
Abstract
The desert locust (Schistocerca gregaria) has been used as material for numerous cytogenetic studies. Its genome size is estimated to be 8.55 Gb of DNA comprised in 11 autosomes and the X chromosome. Its X0/XX sex chromosome determinism therefore results in females having 24 chromosomes whereas males have 23. Surprisingly, little is known about the DNA content of this locust’s huge chromosomes. Here, we use the Feulgen Image Analysis Densitometry and C-banding techniques to respectively estimate the DNA quantity and heterochromatin content of each chromosome. We also identify three satellite DNAs using both restriction endonucleases and next-generation sequencing. We then use fluorescent in situ hybridization to determine the chromosomal location of these satellite DNAs as well as that of six tandem repeat DNA gene families. The combination of the results obtained in this work allows distinguishing between the different chromosomes not only by size, but also by the kind of repetitive DNAs that they contain. The recent publication of the draft genome of the migratory locust (Locusta migratoria), the largest animal genome hitherto sequenced, invites for sequencing even larger genomes. S. gregaria is a pest that causes high economic losses. It is thus among the primary candidates for genome sequencing. But this species genome is about 50 % larger than that of L. migratoria, and although next-generation sequencing currently allows sequencing large genomes, sequencing it would mean a greater challenge. The chromosome sizes and markers provided here should not only help planning the sequencing project and guide the assembly but would also facilitate assigning assembled linkage groups to actual chromosomes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Several are the particularities that make species of the Orthoptera order worth studying. They, for instance, show some of the most striking morphologies, like the armored-looking Plagiotriptus carli (Thericleidae) and the leaf-looking Systella dusmeti (Trigonopterygidae). They also exhibit some of the most astonishing population dynamics and behaviors. Of these, the gregariousness and swarming of some Orthoptera pest species are notorious cases of behavioral adaptations that give some of these species the status of ‘economically interesting’ (in the negative sense of the word). Among these, the desert locust (Schistocerca gregaria) is infamous for the recurring outbreaks its populations experience in Africa and parts of Asia.
With known genome sizes ranging from 1.52 Gb, estimated for the cave cricket Hadenoecus subterraneus (Rasch and Rasch 1981), to the enormous 16.56 Gb, estimated for the mountain grasshopper Podisma pedestris (Westerman et al. 1987), orthopterans exhibit a high degree of genome size variability while containing some of the largest genome-containing species. This seems in agreement with the positive correlation between genome size and the rate of genome size evolution proposed by Oliver et al. (2007). In such a case, the observed diversity in orthopteran genome sizes might be due to a high rate of genome size evolution in this group facilitated by the large genome sizes of its ancestral members. There is an apparent absence of frequent whole genome duplication events in orthoptera—for instance, only two out of the over 100 polyploid species reported in Otto and Whitton (2000) are orthopteran. A key factor for both the size and variation of the orthopteran genomes should thus be the contribution of tandem repeat DNAs and transposable elements (TEs). These were proven to increase genomic sizes and diversity in other organisms (e.g., Bennetzen 2005; Gregory 2005; Oliver et al. 2007). In fact, the extensive occurrence of repetitive elements in grasshopper genomes has recently been corroborated after whole genome sequencing of the migratory locust (Locusta migratoria) genome, whose ∼6.3-Gb size makes it the largest animal genome hitherto sequenced (Wang et al. 2014).
Since 2010, we are engaged in a research program aiming at the analysis of several aspects related to the S. gregaria genetics (see Bakkali 2013). A first Sanger-based sequencing of a S. gregaria transcriptome was carried out for the central nervous system (CNS) by Badisco et al. (2011). To complement it, we carried out comparative next-generation sequencings of different tissues and physiological states, with results that should allow for deep insights into the molecular basis of locust gregariousness (in preparation). Still, on top of whatever differentially expressed genes will be the epigenetics—meaning that, sooner or later, epigenetics studies (including bisulfite sequencing) need to be done (Bakkali 2013). For that, the availability of a sequenced genome is more than helpful. S. gregaria genome has an estimated size of 8.55 Gb (Fox 1970; John and Hewitt 1966; Wilmore and Brown 1975), almost three times the size of the human genome. Its sequencing and assembly would thus be a difficult task due to the inability of the currently available sequence assemblers to efficiently deal with such repeat-enriched genomes. Still, the S. gregaria genome is no doubt worth sequencing, since it would not only produce useful data for understanding the genetics of gregariousness, and for the fight against outbreaks, but would also provide sequences for comparative studies.
With this in mind, and in line with the workflow and genetic analyses suggested in Bakkali (2013) for applying to pest locusts, here we start by a study of the chromosomes. Of course, ours is not the first chromosome-level work on S. gregaria. This species has previously been studied for several purposes, such as the influence of several factors on chiasma frequency (Craig-Cameron 1970; Csik and Koller 1939; Henderson 1962, 1963, 1964, 1988; White 1934), chiasma distribution (Fox 1973), and meiotic behavior (John and Naylor 1961), the effect of X-rays (Fox 1966a, b, 1967a, b; Westerman 1967, 1968) or actinomycin-D (Jain and Singh 1967) on chromosomes, the nature of crossing-over (Craig-Cameron and Jones 1970; Jones 1977), chromosome ultrastructure (Wolf 1996; Wolf and Sumner 1996), chromosome histone H4 acetylation (Wolf and Turner 1996), presence of mitochondrial DNA on chromosomes (Bensasson et al. 2000; Vaughan et al. 1999), and the location of some Hox genes (Ferrier and Akam 1996).
