers and noncrossovers. Crossovers resulting from these nucleases are interference independent and account for ~ 20% of crossovers in S. cerevisiae (de los Santos et al. 2003; Zakharyevich et al. 2012). In contrast, Mus81-Eme1 generates most meiotic crossovers in the budding yeast Schizosaccharomyces pombe, while the MEI-9, an XPF-like protein, resolves meiotic joint molecules in Drosophila (Boddy et al. 2001; Yildiz et al. 2002).
Role of recombination proteins in promoting the stability of DNA replication forks
In addition to repairing DSBs, recombination proteins have critical functions in other pathways of DNA metabolism. Specifically, recombination is one of the pathways that contribute to the repair of DNA cross-links, which has been covered elsewhere (Hinz 2010). Furthermore, recombination in multiple ways promotes the stability of replicating DNA (Branzei and Szakal 2017). Abnormalities such as protein blocks, chemical modifications, abnormal secondary structures, or collisions of replication forks with the transcription machinery can impede DNA replication. Depending on the nature of the replication stress, cells utilize various mechanisms that help them deal with these situations (Fig. 10a–d). This includes repriming of DNA replication and/or translesion DNA synthesis, which lead to lesion bypass. Postreplicative repair can fill any gaps left behind by the replication machinery (Bianchi et al. 2013; Garcia-Gomez et al. 2013; Mouron et al. 2013; Branzei and Szakal 2016; Guilliam and Doherty 2017; Vaisman and Woodgate 2017). In some cases, the replication fork can fall apart, resulting in a single-ended DSB, which is repaired as described in the “End-joining and recombination processes involve several subpathways” section. Here, we will focus on two related functions of recombination proteins that were uncovered only in recent years, namely in the replication fork reversal and the protection of DNA from nucleolytic degradation at stalled replication forks.

An overview of pathways that relieve DNA replication stress in human cells. a When a DNA replication fork encounters an obstacle, it may be bypassed by translesion synthesis. b A DNA replication fork can also reverse, which may temporarily protect DNA from breaking. Replication fork reversal can also lead to lesion bypass. Factors that may regulate the various steps of replication fork reversal and restart are indicated. RAD51 likely mediates replication fork reversal and protects reversed replication forks from degradation by the MRE11 nuclease. c Replication repriming downstream of a lesion is another mechanism of DNA damage tolerance. d Replication forks can also break and become a substrate for homologous recombination as indicated in Fig. 4
Replication fork reversal is the conversion of a stalled DNA replication fork into a four-way junction (Fig. 10b). Fork reversal is achieved by the annealing of the two nascent DNA strands and the reannealing of the two parental strands, which resembles a DNA double-strand break. Fork reversal can thus lead to DSB formation in the absence of template breakage (Fig. 10b). Although arising through a very different mechanism, the resulting four-way junction is structurally identical to a Holliday junction that forms during DNA recombination (Fig. 10b). The concept of replication fork reversal has been proposed a long time ago (Higgins et al. 1976). In bacteria, it has been established as a way that contributes to the restart of stalled replication forks. In E. coli, the actions of RecA (a homolog of RAD51), followed by branch migration by RuvAB, or the motor activity of RecG, can drive replication fork reversal (Seigneur et al. 1998; McGlynn and Lloyd 2001; Gupta et al. 2014). For a long time, this process was believed to represent only a pathological reaction in eukaryotic cells (Sogo et al. 2002; Hu et al. 2012). Specifically, fork reversal in yeast was described to occur in checkpoint-defective cells (Lopes et al. 2001). Only recently, it has been demonstrated that replication fork reversal can be beneficial to prevent DNA template breakage in human cells, and that it is a global response to DNA replication stress (Ray Chaudhuri et al. 2012; Neelsen and Lopes 2015; Zellweger et al. 2015). Electron microscopy revealed that the regressed arms of the replication forks can reach and occasionally exceed 1 kb in length, implicating that they form in the course of an active and enzyme-driven process.
One of the first proteins shown to promote replication fork reversal in vivo was RAD51 (Zellweger et al. 2015). In accord, RAD51, to a limited extent, can drive migration of Holliday junctions via polymerization on DNA (Rossi et al. 2011). However, RAD51 per se has no motor activity, so it is unlikely that RAD51 can drive fork reversal on its own. The best candidates for this function are motor proteins such as DNA helicases or translocases. To this point, the motor protein RAD54 was shown to catalyze fork reversal in vitro in a way that was stimulated by RAD51 (Bugreev et al. 2011). Other proteins, including SMARCAL1, ZRANB3, HLTF, FBH1, RECQ5, and FANCM, were proposed to function in fork reversal in vitro or in vivo (Kanagaraj et al. 2006; Gari et al. 2008; Betous et al. 2012; Ciccia et al. 2012; Bhat et al. 2015; Fugger et al. 2015; Kile et al. 2015; Kolinjivadi et al. 2017; Taglialatela et al. 2017; Vujanovic et al. 2017). However, the interplay of these factors with RAD51 is not defined, and it is not clear to which extent these motor proteins can function in a redundant manner. Interestingly, fork reversal likely does not require BRCA2, showing that, unlike in DSB repair, the function of RAD51 in promoting fork reversal is BRCA2 independent (Prakash et al. 2015; Kolinjivadi et al. 2017; Mijic et al. 2017). Fork reversal allows time for DNA repair ahead of the fork, or may result in DNA damage bypass (i.e. damage tolerance, in case when one reversed arm templates DNA synthesis of the complementary strand). Once this is completed, DNA replication must resume. Several mechanisms have been described that promote the restoration of reversed replication forks, which involve motor proteins such as RECQ1, RAD54 or the nucleolytic degradation of the reversed arms by MRE11, EXO1, the DNA2-WRN nuclease-helicase pair or by the MUS81-dependent cleavage, with different implication for genome stability (Bugreev et al. 2011; Berti et al. 2013; Pepe and West 2014a; Thangavel et al. 2015; Lemacon et al. 2017). While replication fork reversal may be pathologic in some cases, it has been demonstrated that inhibition of fork reversal results in increased DNA breakage and genome instability, underpinning the role of this process in preventing genome rearrangements (Neelsen and Lopes 2015; Mijic et al. 2017).
As described in the “Formation of RAD51-ssDNA filament on resected DNA” section, RAD51 is the main DNA strand exchange protein in eukaryotes. It remains to be defined whether RAD51 has a structural or a catalytic function in replication fork reversal. There is, however, one key function of RAD51 that is DNA repair and recombination independent. It has been demonstrated that BRCA1, BRCA2, and RAD51 function together to protect stalled—and possibly reversed—DNA replication forks from degradation by the MRE11 nuclease (Hashimoto et al. 2010; Schlacher et al. 2011, 2012; Mijic et al. 2017). It has been shown that a stable RAD51 nucleoprotein filament is required to protect DNA from MRE11 (Kolinjivadi et al. 2017; Zadorozhny et al. 2017). The BRCA1 and BRCA2 factors likely act directly or indirectly to stabilize the RAD51 nucleoprotein filament. It is being discussed whether the essential function of BRCA2 is to prevent DNA degradation at stalled forks by stabilizing RAD51, or to promote canonical DSB repair (Ray Chaudhuri et al. 2016; Feng and Jasin 2017). At the same time, unrestricted RAD51 activity at stalled forks is equally detrimental: the RADX factor was recently identified as a negative regulator of RAD51 at stalled forks (Dungrawala et al. 2017). These experiments collectively demonstrated that in DNA replication, the RAD51 protein, in addition to performing canonical “recombination” functions has other, previously unrecognized roles in replication fork reversal and protection from nucleolytic degradation. To elucidate the underlying molecular mechanisms of these processes represents a challenge to researchers in the coming years.
References
Akamatsu Y, Jasin M (2010) Role for the mammalian Swi5-Sfr1 complex in DNA strand break repair through homologous recombination. PLoS Genet 6:e1001160
Allers T, Lichten M (2001) Differential timing and control of noncrossover and crossover recombination during meiosis. Cell 106:47–57
Anand R, Beach A, Li K, Haber J (2017) Rad51-mediated double-strand break repair and mismatch correction of divergent substrates. Nature 544:377–380
Anand R, Ranjha L, Cannavo E, Cejka P (2016) Phosphorylated CtIP functions as a co-factor of the MRE11-RAD50-NBS1 endonuclease in DNA end resection. Mol Cell 64:940–950
Arakawa H, Hauschild J, Buerstedde JM (2002) Requirement of the activation-induced deaminase (AID) gene for immunoglobulin gene conversion. Science 295:1301–1306
Arya R, Bassing CH (2017) V(D)J recombination exploits DNA damage responses to promote immunity. Trends Genet 33:479–489
Audebert M, Salles B, Calsou P (2004) Involvement of poly(ADP-ribose) polymerase-1 and XRCC1/DNA ligase III in an alternative route for DNA double-strand breaks rejoining. J Biol Chem 279:55117–55126
Bae SH, Choi E, Lee KH, Park JS, Lee SH, Seo YS (1998) Dna2 of Saccharomyces cerevisiae possesses a single-stranded DNA-specific endonuclease activity that is able to act on double-stranded DNA in the presence of ATP. J Biol Chem 273:26880–26890
Barber LJ, Youds JL, Ward JD, McIlwraith MJ, O’Neil NJ, Petalcorin MI, Martin JS, Collis SJ, Cantor SB, Auclair M et al (2008) RTEL1 maintains genomic stability by suppressing homologous recombination. Cell 135:261–271
Bardwell AJ, Bardwell L, Tomkinson AE, Friedberg EC (1994) Specific cleavage of model recombination and repair intermediates by the yeast Rad1-Rad10 DNA endonuclease. Science 265:2082–2085
Baskar R, Dai J, Wenlong N, Yeo R, Yeoh KW (2014) Biological response of cancer cells to radiation treatment. Front Mol Biosci 1:24
Baudat F, Manova K, Yuen JP, Jasin M, Keeney S (2000) Chromosome synapsis defects and sexually dimorphic meiotic progression in mice lacking Spo11. Mol Cell 6:989–998
Baumann P, Benson FE, West SC (1996) Human Rad51 protein promotes ATP-dependent homologous pairing and strand transfer reactions in vitro. Cell 87:757–766
Bebenek K, Pedersen LC, Kunkel TA (2014) Structure-function studies of DNA polymerase lambda. Biochemistry 53:2781–2792
Bedell VM, Wang Y, Campbell JM, Poshusta TL, Starker CG, Krug RG 2nd, Tan W, Penheiter SG, Ma AC, Leung AY et al (2012) In vivo genome editing using a high-efficiency TALEN system. Nature 491:114–118
Bennardo N, Cheng A, Huang N, Stark JM (2008) Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet 4:e1000110
Benson FE, Baumann P, West SC (1998) Synergistic actions of Rad51 and Rad52 in recombination and DNA repair. Nature 391:401–404
Benson FE, Stasiak A, West SC (1994) Purification and characterization of the human Rad51 protein, an analogue of E. coli RecA. EMBO J 13:5764–5771
Berti M, Ray Chaudhuri A, Thangavel S, Gomathinayagam S, Kenig S, Vujanovic M, Odreman F, Glatter T, Graziano S, Mendoza-Maldonado R et al (2013) Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition. Nat Struct Mol Biol 20:347–354
Betous R, Mason AC, Rambo RP, Bansbach CE, Badu-Nkansah A, Sirbu BM, Eichman BF, Cortez D (2012) SMARCAL1 catalyzes fork regression and Holliday junction migration to maintain genome stability during DNA replication. Genes Dev 26:151–162
Bhargava R, Onyango DO, Stark JM (2016) Regulation of single-strand annealing and its role in genome maintenance. Trends Genet 32:566–575
Bhat KP, Betous R, Cortez D (2015) High-affinity DNA-binding domains of replication protein A (RPA) direct SMARCAL1-dependent replication fork remodeling. J Biol Chem 290:4110–4117
Bianchi J, Rudd SG, Jozwiakowski SK, Bailey LJ, Soura V, Taylor E, Stevanovic I, Green AJ, Stracker TH, Lindsay HD et al (2013) PrimPol bypasses UV photoproducts during eukaryotic chromosomal DNA replication. Mol Cell 52:566–573
Biehs R, Steinlage M, Barton O, Juhasz S, Kunzel J, Spies J, Shibata A, Jeggo PA, Lobrich M (2017) DNA double-strand break resection occurs during non-homologous end joining in G1 but is distinct from resection during homologous recombination. Mol Cell 65:671–684 e675
Bishop DK, Park D, Xu L, Kleckner N (1992) DMC1: a meiosis-specific yeast homolog of E. coli recA required for recombination, synaptonemal complex formation, and cell cycle progression. Cell 69:439–456
Bishop DK, Zickler D (2004) Early decision; meiotic crossover interference prior to stable strand exchange and synapsis. Cell 117:9–15
Blanco MG, Matos J, West SC (2014) Dual control of Yen1 nuclease activity and cellular localization by Cdk and Cdc14 prevents genome instability. Mol Cell 54:94–106
Bocquet N, Bizard AH, Abdulrahman W, Larsen NB, Faty M, Cavadini S, Bunker RD, Kowalczykowski SC, Cejka P, Hickson ID et al (2014) Structural and mechanistic insight into Holliday-junction dissolution by topoisomerase IIIalpha and RMI1. Nat Struct Mol Biol 21:261–268
Boddy MN, Gaillard PH, McDonald WH, Shanahan P, Yates JR 3rd, Russell P (2001) Mus81-Eme1 are essential components of a Holliday junction resolvase. Cell 107:537–548
Bonetti D, Clerici M, Manfrini N, Lucchini G, Longhese MP (2010) The MRX complex plays multiple functions in resection of Yku- and Rif2-protected DNA ends. PLoS One 5:e14142
Borde V, Lin W, Novikov E, Petrini JH, Lichten M, Nicolas A (2004) Association of Mre11p with double-strand break sites during yeast meiosis. Mol Cell 13:389–401
Branzei D, Szakal B (2016) DNA damage tolerance by recombination: molecular pathways and DNA structures. DNA Repair (Amst) 44:68–75
Branzei D, Szakal B (2017) Building up and breaking down: mechanisms controlling recombination during replication. Crit Rev Biochem Mol Biol 52:381–394
Bregenhorn S, Kallenberger L, Artola-Boran M, Pena-Diaz J, Jiricny J (2016) Non-canonical uracil processing in DNA gives rise to double-strand breaks and deletions: relevance to class switch recombination. Nucleic Acids Res 44:2691–2705
Broderick R, Nieminuszczy J, Baddock HT, Deshpande RA, Gileadi O, Paull TT, McHugh PJ, Niedzwiedz W (2016) EXD2 promotes homologous recombination by facilitating DNA end resection. Nat Cell Biol 18:271–280
Brosh RM, Li JL, Kenny MK, Karow JK, Cooper MP, Kureekattil RP, Hickson ID, Bohr VA (2000) Replication protein A physically interacts with the Bloom’s syndrome protein and stimulates its helicase activity. J Biol Chem 275:23500–23508
Bugreev DV, Brosh RM Jr, Mazin AV (2008) RECQ1 possesses DNA branch migration activity. J Biol Chem 283:20231–20242
Bugreev DV, Hanaoka F, Mazin AV (2007a) Rad54 dissociates homologous recombination intermediates by branch migration. Nat Struct Mol Biol 14:746–753
Bugreev DV, Rossi MJ, Mazin AV (2011) Cooperation of RAD51 and RAD54 in regression of a model replication fork. Nucleic Acids Res 39:2153–2164
Bugreev DV, Yu X, Egelman EH, Mazin AV (2007b) Novel pro- and anti-recombination activities of the Bloom’s syndrome helicase. Genes Dev 21:3085–3094
Buisson R, Dion-Cote AM, Coulombe Y, Launay H, Cai H, Stasiak AZ, Stasiak A, Xia B, Masson JY (2010) Cooperation of breast cancer proteins PALB2 and piccolo BRCA2 in stimulating homologous recombination. Nat Struct Mol Biol 17:1247–1254
Canela A, Maman Y, Jung S, Wong N, Callen E, Day A, Kieffer-Kwon KR, Pekowska A, Zhang H, Rao SSP et al (2017) Genome organization drives chromosome fragility. Cell 170:507–521 e518
Cannavo E, Cejka P (2014) Sae2 promotes dsDNA endonuclease activity within Mre11-Rad50-Xrs2 to resect DNA breaks. Nature 514:122–125
Carballo JA, Johnson AL, Sedgwick SG, Cha RS (2008) Phosphorylation of the axial element protein Hop1 by Mec1/Tel1 ensures meiotic interhomolog recombination. Cell 132:758–770
Carreira A, Hilario J, Amitani I, Baskin RJ, Shivji MK, Venkitaraman AR, Kowalczykowski SC (2009) The BRC repeats of BRCA2 modulate the DNA-binding selectivity of RAD51. Cell 136:1032–1043
Carson CT, Schwartz RA, Stracker TH, Lilley CE, Lee DV, Weitzman MD (2003) The Mre11 complex is required for ATM activation and the G2/M checkpoint. EMBO J 22:6610–6620
Casellas R, Nussenzweig A, Wuerffel R, Pelanda R, Reichlin A, Suh H, Qin XF, Besmer E, Kenter A, Rajewsky K et al (1998) Ku80 is required for immunoglobulin isotype switching. EMBO J 17:2404–2411
Castor D, Nair N, Declais AC, Lachaud C, Toth R, Macartney TJ, Lilley DM, Arthur JS, Rouse J (2013) Cooperative control of Holliday junction resolution and DNA repair by the SLX1 and MUS81-EME1 nucleases. Mol Cell 52:221–233