Still, almost nothing is known about the DNA content and molecular composition of the S. gregaria chromosomes. So, here, we provide a number of chromosome-identifying markers. For that, we first estimated the DNA content of every chromosome to get a quantitative idea of the differences between them. We also searched for satellite DNAs (satDNA) both after restriction endonuclease digestion of the genomic DNA as well as after Illumina genome sequencing and in silico search for tandemly repeated sequences. We then carried out chromosome banding and in situ mapping of the three resulting satDNAs as well as six genomic markers. In addition, we estimated the expression levels of the satDNAs in Illumina-sequenced transcriptomes from four different tissues.
This work should be useful to any future sequencing project on the S. gregaria genome, as it might be useful for mapping some of the assembled genomic contigs to their corresponding chromosomes and would be yet another step in the characterization of the chromosomes and genome of this important pest species.
Materials and methods
Biological samples, chromosome banding, and DNA extraction
Males and females of the desert locust (S. gregaria) from our laboratory colony, whose origin were four egg pods kindly offered to us in 2009 by Prof. Jozef Vanden Broeck (KU Leuven, Belgium), were crossed to obtain 5-day embryos for cytological analysis. Eggs were incubated at 27 °C. Three males were anesthetized, and testes were dissected out and fixed in 3:1 absolute ethanol–acetic acid. Meiotic and mitotic chromosomes were obtained from testes and embryos following the protocols described in Camacho et al. (1991). C-banding and fluorochrome banding were performed as described in Camacho et al. (1984). Genomic DNA (gDNA) was obtained from an inbred female using the method described for single flies (Bakkali 2011).
Chromosome size estimation
The DNA content of each autosome was assessed using the Feulgen Image Analysis Densitometry (FIAD) technique as described in Ruiz-Ruano et al. (2011). For this purpose, and for each male, we measured the chromosome bivalents from eight primary spermatocytes at diplotene or metaphase I. We restricted the analysis to cells where all the 11 autosomal bivalents and the X chromosome univalent were well separated. The obtained Integrated Optical Density (IOD) values were arranged in order of decreasing value and were assigned to the autosomes in order of increasing number (L1 to S11). We then calculated the IOD ratio for each autosome (with respect to the total autosomal IOD) by dividing each IOD value by the sum of the IOD values of all the 11 autosomes of the cell.
We did not quantify the DNA content of the X chromosome in the same way as we did for the 11 autosomes, since its different heteropycnosis state during diplotene and metaphase I, compared to the autosomes, could distort measurement (Hardie et al. 2002). Instead, we took advantage of the fact that grasshoppers show an X0/XX sex chromosome determinism, which implies that a male produces two types of spermatids, ones carrying the X chromosome and the others lacking it. We measured the whole cell (nucleus) IOD in 50 spermatids from the same three males. This yielded a bimodal distribution with two peaks corresponding to the X-carrying (X+) and X-lacking (X−) spermatids, the latter logically showing lower IOD. The DNA content of the X chromosome was thus inferred from the difference between the two IOD peaks (see Ruiz-Ruano et al. 2011). The IOD ratio of the X chromosome was calculated as the difference between the two IOD peaks divided by the IOD of the X+ peak. It was then multiplied by the C-value (8.74 pg, on average (Fox 1970; John and Hewitt 1966; Wilmore and Brown 1975)) to convert it to DNA quantity (in pg). The total quantity of DNA in the autosomes was inferred by subtracting the DNA quantity in the X chromosome from the C-value. The DNA amount (in pg) in each autosome was then calculated by multiplying its IOD ratio by the total pg of DNA in all the autosomes. Finally, the mass was converted to gigabase pairs (Gbp) taking into account that 1 pg of DNA corresponds to about 0.978 Gbp (Dolezel et al. 2003).
Satellite DNA identification
To search for satDNAs in the S. gregaria genome, we first separately digested the gDNA using a set of 14 different common six-cutter restriction endonucleases that we usually use for such purposes (given that we had no prior information on this species genome, we used all the endonucleases available to us with no prior selection). We then isolated the appropriate bands from the electrophoresis gels of the digestions that gave ladder patterns. Identification of the satDNA sequences was then reached after cloning and sequencing (see protocols in Lorite et al. (2013)). This allowed us to isolate a satDNA with a repeat unit of about 350 bp. A second approach, based on Illumina HiSeq 2000 Paired-end sequencing of the gDNA, was used in order to detect other satDNAs. The paired-end reads were joined using the “fastq-join” software of the FASTX-Toolkit suit (Gordon and Hannon 2010) with default options. Of the joined reads, 500,000 sequences were randomly selected for clustering, using the RepeatExplorer (Novak et al. 2013) server at http://www.repeatexplorer.org (as options; the paired-end reads were considered, and 55 and 40 bp were respectively taken as minimum overlap length for clustering and assembly). We then searched for clusters that show high graph density, a typical characteristic of satDNA families (Novak et al. 2010). We manually processed the assembled contigs using Geneious v4.8 (Drummond et al. 2009) using the High Sensitivity/Slow option, and we visualized the dotplot graphics in order to determine tandem repetitions, split them in monomers, then aligned and got a consensus sequence of the monomeric units. We then used the assembled DNA sequences to design primers (Table S1), using the Primer3 software (Rozen and Skaletsky 1999), and we polymerase chain reaction (PCR)-amplified the three satDNAs detected in this work.