Ceballos SJ, Heyer WD (2011) Functions of the Snf2/Swi2 family Rad54 motor protein in homologous recombination. Biochim Biophys Acta 1809:509–523
Cejka P (2015) DNA end resection: nucleases team up with the right partners to initiate homologous recombination. J Biol Chem 290(38):22931–22938
Cejka P, Cannavo E, Polaczek P, Masuda-Sasa T, Pokharel S, Campbell JL, Kowalczykowski SC (2010a) DNA end resection by Dna2-Sgs1-RPA and its stimulation by Top3-Rmi1 and Mre11-Rad50-Xrs2. Nature 467:112–116
Cejka P, Plank JL, Bachrati CZ, Hickson ID, Kowalczykowski SC (2010b) Rmi1 stimulates decatenation of double Holliday junctions during dissolution by Sgs1-Top3. Nat Struct Mol Biol 17:1377–1382
Cejka P, Plank JL, Dombrowski CC, Kowalczykowski SC (2012) Decatenation of DNA by the S. cerevisiae Sgs1-Top3-Rmi1 and RPA complex: a mechanism for disentangling chromosomes. Mol Cell 47:886–896
Chaganti RS, Schonberg S, German J (1974) A manyfold increase in sister chromatid exchanges in Bloom’s syndrome lymphocytes. Proc Natl Acad Sci U S A 71:4508–4512
Chan YW, West SC (2014) Spatial control of the GEN1 Holliday junction resolvase ensures genome stability. Nat Commun 5:4844
Chang HHY, Pannunzio NR, Adachi N, Lieber MR (2017) Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat Rev Mol Cell Biol 18:495–506
Chanut P, Britton S, Coates J, Jackson SP, Calsou P (2016) Coordinated nuclease activities counteract Ku at single-ended DNA double-strand breaks. Nat Commun 7:12889
Chapman JR, Taylor MR, Boulton SJ (2012) Playing the end game: DNA double-strand break repair pathway choice. Mol Cell 47:497–510
Chappell C, Hanakahi LA, Karimi-Busheri F, Weinfeld M, West SC (2002) Involvement of human polynucleotide kinase in double-strand break repair by non-homologous end joining. EMBO J 21:2827–2832
Chen L, Trujillo K, Ramos W, Sung P, Tomkinson AE (2001) Promotion of Dnl4-catalyzed DNA end-joining by the Rad50/Mre11/Xrs2 and Hdf1/Hdf2 complexes. Mol Cell 8:1105–1115
Chen SH, Plank JL, Willcox S, Griffith JD, Hsieh TS (2014) Top3alpha is required during the convergent migration step of double Holliday junction dissolution. PLoS One 9:e83582
Chi P, San Filippo J, Sehorn MG, Petukhova GV, Sung P (2007) Bipartite stimulatory action of the Hop2-Mnd1 complex on the Rad51 recombinase. Genes Dev 21:1747–1757
Chiolo I, Minoda A, Colmenares SU, Polyzos A, Costes SV, Karpen GH (2011) Double-strand breaks in heterochromatin move outside of a dynamic HP1a domain to complete recombinational repair. Cell 144:732–744
Chu VT, Weber T, Wefers B, Wurst W, Sander S, Rajewsky K, Kuhn R (2015) Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat Biotechnol 33:543–548
Ciccia A, Elledge SJ (2010) The DNA damage response: making it safe to play with knives. Mol Cell 40:179–204
Ciccia A, Nimonkar AV, Hu Y, Hajdu I, Achar YJ, Izhar L, Petit SA, Adamson B, Yoon JC, Kowalczykowski SC et al (2012) Polyubiquitinated PCNA recruits the ZRANB3 translocase to maintain genomic integrity after replication stress. Mol Cell 47:396–409
Cloud V, Chan YL, Grubb J, Budke B, Bishop DK (2012) Rad51 is an accessory factor for Dmc1-mediated joint molecule formation during meiosis. Science 337:1222–1225
Daley JM, Niu H, Sung P (2013) Roles of DNA helicases in the mediation and regulation of homologous recombination. Adv Exp Med Biol 767:185–202
de los Santos T, Hunter N, Lee C, Larkin B, Loidl J, Hollingsworth NM (2003) The Mus81/Mms4 endonuclease acts independently of double-Holliday junction resolution to promote a distinct subset of crossovers during meiosis in budding yeast. Genetics 164:81–94
Dehe PM, Gaillard PH (2017) Control of structure-specific endonucleases to maintain genome stability. Nat Rev Mol Cell Biol 18:315–330
Deng SK, Gibb B, de Almeida MJ, Greene EC, Symington LS (2014) RPA antagonizes microhomology-mediated repair of DNA double-strand breaks. Nat Struct Mol Biol 21:405–412
Deshpande RA, Lee JH, Arora S, Paull TT (2016) Nbs1 converts the human Mre11/Rad50 nuclease complex into an endo/exonuclease machine specific for protein-DNA adducts. Mol Cell 64:593–606
Dinkelmann M, Spehalski E, Stoneham T, Buis J, Wu Y, Sekiguchi JM, Ferguson DO (2009) Multiple functions of MRN in end-joining pathways during isotype class switching. Nat Struct Mol Biol 16:808–813
Dion V, Kalck V, Horigome C, Towbin BD, Gasser SM (2012) Increased mobility of double-strand breaks requires Mec1, Rad9 and the homologous recombination machinery. Nat Cell Biol 14:502–509
Donnianni RA, Symington LS (2013) Break-induced replication occurs by conservative DNA synthesis. Proc Natl Acad Sci U S A 110:13475–13480
Duckett DR, Murchie AI, Diekmann S, von Kitzing E, Kemper B, Lilley DM (1988) The structure of the Holliday junction, and its resolution. Cell 55:79–89
Dunderdale HJ, Benson FE, Parsons CA, Sharples GJ, Lloyd RG, West SC (1991) Formation and resolution of recombination intermediates by E. coli RecA and RuvC proteins. Nature 354:506–510
Dungrawala H, Bhat KP, Le Meur R, Chazin WJ, Ding X, Sharan SK, Wessel SR, Sathe AA, Zhao R, Cortez D (2017) RADX promotes genome stability and modulates chemosensitivity by regulating RAD51 at replication forks. Mol Cell 67:374–386 e375
Eggler AL, Inman RB, Cox MM (2002) The Rad51-dependent pairing of long DNA substrates is stabilized by replication protein A. J Biol Chem 277:39280–39288
Ehmsen KT, Heyer WD (2008) Saccharomyces cerevisiae Mus81-Mms4 is a catalytic, DNA structure-selective endonuclease. Nucleic Acids Res 36:2182–2195
Ehrenstein MR, Neuberger MS (1999) Deficiency in Msh2 affects the efficiency and local sequence specificity of immunoglobulin class-switch recombination: parallels with somatic hypermutation. EMBO J 18:3484–3490
Fasching CL, Cejka P, Kowalczykowski SC, Heyer WD (2015) Top3-Rmi1 dissolve Rad51-mediated D loops by a topoisomerase-based mechanism. Mol Cell 57:595–606
Fearon ER, Vogelstein B (1990) A genetic model for colorectal tumorigenesis. Cell 61:759–767
Fekairi S, Scaglione S, Chahwan C, Taylor ER, Tissier A, Coulon S, Dong MQ, Ruse C, Yates JR 3rd, Russell P et al (2009) Human SLX4 is a Holliday junction resolvase subunit that binds multiple DNA repair/recombination endonucleases. Cell 138:78–89
Feng W, Jasin M (2017) BRCA2 suppresses replication stress-induced mitotic and G1 abnormalities through homologous recombination. Nat Commun 8:525
Feng Z, Scott SP, Bussen W, Sharma GG, Guo G, Pandita TK, Powell SN (2011) Rad52 inactivation is synthetically lethal with BRCA2 deficiency. Proc Natl Acad Sci U S A 108:686–691
Forget AL, Kowalczykowski SC (2012) Single-molecule imaging of DNA pairing by RecA reveals a three-dimensional homology search. Nature 482:423–427
Fricke WM, Brill SJ (2003) Slx1-Slx4 is a second structure-specific endonuclease functionally redundant with Sgs1-Top3. Genes Dev 17:1768–1778
Fugger K, Mistrik M, Danielsen JR, Dinant C, Falck J, Bartek J, Lukas J, Mailand N (2009) Human Fbh1 helicase contributes to genome maintenance via pro- and anti-recombinase activities. J Cell Biol 186:655–663
Fugger K, Mistrik M, Neelsen KJ, Yao Q, Zellweger R, Kousholt AN, Haahr P, Chu WK, Bartek J, Lopes M et al (2015) FBH1 catalyzes regression of stalled replication forks. Cell Rep. https://doi.org/10.1016/j.celrep.2015.02.028
Gaillard PHL, Noguchi E, Shanahan P, Russell P (2003) The endogenous Mus81-Eme1 complex resolves Holliday junctions by a nick and counternick mechanism. Mol Cell 12:747–759
Gaines WA, Godin SK, Kabbinavar FF, Rao T, VanDemark AP, Sung P, Bernstein KA (2015) Promotion of presynaptic filament assembly by the ensemble of S. cerevisiae Rad51 paralogues with Rad52. Nat Commun 6:7834
Garcia V, Gray S, Allison RM, Cooper TJ, Neale MJ (2015) Tel1(ATM)-mediated interference suppresses clustered meiotic double-strand-break formation. Nature 520:114–118
Garcia V, Phelps SE, Gray S, Neale MJ (2011) Bidirectional resection of DNA double-strand breaks by Mre11 and Exo1. Nature 479:241–244
Garcia-Gomez S, Reyes A, Martinez-Jimenez MI, Chocron ES, Mouron S, Terrados G, Powell C, Salido E, Mendez J, Holt IJ et al (2013) PrimPol, an archaic primase/polymerase operating in human cells. Mol Cell 52:541–553
Gari K, Decaillet C, Delannoy M, Wu L, Constantinou A (2008) Remodeling of DNA replication structures by the branch point translocase FANCM. Proc Natl Acad Sci U S A 105:16107–16112
Gottlieb TM, Jackson SP (1993) The DNA-dependent protein kinase: requirement for DNA ends and association with Ku antigen. Cell 72:131–142
Gravel S, Chapman JR, Magill C, Jackson SP (2008) DNA helicases Sgs1 and BLM promote DNA double-strand break resection. Genes Dev 22:2767–2772
Guilliam TA, Doherty AJ (2017) PrimPol—prime time to reprime. Genes (Basel) 8(1):20
Gupta S, Yeeles JT, Marians KJ (2014) Regression of replication forks stalled by leading-strand template damage: I. Both RecG and RuvAB catalyze regression, but RuvC cleaves the holliday junctions formed by RecG preferentially. J Biol Chem 289:28376–28387
Hasham MG, Donghia NM, Coffey E, Maynard J, Snow KJ, Ames J, Wilpan RY, He Y, King BL, Mills KD (2010) Widespread genomic breaks generated by activation-induced cytidine deaminase are prevented by homologous recombination. Nat Immunol 11:820–826
Hashimoto Y, Ray Chaudhuri A, Lopes M, Costanzo V (2010) Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat Struct Mol Biol 17:1305–1311
Helmink BA, Bredemeyer AL, Lee BS, Huang CY, Sharma GG, Walker LM, Bednarski JJ, Lee WL, Pandita TK, Bassing CH et al (2009) MRN complex function in the repair of chromosomal Rag-mediated DNA double-strand breaks. J Exp Med 206:669–679
Heyer WD (2015) Regulation of recombination and genomic maintenance. Cold Spring Harb Perspect Biol 7:a016501
Hicks WM, Kim M, Haber JE (2010) Increased mutagenesis and unique mutation signature associated with mitotic gene conversion. Science 329:82–85
Hickson ID, Mankouri HW (2011) Processing of homologous recombination repair intermediates by the Sgs1-Top3-Rmi1 and Mus81-Mms4 complexes. Cell Cycle 10:3078–3085
Higgins NP, Kato K, Strauss B (1976) A model for replication repair in mammalian cells. J Mol Biol 101:417–425
Hinz JM (2010) Role of homologous recombination in DNA interstrand crosslink repair. Environ Mol Mutagen 51:582–603
Holm C, Covey JM, Kerrigan D, Pommier Y (1989) Differential requirement of DNA replication for the cytotoxicity of DNA topoisomerase I and II inhibitors in Chinese hamster DC3F cells. Cancer Res 49:6365–6368
Horigome C, Oma Y, Konishi T, Schmid R, Marcomini I, Hauer MH, Dion V, Harata M, Gasser SM (2014) SWR1 and INO80 chromatin remodelers contribute to DNA double-strand break perinuclear anchorage site choice. Mol Cell 55:626–639
Howard SM, Yanez DA, Stark JM (2015) DNA damage response factors from diverse pathways, including DNA crosslink repair, mediate alternative end joining. PLoS Genet 11:e1004943
Hu J, Sun L, Shen F, Chen Y, Hua Y, Liu Y, Zhang M, Hu Y, Wang Q, Xu W et al (2012) The intra-S phase checkpoint targets Dna2 to prevent stalled replication forks from reversing. Cell 149:1221–1232
Hu Y, Raynard S, Sehorn MG, Lu X, Bussen W, Zheng L, Stark JM, Barnes EL, Chi P, Janscak P et al (2007) RECQL5/Recql5 helicase regulates homologous recombination and suppresses tumor formation via disruption of Rad51 presynaptic filaments. Genes Dev 21:3073–3084
Huang J, Dynan WS (2002) Reconstitution of the mammalian DNA double-strand break end-joining reaction reveals a requirement for an Mre11/Rad50/NBS1-containing fraction. Nucleic Acids Res 30:667–674
Huertas P, Cortes-Ledesma F, Sartori AA, Aguilera A, Jackson SP (2008) CDK targets Sae2 to control DNA-end resection and homologous recombination. Nature 455:689–692
Huertas P, Jackson SP (2009) Human CtIP mediates cell cycle control of DNA end resection and double strand break repair. J Biol Chem 284:9558–9565
Hunter N (2015) Meiotic recombination: the essence of heredity. Cold Spring Harb Perspect Biol 7
Hunter N, Kleckner N (2001) The single-end invasion: an asymmetric intermediate at the double-strand break to double-Holliday junction transition of meiotic recombination. Cell 106:59–70
Imai K, Slupphaug G, Lee WI, Revy P, Nonoyama S, Catalan N, Yel L, Forveille M, Kavli B, Krokan HE et al (2003) Human uracil-DNA glycosylase deficiency associated with profoundly impaired immunoglobulin class-switch recombination. Nat Immunol 4:1023–1028
Ip SC, Rass U, Blanco MG, Flynn HR, Skehel JM, West SC (2008) Identification of Holliday junction resolvases from humans and yeast. Nature 456:357–361
Ira G, Pellicioli A, Balijja A, Wang X, Fiorani S, Carotenuto W, Liberi G, Bressan D, Wan L, Hollingsworth NM et al (2004) DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature 431:1011–1017
Ivanov EL, Sugawara N, Fishman-Lobell J, Haber JE (1996) Genetic requirements for the single-strand annealing pathway of double-strand break repair in Saccharomyces cerevisiae. Genetics 142:693–704
Jackson SP, Bartek J (2009) The DNA-damage response in human biology and disease. Nature 461:1071–1078
Jensen RB, Carreira A, Kowalczykowski SC (2010) Purified human BRCA2 stimulates RAD51-mediated recombination. Nature 467:678–683
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337:816–821
Johzuka K, Ogawa H (1995) Interaction of Mre11 and Rad50: two proteins required for DNA repair and meiosis-specific double-strand break formation in Saccharomyces cerevisiae. Genetics 139:1521–1532
Kanagaraj R, Saydam N, Garcia PL, Zheng L, Janscak P (2006) Human RECQ5beta helicase promotes strand exchange on synthetic DNA structures resembling a stalled replication fork. Nucleic Acids Res 34:5217–5231
Kawamoto T, Araki K, Sonoda E, Yamashita YM, Harada K, Kikuchi K, Masutani C, Hanaoka F, Nozaki K, Hashimoto N et al (2005) Dual roles for DNA polymerase eta in homologous DNA recombination and translesion DNA synthesis. Mol Cell 20:793–799
Keeney S (2008) Spo11 and the formation of DNA double-strand breaks in meiosis. Genome Dyn Stab 2:81–123
Keeney S, Giroux CN, Kleckner N (1997) Meiosis-specific DNA double-strand breaks are catalyzed by Spo11, a member of a widely conserved protein family. Cell 88:375–384
Keeney S, Kleckner N (1995) Covalent protein-DNA complexes at the 5′ strand termini of meiosis-specific double-strand breaks in yeast. Proc Natl Acad Sci U S A 92:11274–11278
Keeney S, Lange J, Mohibullah N (2014) Self-organization of meiotic recombination initiation: general principles and molecular pathways. Annu Rev Genet 48:187–214
Kile AC, Chavez DA, Bacal J, Eldirany S, Korzhnev DM, Bezsonova I, Eichman BF, Cimprich KA (2015) HLTF’s ancient HIRAN domain binds 3′ DNA ends to drive replication fork reversal. Mol Cell 58:1090–1100
Kim JH, Grosbart M, Anand R, Wyman C, Cejka P, Petrini JH (2017) The Mre11-Nbs1 interface is essential for viability and tumor suppression. Cell Rep 18:496–507
Kim KP, Weiner BM, Zhang L, Jordan A, Dekker J, Kleckner N (2010) Sister cohesion and structural axis components mediate homolog bias of meiotic recombination. Cell 143:924–937
Kolinjivadi AM, Sannino V, De Antoni A, Zadorozhny K, Kilkenny M, Techer H, Baldi G, Shen R, Ciccia A, Pellegrini L et al (2017) Smarcal1-mediated fork reversal triggers Mre11-dependent degradation of nascent DNA in the absence of Brca2 and stable Rad51 nucleofilaments. Mol Cell 67:867–881
Kosugi S, Hasebe M, Tomita M, Yanagawa H (2009) Systematic identification of cell cycle-dependent yeast nucleocytoplasmic shuttling proteins by prediction of composite motifs. Proc Natl Acad Sci U S A 106:10171–10176
Kowalczykowski SC (2015) An overview of the molecular mechanisms of recombinational DNA repair. Cold Spring Harb Perspect Biol 7
Krejci L, Van Komen S, Li Y, Villemain J, Reddy MS, Klein H, Ellenberger T, Sung P (2003) DNA helicase Srs2 disrupts the Rad51 presynaptic filament. Nature 423:305–309
Lam I, Keeney S (2014) Mechanism and regulation of meiotic recombination initiation. Cold Spring Harb Perspect Biol 7:a016634
Lange J, Yamada S, Tischfield SE, Pan J, Kim S, Zhu X, Socci ND, Jasin M, Keeney S (2016) The landscape of mouse meiotic double-strand break formation, processing, and repair. Cell 167:695–708 e616
Langerak P, Mejia-Ramirez E, Limbo O, Russell P (2011) Release of Ku and MRN from DNA ends by Mre11 nuclease activity and Ctp1 is required for homologous recombination repair of double-strand breaks. PLoS Genet 7:e1002271
Lavery PE, Kowalczykowski SC (1992) A postsynaptic role for single-stranded DNA-binding protein in recA protein-promoted DNA strand exchange. J Biol Chem 267:9315–9320
Lee JH, Paull TT (2004) Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science 304:93–96
Lee JH, Paull TT (2005) ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science 308:551–554