Transcription of satellite DNAs
Sets of Illumina sequencing reads, belonging to S. gregaria CNS, muscle, testis, and ovary transcriptome projects that are in preparation in our lab (unpublished data), were used to estimate the expression levels of some of the repeated sequences in each of the abovementioned tissues. For this purpose, all the raw sequencing reads from each tissue-sequencing library were separately aligned to each of the repeated sequences using BWA (Li and Durbin 2009). The succession of commands for BWA were as follows: bwa index -a is file.fasta, then bwa aln -t 2 file.fasta reads1.fastq (for the file.sai1) then bwa aln -t 2 file.fasta reads2.fastq (for the file.sai2) then bwa sampe -s file.fasta file.sai1 file.sai2 (for the file.sam). The aligned reads were counted using the htseq-count script of the HTSeq (Anders et al. 2014) program with -q -s no -m intersection-nonempty as options. The read counts were then compared between sequences and tissues after normalization (i.e., division by the total number of reads in each tissue sequencing library). For comparative purposes, the normalized numbers were taken as indicative of the relative degree of expression of each satDNA in each tissue. The relative abundance of the respective sequences in the genome was calculated by dividing the number of reads from the genomic sequencing that align to the respective satDNA by the total number of reads in the genome sequencing library. The ratio of the normalized numbers of read counts corresponding to a satDNA in a transcriptome, and the relative abundance of the same satDNA in the genome gave us an indication of the degree of expression of that satDNA in the tissue in question relative to its abundance in the genome.
Fluorescent in situ hybridization
Mapping of the DNAs on S. gregaria chromosomes was carried out following the fluorescent in situ hybridization (FISH) protocol described in Cabrero et al. (2003). In addition to the three satDNAs detected in this work, we took advantage of the conservation of primers that work for us in other orthopteran species, to amplify sequences of multi-copy genes that are often used as chromosomal markers (they usually form FISH-detectable clusters). We thus amplified, labelled, and mapped the 18S and 5S rRNA genes, the histone H3 genes, U1 and U2 snRNA genes, and the telomeric DNA. The 18S probe was obtained by PCR amplification of a 1,113-bp fragment of the Eyprepocnemis plorans 18S ribosomal gene using the 18S-E and 1100R primers designed by Timothy et al. (2000). The PCR consisted of an initial denaturation at 94 °C for 3 min, followed by 30 cycles of 94 °C for 30 s, 45 °C for 1 min, 72 °C for 2 min, and a final extension of 72 °C for 7 min. 5S rRNA and H3 histone genes probes were obtained as described in Cabrero et al. (2003) and Cabrero et al. (2009), respectively. For amplification of the U1 and U2 snDNA fragments, we used the primers and conditions described by Cabral-de-Mello et al. (2012) and Bueno et al. (2013), respectively. As telomeric probe, we used the fluorescein-labelled synthetic deoxyoligomers (GGTTA)x7 and (TAACC)x7 described in Meyne et al. (1995). Probes of the three satDNAs detected in this work were obtained after PCR amplification using the primers designed as described above (see Table S1) and the following reaction conditions: initial denaturation for 5 min at 94 °C, 35 cycles of 20 s at 94 °C, 30 s at 62 °C and 15 s at 72 °C, and a final extension step at 72 °C for 7 min. PCR products of the three satDNA amplifications were visualized in a 1.5 % agarose gel. Ladder-like band patterns were obtained in the agarose gel column corresponding to each of the three PCRs as expected for our satDNAs whose previously determined monomer lengths (see above) are 171, 352, and 170 bp, respectively. After excision from the gel and PCR re-amplification of 10 ng of DNA using the same PCR primers and cycling as the first PCR, the reamplified DNA from each monomer gel band was sequenced by Macrogen Inc. For direct detection of the hybridization signals, probes were labelled by nick-translation using tetramethylrhodamine-11-dUTP (5S, histone H3, SG1 and SG3) or fluorescein-11-dUTP (18S, SG2-alpha, SG1). The U1 and U2 snDNA probes, however, were PCR-labelled using digoxigenin-11-dUTP and detected using anti-digoxigenin rhodamine (Roche).
Double FISH was carried out by simultaneously combining two DNA probes labelled with different fluorochromes. In order to show the three satDNAs in a same cell, double FISH was first used for two of the differently labelled probes and the result documented by photographing cells and taking their coordinates in the x–y axes of the microscope stage. Slide washing in 4XSSC/Tween 20 for 0.5 h at room temperature and in 2XSSC at 42 °C for 1 h removed the hybridized probes from the chromosomes. This allowed us to carry out a new hybridization with the third remaining satDNA. After photographing the same cells as in the earlier double-FISH and replacing the red color of the third probe by a yellow color, the images with the results of the three satDNA hybridizations were merged using the Gimp software. For photography, slides were counterstained using DAPI and mounted in Vectashield (Vector, USA). Fiber-FISH was carried out as described in Muñoz-Pajares et al. (2011). Hybridization signals were observed under a BX41 epifluorescence Olympus microscope equipped with a DP70 cooled digital camera for photography and after application of the appropriate filters. Images were merged and optimized for brightness and contrast using the Gimp software.