Lee-Theilen M, Matthews AJ, Kelly D, Zheng S, Chaudhuri J (2011) CtIP promotes microhomology-mediated alternative end joining during class-switch recombination. Nat Struct Mol Biol 18:75–79
Lemacon D, Jackson J, Quinet A, Brickner JR, Li S, Yazinski S, You Z, Ira G, Zou L, Mosammaparast N et al (2017) MRE11 and EXO1 nucleases degrade reversed forks and elicit MUS81-dependent fork rescue in BRCA2-deficient cells. Nat Commun 8:860
Lengsfeld BM, Rattray AJ, Bhaskara V, Ghirlando R, Paull TT (2007) Sae2 is an endonuclease that processes hairpin DNA cooperatively with the Mre11/Rad50/Xrs2 complex. Mol Cell 28:638–651
Leu JY, Chua PR, Roeder GS (1998) The meiosis-specific Hop2 protein of S. cerevisiae ensures synapsis between homologous chromosomes. Cell 94:375–386
Levikova M, Klaue D, Seidel R, Cejka P (2013) Nuclease activity of Saccharomyces cerevisiae Dna2 inhibits its potent DNA helicase activity. Proc Natl Acad Sci U S A 110:E1992–E2001
Levikova M, Pinto C, Cejka P (2017) The motor activity of DNA2 functions as an ssDNA translocase to promote DNA end resection. Genes Dev 31:493–502
Li X, Stith CM, Burgers PM, Heyer WD (2009) PCNA is required for initiation of recombination-associated DNA synthesis by DNA polymerase delta. Mol Cell 36:704–713
Li Z, Otevrel T, Gao Y, Cheng HL, Seed B, Stamato TD, Taccioli GE, Alt FW (1995) The XRCC4 gene encodes a novel protein involved in DNA double-strand break repair and V(D)J recombination. Cell 83:1079–1089
Lipkin SM, Moens PB, Wang V, Lenzi M, Shanmugarajah D, Gilgeous A, Thomas J, Cheng J, Touchman JW, Green ED et al (2002) Meiotic arrest and aneuploidy in MLH3-deficient mice. Nat Genet 31:385–390
Liu J, Doty T, Gibson B, Heyer WD (2010) Human BRCA2 protein promotes RAD51 filament formation on RPA-covered single-stranded DNA. Nat Struct Mol Biol 17:1260–1262
Liu J, Renault L, Veaute X, Fabre F, Stahlberg H, Heyer WD (2011a) Rad51 paralogues Rad55-Rad57 balance the antirecombinase Srs2 in Rad51 filament formation. Nature 479:245–248
Liu T, Wan L, Wu Y, Chen J, Huang J (2011b) hSWS1.SWSAP1 is an evolutionarily conserved complex required for efficient homologous recombination repair. J Biol Chem 286:41758–41766
Llorente B, Smith CE, Symington LS (2008) Break-induced replication: what is it and what is it for? Cell Cycle 7:859–864
Lobrich M, Jeggo P (2017) A process of resection-dependent nonhomologous end joining involving the goddess Artemis. Trends Biochem Sci 42(9):690–701
Lombardo A, Genovese P, Beausejour CM, Colleoni S, Lee YL, Kim KA, Ando D, Urnov FD, Galli C, Gregory PD et al (2007) Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery. Nat Biotechnol 25:1298–1306
Lopes M, Cotta-Ramusino C, Pellicioli A, Liberi G, Plevani P, Muzi-Falconi M, Newlon CS, Foiani M (2001) The DNA replication checkpoint response stabilizes stalled replication forks. Nature 412:557–561
Lynn A, Soucek R, Borner GV (2007) ZMM proteins during meiosis: crossover artists at work. Chromosom Res 15:591–605
Ma Y, Pannicke U, Schwarz K, Lieber MR (2002) Hairpin opening and overhang processing by an Artemis/DNA-dependent protein kinase complex in nonhomologous end joining and V(D)J recombination. Cell 108:781–794
Mahadevaiah SK, Turner JM, Baudat F, Rogakou EP, de Boer P, Blanco-Rodriguez J, Jasin M, Keeney S, Bonner WM, Burgoyne PS (2001) Recombinational DNA double-strand breaks in mice precede synapsis. Nat Genet 27:271–276
Makharashvili N, Tubbs AT, Yang SH, Wang H, Barton O, Zhou Y, Deshpande RA, Lee JH, Lobrich M, Sleckman BP et al (2014) Catalytic and noncatalytic roles of the CtIP endonuclease in double-strand break end resection. Mol Cell 54:1022–1033
Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM (2013) RNA-guided human genome engineering via Cas9. Science 339:823–826
Martin V, Chahwan C, Gao H, Blais V, Wohlschlegel J, Yates JR 3rd, McGowan CH, Russell P (2006) Sws1 is a conserved regulator of homologous recombination in eukaryotic cells. EMBO J 25:2564–2574
Mason JM, Dusad K, Wright WD, Grubb J, Budke B, Heyer WD, Connell PP, Weichselbaum RR, Bishop DK (2015) RAD54 family translocases counter genotoxic effects of RAD51 in human tumor cells. Nucleic Acids Res 43:3180–3196
Masson JY, Tarsounas MC, Stasiak AZ, Stasiak A, Shah R, McIlwraith MJ, Benson FE, West SC (2001) Identification and purification of two distinct complexes containing the five RAD51 paralogs. Genes Dev 15:3296–3307
Mateos-Gomez PA, Gong F, Nair N, Miller KM, Lazzerini-Denchi E, Sfeir A (2015) Mammalian polymerase theta promotes alternative NHEJ and suppresses recombination. Nature 518:254–257
Mateos-Gomez PA, Kent T, Deng SK, McDevitt S, Kashkina E, Hoang TM, Pomerantz RT, Sfeir A (2017) The helicase domain of Poltheta counteracts RPA to promote alt-NHEJ. Nat Struct Mol Biol 24(12):1116–1123
Matos J, Blanco MG, Maslen S, Skehel JM, West SC (2011) Regulatory control of the resolution of DNA recombination intermediates during meiosis and mitosis. Cell 147:158–172
Matos J, Blanco MG, West SC (2013) Cell-cycle kinases coordinate the resolution of recombination intermediates with chromosome segregation. Cell Rep 4:76–86
Matos J, West SC (2014) Holliday junction resolution: regulation in space and time. DNA Repair (Amst) 19:176–181
Mazina OM, Mazin AV, Nakagawa T, Kolodner RD, Kowalczykowski SC (2004) Saccharomyces cerevisiae Mer3 helicase stimulates 3′-5′ heteroduplex extension by Rad51; implications for crossover control in meiotic recombination. Cell 117:47–56
McBlane JF, van Gent DC, Ramsden DA, Romeo C, Cuomo CA, Gellert M, Oettinger MA (1995) Cleavage at a V(D)J recombination signal requires only RAG1 and RAG2 proteins and occurs in two steps. Cell 83:387–395
McGlynn P, Lloyd RG (2001) Rescue of stalled replication forks by RecG: simultaneous translocation on the leading and lagging strand templates supports an active DNA unwinding model of fork reversal and Holliday junction formation. Proc Natl Acad Sci U S A 98:8227–8234
McIlwraith MJ, Vaisman A, Liu Y, Fanning E, Woodgate R, West SC (2005) Human DNA polymerase eta promotes DNA synthesis from strand invasion intermediates of homologous recombination. Mol Cell 20:783–792
McIlwraith MJ, West SC (2008) DNA repair synthesis facilitates RAD52-mediated second-end capture during DSB repair. Mol Cell 29:510–516
Methot SP, Di Noia JM (2017) Molecular mechanisms of somatic hypermutation and class switch recombination. Adv Immunol 133:37–87
Mijic S, Zellweger R, Chappidi N, Berti M, Jacobs K, Mutreja K, Ursich S, Ray Chaudhuri A, Nussenzweig A, Janscak P et al (2017) Replication fork reversal triggers fork degradation in BRCA2-defective cells. Nat Commun 8:859
Miller AS, Daley JM, Pham NT, Niu H, Xue X, Ira G, Sung P (2017) A novel role of the Dna2 translocase function in DNA break resection. Genes Dev 31:503–510
Mimitou EP, Symington LS (2008) Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature 455:770–774
Mimitou EP, Symington LS (2010) Ku prevents Exo1 and Sgs1-dependent resection of DNA ends in the absence of a functional MRX complex or Sae2. EMBO J 29:3358–3369
Mimitou EP, Yamada S, Keeney S (2017) A global view of meiotic double-strand break end resection. Science 355:40–45
Mine-Hattab J, Rothstein R (2012) Increased chromosome mobility facilitates homology search during recombination. Nat Cell Biol 14:510–517
Mitchel K, Lehner K, Jinks-Robertson S (2013) Heteroduplex DNA position defines the roles of the Sgs1, Srs2, and Mph1 helicases in promoting distinct recombination outcomes. PLoS Genet 9:e1003340
Moldovan GL, Dejsuphong D, Petalcorin MI, Hofmann K, Takeda S, Boulton SJ, D'Andrea AD (2012) Inhibition of homologous recombination by the PCNA-interacting protein PARI. Mol Cell 45:75–86
Moon AF, Pryor JM, Ramsden DA, Kunkel TA, Bebenek K, Pedersen LC (2014) Sustained active site rigidity during synthesis by human DNA polymerase mu. Nat Struct Mol Biol 21:253–260
Mortensen UH, Bendixen C, Sunjevaric I, Rothstein R (1996) DNA strand annealing is promoted by the yeast Rad52 protein. Proc Natl Acad Sci U S A 93:10729–10734
Mouron S, Rodriguez-Acebes S, Martinez-Jimenez MI, Garcia-Gomez S, Chocron S, Blanco L, Mendez J (2013) Repriming of DNA synthesis at stalled replication forks by human PrimPol. Nat Struct Mol Biol 20:1383–1389
Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T (2000) Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell 102:553–563