Statistical analyses
Shapiro-Wilk’s W test was used to check whether the IOD ratio variable fitted a normal distribution, and then non-parametric Kruskal–Wallis ANOVA and t test for dependent samples were applied to compare the IOD ratio for each chromosome between individuals and IOD ratio between consecutive autosomes, respectively. All analyses were carried out using the Statistica 6.0 software (Statsoft Inc.).
Results
DNA content of each S. gregaria chromosome
Likewise most acridid species, S. gregaria X0/XX sex chromosome determinism results in a chromosome complement consisting of 23 apparently telocentric chromosomes in males and 24 in females. The autosomes can be classified into three size groups: long (L1–L3), medium (M4–M8), and short (S9–S11), the X chromosome being the second element in size (Fig. 1a). After measuring the DNA content of each chromosome, using the FIAD technique, the combined IOD ratio variable failed to fit a normal distribution (Shapiro–Wilk’s W test, p < 0.05), for which reason we used nonparametric Kruskal–Wallis ANOVA to compare the IOD ratios between males and chromosomes. While the differences between the three analyzed males were not significant (p = 0.992), there were highly significant differences between the different chromosomes (p < 2.2−16). The results of the technique employed were therefore consistent between the different males, and they discriminated very well between the chromosome sets of the complement. Within individual chromosomes, Shapiro–Wilk’s W test suggested that the IOD values of each single chromosome follow a normal distribution (p > 0.05) except for chromosome S9, whose p = 0.041 could be attributed to type I error. To compare IOD values between consecutive autosomes, we performed t tests for dependent samples which showed significant differences in all cases (Table 1). This indicates that the FIAD technique discriminates very well even between the autosomes showing the smallest size difference. The X chromosome was excluded from the t test because its IOD value was indirectly inferred (see “Material and methods”). The coefficients of variation of the IOD ratios were lower than 10 % for 30 out of the 33 autosomes of the three analyzed males (the S9, S10, and S11 chromosomes of the third male being the only exception). Our results therefore meet Hardie et al. (2002) criterion for the FIAD technique. As a whole, the measurements in the three males allowed us to calculate the proportion that each autosome represents with respect to the total autosomal DNA (IOD ratio) (Table 1). Our estimates of the DNA content in the X chromosome showed that it comprises about 13.72 % of the C-value (i.e., 1.2 pg, equivalent to 1.17 Gbp), the total DNA content of the 11 autosomes thus being 7.54 pg (Table 1). Our results therefore provide quantitative measurements of the sizes of each chromosome of the S. gregaria genome and confirm that the X chromosome is the second largest element in size.

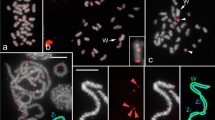
Characterization of the S. gregaria chromosomes by C-banding (a), DAPI and CMA3 banding (b–d), and FISH (e) and fiber-FISH (f–i) with 18S and 5S rDNA probes. Note the C-bands at the centromeric regions of all the chromosomes and the distal parts of the three smallest ones. Also note the CMA3 bands of the chromosomes L3, M6, and M8 as well as the coincidence between the first two and some of the 18S and 5S rDNA FISH signals. The two 18S rDNA signals detected on chromosomes L3 and M6 coincide in location with two of the five 5S rDNA signals (e), and fiber FISH reveals that the co-localization actually corresponds to intermingled clusters of both rDNAs (f–i). The 5S rDNA also appears alone on the distal region of the chromosome L3 and the proximal and interstitial parts of the chromosome M5 (e). The bar in figure (a) corresponds to 5 μm, and the asterisks in figure h mark some of the 18S and 5S rDNA hybridization sites as revealed by fiber FISH
C-banding and DA-DAPI-CMA3 banding
The C-banding pattern of the S. gregaria chromosome complement consists of paracentromeric C-positive bands in all chromosomes and distal bands in the three smallest autosomes (Fig. 1a). The fluorescence pattern revealed by the triple DA-DAPI-CMA3 staining revealed the absence of differential staining for DAPI (Fig. 1b) and the presence of conspicuous CMA3+ interstitial bands in three chromosome pairs, i.e., L3, M6, and M8 (Fig. 1c).
Chromosome mapping of the rDNA, histone, U snDNA, and telomeric sequences
FISH with the 18S and 5S rDNA probes showed the presence of 18S rDNA at interstitial locations of both L3 and M6 chromosomes (Fig. 1e). The 5S rDNA is also interstitially located in these two chromosomes. In addition, other FISH signals for the 5S rDNA are located on a distal region of the L3 chromosome as well as in a proximal and an interstitial region of the M5 chromosome (Fig. 1e). Remarkably, merging of the two FISH images suggests that the two interstitial 18S rDNA bands in chromosomes L3 and M6 coincide in location with 5S rDNA bands. Indeed, fiber FISH analysis showed that these chromosomal regions harbor mixed clusters of the two types of rDNA, meaning that the overlap of the FISH signals in the merged image is due to genuine co-localization rather than imprecision or optical effect of the FISH or image merging (Fig. 1f–i).
FISH with the H3 histone gene probe showed the presence of a single interstitial cluster in the M8 chromosome (Fig. 2a), a conserved location in most acridid grasshoppers whose karyotype is composed of 11 autosomal pairs and the X chromosome (Cabrero and Camacho 2008). The U1 and U2 snDNAs are both interstitially located in the longest chromosome (L1), but they do not co-localize since U1 is located in the distal half of that chromosome (Fig. 2b) whereas U2 is located in the proximal half (Fig. 2c). FISH with the telomeric DNA probe showed its presence in all chromosome ends. Interestingly, the two smallest autosomes (S10 and S11) carried additional interstitial telomeric DNA (Fig. 2d).

FISH mapping of the histone H3 (a), U1 and U2 snDNA (b and c), telomeric DNA (d), SG1 and SG2 satDNAs (e), SG1 and SG3 satDNAs (f), SG2-alpha and SG3 satDNAs (g) and SG1, SG2-alpha and SG3 satDNAs (h). Fiber-FISH reveals the intermingling between some of the telomeric DNA and the SG1 and SG3 satDNAs (i and j). Chromosomes of the FISH preparations were counterstained using DAPI. Note that the histone H3 signal is apparent only on chromosome M8, coinciding with the only CMA3+ band that do not coincide with the rDNAs in Fig. 1. U1 and U2 snDNAs both locate to different interstitial regions of the largest chromosome (L1) and, for its part, the telomeric DNA locates to telomeres of all chromosomes as well as to interstitial regions of the two smallest chromosomes (S10 and S11). SG1 satDNA is present at the pericentromeric regions of all the chromosomes of the complement; SG2-alpha is distally located on the three shortest chromosomes (coinciding with the distal C-bands in Fig. 1), whereas SG3 is restricted to the interstitial part of the chromosome S10. The little c next the L1 chromosomes in subpanel c marks the centromer
Satellite DNA detection
Out of the 14 restriction endonucleases used for digesting the genomic DNA, only HindIII generated the typical pattern expected for tandemly repeated DNAs (Fig. S1). After gel extraction, cloning, and sequencing, the monomeric band (350 bp) happened to contain two different repetitive DNA families. The sequence of one of these families shows high similarity (more than 80 %) to a region of the EploRTE5 and EploRT20 non-LTR retrotransposons from the grasshopper E. plorans (GenBank accession nos. JN604090 and JN604091). It was excluded from the current work whose aim does not cover the analysis of TEs. These are not restricted to a specific chromosome and thus would not provide potential chromosomal markers. The sequence of the second repetitive DNA family (Fig. S2) was highly similar to a sequence defined as S. gregaria alpha repetitive DNA (GenBank accession no. X52936). To our knowledge, nothing has been published yet about this repetitive DNA (which we will henceforth call SG2-alpha), although its sequence was deposited in GenBank in 1992. All the sequenced clones from the dimeric 700 bp band obtained after HindIII digestion of the gDNA harbored the same sequence. This latter was also highly similar (more than 80 %) to the EploRTE5 and EploRT20 non-LTR retrotransposons from the grasshopper E. plorans. The 350 and the 700 bp non-LTR retrotransposon sequences are thus products of the differential cuts of an abundant S. gregaria retrotransposon at three equidistant HindIII sites. Southern blot hybridization of the HindIII digested gDNA using the SG2-alpha sequence as probe generated a ladder of multimers with a basic unit of 350 bp (Fig. S1). Therefore, the SG2-alpha repeat belongs indeed to a tandem repeat DNA family.
Given the difficulty and potential limitations of the conventional restriction endonuclease digestion as method for identifying satDNAs (see “Discussion”), we complemented our results by searching for tandem repeats in a 5 Gb set of Illumina Hiseq2000 Paired-End reads from a whole S. gregaria genome sequencing run (i.e., about 0.5× coverage). As proof of the reliability of the method, we recovered the SG2-alpha satDNA (previously identified by HindIII digestion of the gDNA). In addition, we found two other satDNA families, henceforth named SG1 and SG3, with consensus sequences of 171 and 170 bp, respectively (Table 2 and Fig. S3). The sequences of eight SG2-alpha clones (Fig. S2) and the consensus sequences of the SG1 and SG3 satDNAs (Fig. S3) were deposited in GenBank under the accession numbers HG965751 to HG965758, KJ649466, and KJ649467, respectively.
Physical mapping of the satellite DNAs
The chromosomal location of the three satDNAs identified in this work was analyzed using single and double FISH. Positive hybridization signals were detected at different chromosomal locations, with SG1 being located at the pericentromeric regions of all the chromosomes (Fig. 2e and f), SG2-alpha being restricted to the distal half of the three smallest autosomal pairs (Fig. 2f, g) whereas SG3 locates only to an interstitial region of one of the smallest autosomes (S10) (Fig. 2e and g). The visualization of the three satDNAs in the S10 autosome clearly showed that they occupy separate locations (Fig. 2h). Finally, the fiber-FISH technique showed that the interstitial telomeric DNA observed in some chromosomes (see Fig. 2d) is intermingled with the SG1 (Fig. 2i) and SG3 (Fig. 2j) satDNAs. It is worth mentioning that none of these satDNA sequences showed evidence of potential hybridization neither with the (GGTTA)x7 repeat nor with its reverse complement.
Transcription of the satellite DNAs
The proportions of reads observed for each satDNA in each of the four transcriptome libraries (i.e., the normalized read counts obtained by dividing the read counts corresponding to each satDNA by the total read counts in each library) indicated transcription of the three satDNAs in most organs (Table 3). The gonads showed higher expression than somatic tissues, and the testis had more satDNA expression than the ovary. To compare the degree of expression between the three satDNAs, we divided the normalized read proportions in Table 3 by the proportion that each satDNA represents in the genome (see Table 2). This showed that, relative to their abundance in the genome, the highest expression is observed for SG3 and the lowest for SG2-alpha. The latter was almost absent from the two somatic tissues (Fig. 3)—only four reads in the CNS and none in the muscles (see Table 3).