Neale MJ, Pan J, Keeney S (2005) Endonucleolytic processing of covalent protein-linked DNA double-strand breaks. Nature 436:1053–1057
Neelsen KJ, Lopes M (2015) Replication fork reversal in eukaryotes: from dead end to dynamic response. Nat Rev Mol Cell Biol 16:207–220
Nicolette ML, Lee K, Guo Z, Rani M, Chow JM, Lee SE, Paull TT (2010) Mre11-Rad50-Xrs2 and Sae2 promote 5′ strand resection of DNA double-strand breaks. Nat Struct Mol Biol 17:1478–1485
Nimonkar AV, Genschel J, Kinoshita E, Polaczek P, Campbell JL, Wyman C, Modrich P, Kowalczykowski SC (2011) BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev 25:350–362
Nimonkar AV, Sica RA, Kowalczykowski SC (2009) Rad52 promotes second-end DNA capture in double-stranded break repair to form complement-stabilized joint molecules. Proc Natl Acad Sci U S A 106:3077–3082
Nishant KT, Plys AJ, Alani E (2008) A mutation in the putative MLH3 endonuclease domain confers a defect in both mismatch repair and meiosis in Saccharomyces cerevisiae. Genetics 179:747–755
Niu H, Chung WH, Zhu Z, Kwon Y, Zhao W, Chi P, Prakash R, Seong C, Liu D, Lu L et al (2010) Mechanism of the ATP-dependent DNA end-resection machinery from Saccharomyces cerevisiae. Nature 467:108–111
Niu H, Wan L, Baumgartner B, Schaefer D, Loidl J, Hollingsworth NM (2005) Partner choice during meiosis is regulated by Hop1-promoted dimerization of Mek1. Mol Biol Cell 16:5804–5818
Oh J, Al-Zain A, Cannavo E, Cejka P, Symington LS (2016) Xrs2 dependent and independent functions of the Mre11-Rad50 complex. Mol Cell 64:405–415
Olive PL (1998) The role of DNA single- and double-strand breaks in cell killing by ionizing radiation. Radiat Res 150:S42–S51
Olive PL, Banath JP, Durand RE (1990) Heterogeneity in radiation-induced DNA damage and repair in tumor and normal cells measured using the “comet” assay. Radiat Res 122:86–94
Orthwein A, Noordermeer SM, Wilson MD, Landry S, Enchev RI, Sherker A, Munro M, Pinder J, Salsman J, Dellaire G et al (2015) A mechanism for the suppression of homologous recombination in G1 cells. Nature 528:422–426
Oza P, Jaspersen SL, Miele A, Dekker J, Peterson CL (2009) Mechanisms that regulate localization of a DNA double-strand break to the nuclear periphery. Genes Dev 23:912–927
Papavasiliou FN, Schatz DG (2000) Cell-cycle-regulated DNA double-stranded breaks in somatic hypermutation of immunoglobulin genes. Nature 408:216–221
Patel DS, Misenko SM, Her J, Bunting SF (2017) BLM helicase regulates DNA repair by counteracting RAD51 loading at DNA double-strand break sites. J Cell Biol 216(11):3521–3534
Paull TT, Gellert M (1998) The 3′ to 5′ exonuclease activity of Mre 11 facilitates repair of DNA double-strand breaks. Mol Cell 1:969–979
Peoples TL, Dean E, Gonzalez O, Lambourne L, Burgess SM (2002) Close, stable homolog juxtaposition during meiosis in budding yeast is dependent on meiotic recombination, occurs independently of synapsis, and is distinct from DSB-independent pairing contacts. Genes Dev 16:1682–1695
Pepe A, West SC (2014a) MUS81-EME2 promotes replication fork restart. Cell Rep 7:1048–1055
Pepe A, West SC (2014b) Substrate specificity of the MUS81-EME2 structure selective endonuclease. Nucleic Acids Res 42:3833–3845
Petalcorin MI, Galkin VE, Yu X, Egelman EH, Boulton SJ (2007) Stabilization of RAD-51-DNA filaments via an interaction domain in Caenorhabditis elegans BRCA2. Proc Natl Acad Sci U S A 104:8299–8304
Petersen-Mahrt SK, Harris RS, Neuberger MS (2002) AID mutates E. coli suggesting a DNA deamination mechanism for antibody diversification. Nature 418:99–103
Petukhova GV, Pezza RJ, Vanevski F, Ploquin M, Masson JY, Camerini-Otero RD (2005) The Hop2 and Mnd1 proteins act in concert with Rad51 and Dmc1 in meiotic recombination. Nat Struct Mol Biol 12:449–453
Pezza RJ, Voloshin ON, Vanevski F, Camerini-Otero RD (2007) Hop2/Mnd1 acts on two critical steps in Dmc1-promoted homologous pairing. Genes Dev 21:1758–1766
Pham P, Bransteitter R, Petruska J, Goodman MF (2003) Processive AID-catalysed cytosine deamination on single-stranded DNA simulates somatic hypermutation. Nature 424:103–107
Pieper K, Tan J, Piccoli L, Foglierini M, Barbieri S, Chen Y, Silacci-Fregni C, Wolf T, Jarrossay D, Anderle M et al (2017) Public antibodies to malaria antigens generated by two LAIR1 insertion modalities. Nature 548:597–601
Pinto C, Kasaciunaite K, Seidel R, Cejka P (2016) Human DNA2 possesses a cryptic DNA unwinding activity that functionally integrates with BLM or WRN helicases. Elife 5. https://doi.org/10.7554/eLife.18574
Piwko W, Mlejnkova LJ, Mutreja K, Ranjha L, Stafa D, Smirnov A, Brodersen MM, Zellweger R, Sturzenegger A, Janscak P et al (2016) The MMS22L-TONSL heterodimer directly promotes RAD51-dependent recombination upon replication stress. EMBO J 35:2584–2601
Plank JL, Wu J, Hsieh TS (2006) Topoisomerase IIIalpha and Bloom’s helicase can resolve a mobile double Holliday junction substrate through convergent branch migration. Proc Natl Acad Sci U S A 103:11118–11123
Prakash R, Satory D, Dray E, Papusha A, Scheller J, Kramer W, Krejci L, Klein H, Haber JE, Sung P et al (2009) Yeast Mph1 helicase dissociates Rad51-made D-loops: implications for crossover control in mitotic recombination. Genes Dev 23:67–79
Prakash R, Zhang Y, Feng W, Jasin M (2015) Homologous recombination and human health: the roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harb Perspect Biol 7:a016600
Qi Z, Redding S, Lee JY, Gibb B, Kwon Y, Niu H, Gaines WA, Sung P, Greene EC (2015) DNA sequence alignment by microhomology sampling during homologous recombination. Cell 160:856–869
Qing Y, Yamazoe M, Hirota K, Dejsuphong D, Sakai W, Yamamoto KN, Bishop DK, Wu X, Takeda S (2011) The epistatic relationship between BRCA2 and the other RAD51 mediators in homologous recombination. PLoS Genet 7:e1002148
Rada C, Williams GT, Nilsen H, Barnes DE, Lindahl T, Neuberger MS (2002) Immunoglobulin isotype switching is inhibited and somatic hypermutation perturbed in UNG-deficient mice. Curr Biol 12:1748–1755
Ramsden DA, Gellert M (1998) Ku protein stimulates DNA end joining by mammalian DNA ligases: a direct role for Ku in repair of DNA double-strand breaks. EMBO J 17:609–614
Ranjha L, Anand R, Cejka P (2014) The Saccharomyces cerevisiae Mlh1-Mlh3 heterodimer is an endonuclease that preferentially binds to Holliday junctions. J Biol Chem 289:5674–5686
Rao HB, Qiao H, Bhatt SK, Bailey LR, Tran HD, Bourne SL, Qiu W, Deshpande A, Sharma AN, Beebout CJ et al (2017) A SUMO-ubiquitin relay recruits proteasomes to chromosome axes to regulate meiotic recombination. Science 355:403–407
Rass E, Grabarz A, Plo I, Gautier J, Bertrand P, Lopez BS (2009) Role of Mre11 in chromosomal nonhomologous end joining in mammalian cells. Nat Struct Mol Biol 16:819–824
Rass U, Compton SA, Matos J, Singleton MR, Ip SC, Blanco MG, Griffith JD, West SC (2010) Mechanism of Holliday junction resolution by the human GEN1 protein. Genes Dev 24:1559–1569
Ray Chaudhuri A, Callen E, Ding X, Gogola E, Duarte AA, Lee JE, Wong N, Lafarga V, Calvo JA, Panzarino NJ et al (2016) Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 535:382–387
Ray Chaudhuri A, Hashimoto Y, Herrador R, Neelsen KJ, Fachinetti D, Bermejo R, Cocito A, Costanzo V, Lopes M (2012) Topoisomerase I poisoning results in PARP-mediated replication fork reversal. Nat Struct Mol Biol 19:417–423
Renkawitz J, Lademann CA, Jentsch S (2014) Mechanisms and principles of homology search during recombination. Nat Rev Mol Cell Biol 15:369–383
Reynolds A, Qiao H, Yang Y, Chen JK, Jackson N, Biswas K, Holloway JK, Baudat F, de Massy B, Wang J et al (2013) RNF212 is a dosage-sensitive regulator of crossing-over during mammalian meiosis. Nat Genet 45:269–278
Robert T, Nore A, Brun C, Maffre C, Crimi B, Bourbon HM, de Massy B (2016) The TopoVIB-like protein family is required for meiotic DNA double-strand break formation. Science 351:943–949
Rogacheva MV, Manhart CM, Chen C, Guarne A, Surtees J, Alani E (2014) Mlh1-Mlh3, a meiotic crossover and DNA mismatch repair factor, is a Msh2-Msh3-stimulated endonuclease. J Biol Chem 289:5664–5673
Rossi MJ, Bugreev DV, Mazina OM, Mazin AV (2011) The RecA/RAD51 protein drives migration of Holliday junctions via polymerization on DNA. Proc Natl Acad Sci 108:6432–6437
Ryu T, Spatola B, Delabaere L, Bowlin K, Hopp H, Kunitake R, Karpen GH, Chiolo I (2015) Heterochromatic breaks move to the nuclear periphery to continue recombinational repair. Nat Cell Biol 17:1401–1411
Sakofsky CJ, Malkova A (2017) Break induced replication in eukaryotes: mechanisms, functions, and consequences. Crit Rev Biochem Mol Biol 52:395–413
Sartori AA, Lukas C, Coates J, Mistrik M, Fu S, Bartek J, Baer R, Lukas J, Jackson SP (2007) Human CtIP promotes DNA end resection. Nature 450:509–514
Schatz DG, Swanson PC (2011) V(D)J recombination: mechanisms of initiation. Annu Rev Genet 45:167–202
Schimmel J, Kool H, van Schendel R, Tijsterman M (2017) Mutational signatures of non-homologous and polymerase theta-mediated end-joining in embryonic stem cells. EMBO J 36:3634–3649
Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M (2011) Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145:529–542
Schlacher K, Wu H, Jasin M (2012) A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 22:106–116
Schrader CE, Guikema JE, Linehan EK, Selsing E, Stavnezer J (2007) Activation-induced cytidine deaminase-dependent DNA breaks in class switch recombination occur during G1 phase of the cell cycle and depend upon mismatch repair. J Immunol 179:6064–6071
Schrader CE, Linehan EK, Mochegova SN, Woodland RT, Stavnezer J (2005) Inducible DNA breaks in Ig S regions are dependent on AID and UNG. J Exp Med 202:561–568
Schwacha A, Kleckner N (1994) Identification of joint molecules that form frequently between homologs but rarely between sister chromatids during yeast meiosis. Cell 76:51–63
Schwacha A, Kleckner N (1997) Interhomolog bias during meiotic recombination: meiotic functions promote a highly differentiated interhomolog-only pathway. Cell 90:1123–1135
Schwendener S, Raynard S, Paliwal S, Cheng A, Kanagaraj R, Shevelev I, Stark JM, Sung P, Janscak P (2010) Physical interaction of RECQ5 helicase with RAD51 facilitates its anti-recombinase activity. J Biol Chem 285:15739–15745
Seigneur M, Bidnenko V, Ehrlich SD, Michel B (1998) RuvAB acts at arrested replication forks. Cell 95:419–430
Seol JH, Shim EY, Lee SE (2017) Microhomology-mediated end joining: good, bad and ugly. Mutat Res. https://doi.org/10.1016/j.mrfmmm.2017.07.002
Sfeir A, Symington LS (2015) Microhomology-mediated end joining: a back-up survival mechanism or dedicated pathway? Trends Biochem Sci 40:701–714
Shah Punatar R, Martin MJ, Wyatt HD, Chan YW, West SC (2017) Resolution of single and double Holliday junction recombination intermediates by GEN1. Proc Natl Acad Sci U S A 114:443–450
Shibata A (2017) Regulation of repair pathway choice at two-ended DNA double-strand breaks. Mutat Res 803–805:51–55
Shibata A, Moiani D, Arvai AS, Perry J, Harding SM, Genois MM, Maity R, van Rossum-Fikkert S, Kertokalio A, Romoli F et al (2014) DNA double-strand break repair pathway choice is directed by distinct MRE11 nuclease activities. Mol Cell 53:7–18
Shinohara A, Shinohara M, Ohta T, Matsuda S, Ogawa T (1998) Rad52 forms ring structures and co-operates with RPA in single-strand DNA annealing. Genes Cells 3:145–156
Shinohara M, Oh SD, Hunter N, Shinohara A (2008) Crossover assurance and crossover interference are distinctly regulated by the ZMM proteins during yeast meiosis. Nat Genet 40:299–309
Sigurdsson S, Van Komen S, Bussen W, Schild D, Albala JS, Sung P (2001) Mediator function of the human Rad51B-Rad51C complex in Rad51/RPA-catalyzed DNA strand exchange. Genes Dev 15:3308–3318
Simsek D, Brunet E, Wong SY, Katyal S, Gao Y, McKinnon PJ, Lou J, Zhang L, Li J, Rebar EJ et al (2011) DNA ligase III promotes alternative nonhomologous end-joining during chromosomal translocation formation. PLoS Genet 7:e1002080
Simsek D, Jasin M (2010) Alternative end-joining is suppressed by the canonical NHEJ component Xrcc4-ligase IV during chromosomal translocation formation. Nat Struct Mol Biol 17:410–416
Singh TR, Ali AM, Busygina V, Raynard S, Fan Q, Du CH, Andreassen PR, Sung P, Meetei AR (2008) BLAP18/RMI2, a novel OB-fold-containing protein, is an essential component of the Bloom helicase-double Holliday junction dissolvasome. Genes Dev 22:2856–2868
Snowden T, Acharya S, Butz C, Berardini M, Fishel R (2004) hMSH4-hMSH5 recognizes Holliday junctions and forms a meiosis-specific sliding clamp that embraces homologous chromosomes. Mol Cell 15:437–451
Sogo JM, Lopes M, Foiani M (2002) Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science 297:599–602
Solinger JA, Kiianitsa K, Heyer WD (2002) Rad54, a Swi2/Snf2-like recombinational repair protein, disassembles Rad51:dsDNA filaments. Mol Cell 10:1175–1188
Song B, Sung P (2000) Functional interactions among yeast Rad51 recombinase, Rad52 mediator, and replication protein A in DNA strand exchange. J Biol Chem 275:15895–15904
Sourirajan A, Lichten M (2008) Polo-like kinase Cdc5 drives exit from pachytene during budding yeast meiosis. Genes Dev 22:2627–2632
Stephens PJ, McBride DJ, Lin ML, Varela I, Pleasance ED, Simpson JT, Stebbings LA, Leroy C, Edkins S, Mudie LJ et al (2009) Complex landscapes of somatic rearrangement in human breast cancer genomes. Nature 462:1005–1010
Sturzenegger A, Burdova K, Kanagaraj R, Levikova M, Pinto C, Cejka P, Janscak P (2014) DNA2 cooperates with the WRN and BLM RecQ helicases to mediate long-range DNA end resection in human cells. J Biol Chem 289:27314–27326
Sugiyama T, Zaitseva EM, Kowalczykowski SC (1997) A single-stranded DNA-binding protein is needed for efficient presynaptic complex formation by the Saccharomyces cerevisiae Rad51 protein. J Biol Chem 272:7940–7945
Sun H, Treco D, Schultes NP, Szostak JW (1989) Double-strand breaks at an initiation site for meiotic gene conversion. Nature 338:87–90
Sun H, Treco D, Szostak JW (1991) Extensive 3′-overhanging, single-stranded DNA associated with the meiosis-specific double-strand breaks at the ARG4 recombination initiation site. Cell 64:1155–1161
Sung P (1994) Catalysis of ATP-dependent homologous DNA pairing and strand exchange by yeast RAD51 protein. Science 265:1241–1243
Sung P (1997) Yeast Rad55 and Rad57 proteins form a heterodimer that functions with replication protein A to promote DNA strand exchange by Rad51 recombinase. Genes Dev 11:1111–1121
Svendsen JM, Smogorzewska A, Sowa ME, O'Connell BC, Gygi SP, Elledge SJ, Harper JW (2009) Mammalian BTBD12/SLX4 assembles a Holliday junction resolvase and is required for DNA repair. Cell 138:63–77
Sy SM, Huen MS, Chen J (2009) PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proc Natl Acad Sci U S A 106:7155–7160
Sym M, Roeder GS (1994) Crossover interference is abolished in the absence of a synaptonemal complex protein. Cell 79:283–292
Symington LS, Gautier J (2011) Double-strand break end resection and repair pathway choice. Annu Rev Genet 45:247–271
Szostak JW, Orr-Weaver TL, Rothstein RJ, Stahl FW (1983) The double-strand-break repair model for recombination. Cell 33:25–35
Taglialatela A, Alvarez S, Leuzzi G, Sannino V, Ranjha L, Huang JW, Madubata C, Anand R, Levy B, Rabadan R et al (2017) Restoration of replication fork stability in BRCA1- and BRCA2-deficient cells by inactivation of SNF2-family fork remodelers. Mol Cell 68:414–430
Tan J, Pieper K, Piccoli L, Abdi A, Perez MF, Geiger R, Tully CM, Jarrossay D, Maina Ndungu F, Wambua J et al (2016) A LAIR1 insertion generates broadly reactive antibodies against malaria variant antigens. Nature 529:105–109
Taylor MR, Spirek M, Chaurasiya KR, Ward JD, Carzaniga R, Yu X, Egelman EH, Collinson LM, Rueda D, Krejci L et al (2015) Rad51 paralogs remodel pre-synaptic Rad51 filaments to stimulate homologous recombination. Cell 162:271–286
Taylor MR, Spirek M, Jian Ma C, Carzaniga R, Takaki T, Collinson LM, Greene EC, Krejci L, Boulton SJ (2016) A polar and nucleotide-dependent mechanism of action for RAD51 paralogs in RAD51 filament remodeling. Mol Cell 64:926–939
Thangavel S, Berti M, Levikova M, Pinto C, Gomathinayagam S, Vujanovic M, Zellweger R, Moore H, Lee EH, Hendrickson EA et al (2015) DNA2 drives processing and restart of reversed replication forks in human cells. J Cell Biol 208:545–562
Thorslund T, McIlwraith MJ, Compton SA, Lekomtsev S, Petronczki M, Griffith JD, West SC (2010) The breast cancer tumor suppressor BRCA2 promotes the specific targeting of RAD51 to single-stranded DNA. Nat Struct Mol Biol 17:1263–1265
Tran PT, Erdeniz N, Dudley S, Liskay RM (2002) Characterization of nuclease-dependent functions of Exo1p in Saccharomyces cerevisiae. DNA Repair (Amst) 1:895–912