Relative degree of expression in different body parts of each of the three satDNAs studied in this work in relation to their abundance in the S. gregaria genome. This was calculated as the ratio between the normalized number (proportion) of a transcriptome reads that align to a satDNA and the relative number (proportion) of reads that align to that satDNA in the genomic library
Discussion
The chromosomes, repetitive DNA distribution, and satellite DNA transcription
Given the importance of the species and the state-of-the-art in the genetics science, one would expect certain chromosome-level aspects of the S. gegaria genome to be established. However, in reality, there are still discrepancies on issues as basic as the size-ordering of the chromosomes and the location of certain commonly used markers, such as the nucleolar organizing regions (NORs).
The acrocentric chromosomes of S. gregaria were first described by White (1934), who classified the autosomes into three size groups: long (L1–L3), medium (M4–M8), and short (S9–S11) pairs, with no mention of the X chromosome. The latter was described as second largest by John and Naylor (1961), whereas Dutt (1966) measurements suggested that it is one of the medium-sized chromosomes. Our results strongly support John and Naylor’s conclusion, since we found the X chromosome to be the second largest chromosome in DNA content and size.
NORs are the chromosomal loci that contain ribosomal DNA (McClintock 1934). The first mention of them in S. gregaria was by John and Henderson (1962), who detected nucleoli attached to interstitial positions of the L3 and M6 bivalents. However, here again, the picture is not clear due to contradicting reports. On the one hand, both the achromatic gaps reported by John and Naylor (1961) and later confirmed by John and Hewitt (1966) and the G-bands reported by Fox et al. (1973) at the same interstitial positions of the L3 and M6 chromosomes agree with John and Henderson’s 1962 finding. On the other hand, NORs were undetectable by C-banding (Fox et al. 1973), and Hagele (1979) concluded that neither the distal N bands on the L2 and M6 chromosomes nor the interstitial ones on the M8 and S9 chromosomes corresponded to NORs. If this was not confusing enough, Rufas and Gosalvez (1982) reported the presence of additional nucleoli attached to the M6 and S9 bivalents at diplotene. Fox and Santos (1985) however showed that S. gregaria consistently expresses interstitial NORs in the L3 and M6 chromosomes, whereas L. migratoria expressed distal NORs in the L2 and M6 chromosomes and an interstitial NOR in the S9 chromosome. The L. migratoria NOR expression pattern is remarkably similar to the one previously reported by Hagele (1979) and Rufas and Gosalvez (1982) for S. gregaria. Furthermore, Fox and Santos (1985) also reported the presence of an interstitial N+ C-band in the M8 chromosome of both S. gregaria and L. migratoria but concluded that this band does not correspond to a NOR. They speculated that the N bands correspond to G + C-rich regions and that the M8 chromosome could harbor the 5S rRNA genes. Finally, they explained the differences between their results and those reported by Hagele (1979) in terms of possible chromosomal polymorphism, although they also remarked the close similarity of Hagele’s results with their own results in L. migratoria. With our present data, the confusion about the relative chromosomal localization and correspondence between the gaps and the NORs is also solved. We clearly show that the gaps observed by some authors in the L3, M6, and M8 chromosomes (which coincide with N bands) are actually G + C-rich regions (see Fig. 1c) harboring 18S and 5S rDNA clusters (L3 and M6; see Fig. 1e) and H3 histone genes (M8; see Fig. 2a).
The C-banding pattern of S. gregaria chromosomes was first described by Fox et al. (1973) and is further confirmed by our present data. It consists of paracentromeric C-bands in all chromosomes and distal ones in the three S chromosome pairs. In consistency, Brown and Wilmore (1974) showed the presence of repetitive DNA at the centromeric regions of all the chromosomes and at the telomeric regions of the S chromosomes. These authors also showed that 30–40 % of the nuclear DNA is composed of repetitive DNA, as deduced from renaturation kinetics. However, CsC1 gradients analyses failed to show the presence of satDNAs.