Tsai SP, Su GC, Lin SW, Chung CI, Xue X, Dunlop MH, Akamatsu Y, Jasin M, Sung P, Chi P (2012) Rad51 presynaptic filament stabilization function of the mouse Swi5-Sfr1 heterodimeric complex. Nucleic Acids Res 40:6558–6569
Tse YC, Kirkegaard K, Wang JC (1980) Covalent bonds between protein and DNA. Formation of phosphotyrosine linkage between certain DNA topoisomerases and DNA. J Biol Chem 255:5560–5565
Tubbs A, Nussenzweig A (2017) Endogenous DNA damage as a source of genomic instability in cancer. Cell 168:644–656
Uziel T, Lerenthal Y, Moyal L, Andegeko Y, Mittelman L, Shiloh Y (2003) Requirement of the MRN complex for ATM activation by DNA damage. EMBO J 22:5612–5621
Vaisman A, Woodgate R (2017) Translesion DNA polymerases in eukaryotes: what makes them tick? Crit Rev Biochem Mol Biol 52:274–303
van Mameren J, Modesti M, Kanaar R, Wyman C, Peterman EJ, Wuite GJ (2009) Counting RAD51 proteins disassembling from nucleoprotein filaments under tension. Nature 457:745–748
Veaute X, Jeusset J, Soustelle C, Kowalczykowski SC, Le Cam E, Fabre F (2003) The Srs2 helicase prevents recombination by disrupting Rad51 nucleoprotein filaments. Nature 423:309–312
Vujanovic M, Krietsch J, Raso MC, Terraneo N, Zellweger R, Schmid JA, Taglialatela A, Huang JW, Holland CL, Zwicky K et al (2017) Replication fork slowing and reversal upon DNA damage require PCNA polyubiquitination and ZRANB3 DNA translocase activity. Mol Cell 67:882–890 e885
Wang H, Li Y, Truong LN, Shi LZ, Hwang PY, He J, Do J, Cho MJ, Li H, Negrete A et al (2014) CtIP maintains stability at common fragile sites and inverted repeats by end resection-independent endonuclease activity. Mol Cell 54:1012–1021
Wang M, Wu W, Wu W, Rosidi B, Zhang L, Wang H, Iliakis G (2006) PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res 34:6170–6182
Ward JF (1988) DNA damage produced by ionizing radiation in mammalian cells: identities, mechanisms of formation, and reparability. Prog Nucleic Acid Res Mol Biol 35:95–125
Wechsler T, Newman S, West SC (2011) Aberrant chromosome morphology in human cells defective for Holliday junction resolution. Nature 471:642–646
Wei K, Clark AB, Wong E, Kane MF, Mazur DJ, Parris T, Kolas NK, Russell R, Hou H Jr, Kneitz B et al (2003) Inactivation of exonuclease 1 in mice results in DNA mismatch repair defects, increased cancer susceptibility, and male and female sterility. Genes Dev 17:603–614
Wilson MA, Kwon Y, Xu Y, Chung WH, Chi P, Niu H, Mayle R, Chen X, Malkova A, Sung P et al (2013) Pif1 helicase and Poldelta promote recombination-coupled DNA synthesis via bubble migration. Nature 502:393–396
Wold MS (1997) Replication protein A: a heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu Rev Biochem 66:61–92
Wolner B, Peterson CL (2005) ATP-dependent and ATP-independent roles for the Rad54 chromatin remodeling enzyme during recombinational repair of a DNA double strand break. J Biol Chem 280:10855–10860
Wright WD, Heyer WD (2014) Rad54 functions as a heteroduplex DNA pump modulated by its DNA substrates and Rad51 during D loop formation. Mol Cell 53:420–432
Wu L, Hickson ID (2003) The Bloom’s syndrome helicase suppresses crossing over during homologous recombination. Nature 426:870–874
Wyatt HD, Laister RC, Martin SR, Arrowsmith CH, West SC (2017) The SMX DNA repair tri-nuclease. Mol Cell 65:848–860 e811
Wyatt HD, Sarbajna S, Matos J, West SC (2013) Coordinated actions of SLX1-SLX4 and MUS81-EME1 for Holliday junction resolution in human cells. Mol Cell 52:234–247
Xie A, Kwok A, Scully R (2009) Role of mammalian Mre11 in classical and alternative nonhomologous end joining. Nat Struct Mol Biol 16:814–818
Xue X, Raynard S, Busygina V, Singh AK, Sung P (2013) Role of replication protein A in double holliday junction dissolution mediated by the BLM-Topo IIIalpha-RMI1-RMI2 protein complex. J Biol Chem 288:14221–14227
Yabuki M, Fujii MM, Maizels N (2005) The MRE11-RAD50-NBS1 complex accelerates somatic hypermutation and gene conversion of immunoglobulin variable regions. Nat Immunol 6:730–736
Yang H, Jeffrey PD, Miller J, Kinnucan E, Sun Y, Thoma NH, Zheng N, Chen PL, Lee WH, Pavletich NP (2002) BRCA2 function in DNA binding and recombination from a BRCA2-DSS1-ssDNA structure. Science 297:1837–1848
Yang H, Li Q, Fan J, Holloman WK, Pavletich NP (2005) The BRCA2 homologue Brh2 nucleates RAD51 filament formation at a dsDNA-ssDNA junction. Nature 433:653–657
Yildiz O, Majumder S, Kramer B, Sekelsky JJ (2002) Drosophila MUS312 interacts with the nucleotide excision repair endonuclease MEI-9 to generate meiotic crossovers. Mol Cell 10:1503–1509
Yuan SS, Lee SY, Chen G, Song M, Tomlinson GE, Lee EY (1999) BRCA2 is required for ionizing radiation-induced assembly of Rad51 complex in vivo. Cancer Res 59:3547–3551
Zadorozhny K, Sannino V, Belan O, Mlcouskova J, Spirek M, Costanzo V, Krejci L (2017) Fanconi-anemia-associated mutations destabilize RAD51 filaments and impair replication fork protection. Cell Rep 21:333–340
Zahn A, Eranki AK, Patenaude AM, Methot SP, Fifield H, Cortizas EM, Foster P, Imai K, Durandy A, Larijani M et al (2014) Activation induced deaminase C-terminal domain links DNA breaks to end protection and repair during class switch recombination. Proc Natl Acad Sci U S A 111:E988–E997
Zaitseva EM, Zaitsev EN, Kowalczykowski SC (1999) The DNA binding properties of Saccharomyces cerevisiae Rad51 protein. J Biol Chem 274:2907–2915
Zakharyevich K, Ma Y, Tang S, Hwang PY, Boiteux S, Hunter N (2010) Temporally and biochemically distinct activities of Exo1 during meiosis: double-strand break resection and resolution of double Holliday junctions. Mol Cell 40:1001–1015
Zakharyevich K, Tang S, Ma Y, Hunter N (2012) Delineation of joint molecule resolution pathways in meiosis identifies a crossover-specific resolvase. Cell 149:334–347
Zan H, Wu X, Komori A, Holloman WK, Casali P (2003) AID-dependent generation of resected double-strand DNA breaks and recruitment of Rad52/Rad51 in somatic hypermutation. Immunity 18:727–738
Zelensky AN, Schimmel J, Kool H, Kanaar R, Tijsterman M (2017) Inactivation of Pol theta and C-NHEJ eliminates off-target integration of exogenous DNA. Nat Commun 8:66
Zellweger R, Dalcher D, Mutreja K, Berti M, Schmid JA, Herrador R, Vindigni A, Lopes M (2015) Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J Cell Biol 208:563–579
Zhang L, Wang S, Yin S, Hong S, Kim KP, Kleckner N (2014) Topoisomerase II mediates meiotic crossover interference. Nature 511:551–556
Zhang Y, Hefferin ML, Chen L, Shim EY, Tseng HM, Kwon Y, Sung P, Lee SE, Tomkinson AE (2007) Role of Dnl4-Lif1 in nonhomologous end-joining repair complex assembly and suppression of homologous recombination. Nat Struct Mol Biol 14:639–646
Zhang Y, Jasin M (2011) An essential role for CtIP in chromosomal translocation formation through an alternative end-joining pathway. Nat Struct Mol Biol 18:80–84
Zhao W, Steinfeld JB, Liang F, Chen X, Maranon DG, Jian Ma C, Kwon Y, Rao T, Wang W, Sheng C et al (2017) BRCA1-BARD1 promotes RAD51-mediated homologous DNA pairing. Nature 550:360–365
Zhao W, Vaithiyalingam S, San Filippo J, Maranon DG, Jimenez-Sainz J, Fontenay GV, Kwon Y, Leung SG, Lu L, Jensen RB et al (2015) Promotion of BRCA2-dependent homologous recombination by DSS1 via RPA targeting and DNA mimicry. Mol Cell 59:176–187
Zhu Z, Chung WH, Shim EY, Lee SE, Ira G (2008) Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell 134:981–994
Zou L, Elledge SJ (2003) Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 300:1542–1548
Acknowledgements
We would like to thank the members of the Cejka Laboratory (IRB Bellinzona), Massimo Lopes (University of Zurich), and Kathrin Pieper (Lanzavecchia Laboratory, IRB Bellinzona) for comments on the manuscript. We apologize to colleagues whose work could not be cited due to length constraint.
Funding
The work was supported by grants from the European Research Council (681630) and the Swiss National Science Foundation (31003A_175444) to P.C.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
This article does not contain studies involving animals or human participants by any of the authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Ranjha, L., Howard, S. & Cejka, P. Main steps in DNA double-strand break repair: an introduction to homologous recombination and related processes. Chromosoma 127, 187–214 (2018). https://doi.org/10.1007/s00412-017-0658-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00412-017-0658-1