Our initial search for S. gregaria satDNAs using restriction endonuclease digestion of the gDNA had relatively poor success. A low efficiency of that conventional method would thus explain the scarcity of satDNAs previously reported for the repetitive DNA-rich genome of this species. Indeed, among the 14 restriction endonucleases tested in this work, only HindIII gave the ladder pattern typical of a satDNA. However, the 352-bp monomer of that satDNA happened to be no new discovery, as it was already available in the GeneBank under the description of ‘S. gregaria alpha repetitive DNA (accession no. X52936). Furthermore, the chromosomal location of that satDNA (here called SG2-alpha) coincides with the distal C-positive bands of the three smallest autosomal pairs, meaning that the restriction endonucleases method did not succeed in isolating the most abundant satDNA. The latter is located at the paracentromeric regions of all the S. gregaria chromosomes, as predicted by autoradiographic methods (Fox 1966a, b), as well as from the C-banding results here and in Fox et al. (1973) and Brown and Wilmore (1974). In contrast, our second approach for identifying satDNAs, i.e., Illumina sequencing of gDNA, further confirmed the presence of the SG2-alpha satellite and added two more satDNAs (SG1 and SG3). This method also revealed that SG1, SG2-alpha, and SG3 are the three most abundant satDNAs. They respectively account for 18 %, 12 %, and 0.22 % of the S. gregaria genome. The presence and relative abundances of these satDNAs, inferred from the Illumina gDNA sequencing results, were in concordance with the FISH results. These revealed a paracentromeric location of the SG1 in all chromosomes, very high amounts of SG2-alpha in the distal regions of the three smallest autosomes, whereas the SG3 FISH signal was restricted to the interstitial region of a single autosome (S10). As a whole, the three satDNAs represent about 30 % of the genome (Table 2), meaning that Brown and Wilmore’s (Brown and Wilmore 1974) prediction that 30–40 % of the S. gregaria genome is made up of repetitive DNA is clearly an underestimate. It would not account for the satDNAs, all the repeated gene families (e.g., rDNA, histone genes and U snDNA), the other repetitive DNAs (including the telomeric), as well as the high variety of mobile elements. The latter are abundant in grasshopper genomes but were excluded from the current work as they do not tend to be located in specific chromosomes (i.e., they cannot be used as chromosomal markers).
Our present results clearly show that in silico search of tandem repeat DNAs among the reads resulting from massive sequencing of the gDNA is a much powerful and reliable approach for isolating satDNAs than the digestion of the gDNA with restriction endonucleases. The former method was suggested to allow detecting even sub-represented satDNA families (Wang et al. 2008), and our results confirm that it can detect satDNAs representing even less than 1 % of the genome (e.g., SG3) based on as little as 0.5× sequencing coverage of the genome. Furthermore, although this sequencing approach is applicable to genomes of any size, we think it is especially useful for large genomes, whose huge amounts of DNA, repeated sequences, and complex organization often render conventional techniques less efficient. The large genome size and complexity, together with the lower efficiency of the conventional detection of satDNAs by restriction endonucleases, would explain the paucity of satDNAs hitherto characterized in grasshoppers—only 12 satDNAs described in seven species (for review, see Palomeque and Lorite (2008)). Of course, when the digestion by the restriction endonucleases method works, it gives reliable results, as demonstrated by our HindIII isolation of a confirmed satDNA (SG2-alpha). It can even be efficient in detecting some abundant elements in the genome, as shown by our HindIII detection of a retrotransposon. However, we think its weakness comes from its dependence not only on the genomic abundance of the repeat (note that we could not detect the most abundant S. gregaria satDNA), but also on the size and composition of the repeat unit (monomer). Small repeat units or unfavorable nucleotide composition mean lower probability of finding an enzyme that cleaves within the units (which was the case for the SG1 and SG3 satDNAs). One solution might be the use of four-cutters instead of the conventional six-cutter restriction endonucleases, which would result in higher probability of cutting within the short satDNA unit. However, four-cutters are also more likely to cut several other genomic DNAs, resulting in a complex pattern of bands in the electrophoresis gels. For instance, Sau3AI cuts GATC sites both in our SG1 and SG2 satDNA monomers.
The observed transcription of the three satDNAs described here is consistent with previous findings in amphibians (Diaz et al. 1981; Epstein et al. 1986; Jamrich et al. 1983; Varley et al. 1980), insects (Lorite et al. 2002; Renault et al. 1999; Rouleux-Bonnin et al. 1996; Varadaraj and Skinner 1994), and mammals (Gaubatz and Cutler 1990). The possible function of satDNA transcripts is mostly unknown, and it has been suggested that satDNA transcription might start from interspersed genes or transposable elements (Hori et al. 1996; Solovei et al. 1996). However, recent research on the beetle Tribolium castaneum has reported satDNA-associated small interfering RNAs (siRNAs) that affect the epigenetic state of the constitutive heterochromatin under heat-shock stress conditions (Pezer and Ugarkovic 2012). In the case of S. gregaria, the observed transcription levels of the three satDNAs were very low. In fact, the highest transcriptome to genome satDNA abundance ratio was only 1.73 × 10−3. It was found for SG3 expression in the testis (Table 3) and means that only one out of each 577 SG3 genomic copies is transcribed in the cells of that organ. The satDNA expression reported for S. gregaria here actually represents a residual level of transcription most likely due to passive transcription driven by the expression of adjacent DNA sequences.
Repeated DNAs and the challenge of sequencing and assembly of the desert locust genome
Our results suggest that as much as about 50 % of the S. gregaria genome might be repeated DNAs, meaning that the magnitude of assembly artefacts would be a real issue for any future genome assembly project on that species. The use of combined paired-end and mate pair sequencing strategies (Belova et al. 2013; Treangen and Salzberg 2012) should attenuate the noise of such mis-assemblies but would not eliminate it, as long as the sequencing depth does not reach prohibitively costly hundreds fold coverage and the length of the longest mate pair library does not match the length of the largest repeated DNA region. Here, our data show that the 8.55-Gb genome of S. gregaria contains repeated DNA regions of tens of kilobases (almost a third of some of the chromosomes—see Fig. 2); both issues have to be taken into account for any future sequencing project.
Furthermore, and in addition to the chimeric sequences (that can form even between sequences from different chromosomes), another challenge would be mapping and attributing linkage groups to actual chromosomes. Here again, having a prior knowledge of the repeated sequences and their physical location should be helpful in two different ways. First, it would help identify the chromosomes between which mis-assembly into chimeric contigs would be more likely (since it would be more likely to have chimeric contigs between sequences of chromosomes that share the same repeated DNA). For instance, SG1 would cause chimeric mis-assemblies between pericentromeric regions of all the chromosomes of the S. gregaria complement, while SG2 would cause mis-assemblies between sequences of the three smallest chromosomes. Second, knowing the distribution of the repeated DNAs should help attribute linkage groups to actual chromosomes (since the different repeated, ribosomal, histone, and U DNAs would serve as markers to discriminate between some of the linkage groups belonging to the different chromosomes). In this way, 18S and 5S rDNA blocks would attribute contigs to the L3 or M6 chromosomes, whereas M5 is the only chromosome to have the 5S rDNA alone. Similarly, a linkage group that contains a H3 histone block would correspond to the M8 chromosome, whereas one that contains U1 or U2 snDNA would correspond to the L1 chromosome and SG3 would correspond to the S10 chromosome. Of course, having an estimate of the relative length and DNA content of each chromosome (Table 1) would always be helpful, at least as a reference datum against which the length of the different final assemblies could be compared with check for potential large deviations.
The recent publication of a L. migratoria draft genome by Wang et al. (2014) involved a huge sequencing and assembly effort. The genuineness of the resulting sequences was extensively tested by the different complementing methods included in the same published work. Still, potential undetected errors aside, and in spite of the titanic work carried out by the research team, L. migratoria chromosome sequences are not completely assembled into 12 single continuous sequences. The currently available L. migratoria draft genome is still a collection of linkage groups consisting of contigs of between few hundreds and tens of thousands bases of size, and the linkage groups are not unequivocally attributed to actual chromosomes yet. The reason for such difficulty is the large size of the L. migratoria genome and the huge amount of its repetitive DNAs (at least 60 %).
Sooner or later, the even larger S. gregaria genome is to be sequenced, and, judging by the sequencing and assembly work needed for producing the hitherto draft L. migratoria genome, the task will not be an easy one. Still, we expect that our data on the sequences and mapping of repetitive DNAs and other chromosomal markers would contribute to a better planning of the sequencing and assembly strategies and to the identification of any potential automated mis-assembly. Like any other prior information on S. gregaria genome, our data should also help with attributing some of the then assembled linkage groups to actual chromosomes.
References
Anders S, Pyl PT, Huber W (2014) HTSeq—a Python framework to work with high-throughput sequencing data. bioRxiv preprint doi:10.1101/002824
Badisco L, Huybrechts J, Simonet G, Verlinden H, Marchal E, Huybrechts R, Schoofs L, De Loof A, Vanden Broeck J (2011) Transcriptome analysis of the desert locust central nervous system: production and annotation of a Schistocerca gregaria EST database. PLoS One 6:e17274
Bakkali M (2011) Microevolution of cis-regulatory elements: an example from the pair-rule segmentation gene fushi tarazu in the Drosophila melanogaster subgroup. PLoS One 6:e27376
Bakkali M (2013) A bird’s-eye view on the modern genetics workflow and its potential applicability to the locust problem. C R Biol 336:375–383
Belova T, Zhan B, Wright J, Caccamo M, Asp T, Simkova H, Kent M, Bendixen C, Panitz F, Lien S, Dolezel J, Olsen OA, Sandve SR (2013) Integration of mate pair sequences to improve shotgun assemblies of flow-sorted chromosome arms of hexaploid wheat. BMC Genomics 14:222
Bennetzen JL (2005) Transposable elements, gene creation and genome rearrangement in flowering plants. Curr Opin Genet Dev 15:621–627
Bensasson D, Zhang DX, Hewitt GM (2000) Frequent assimilation of mitochondrial DNA by grasshopper nuclear genomes. Mol Biol Evol 17:406–415
Brown AK, Wilmore PJ (1974) Location of repetitious DNA in the chromosomes of the desert locust (Schistocerca gregaria). Chromosoma 47:379–383
Bueno D, Palacios-Gimenez OM, Cabral-de-Mello DC (2013) Chromosomal mapping of repetitive DNAs in the grasshopper reveal possible ancestry of the B chromosome and H3 histone spreading. PLoS One 8:e66532
Cabral-de-Mello DC, Valente GT, Nakajima RT, Martins C (2012) Genomic organization and comparative chromosome mapping of the U1 snRNA gene in cichlid fish, with an emphasis in Oreochromis niloticus. Chromosome Res 20:279–292
Cabrero J, Camacho JP (2008) Location and expression of ribosomal RNA genes in grasshoppers: abundance of silent and cryptic loci. Chromosome Res 16:595–607
Cabrero J, Bakkali M, Bugrov A, Warchalowska-Sliwa E, Lopez-Leon MD, Perfectti F, Camacho JP (2003) Multiregional origin of B chromosomes in the grasshopper Eyprepocnemis plorans. Chromosoma 112:207–211
Cabrero J, Lopez-Leon MD, Teruel M, Camacho JP (2009) Chromosome mapping of H3 and H4 histone gene clusters in 35 species of acridid grasshoppers. Chromosome Res 17:397–404
Camacho JPM, Viseras E, Navas-Castillo J, Cabrero J (1984) C-heterochromatin content of supernumerary chromosome segments of grasshoppers: detection of an euchromatic extra segment. Heredity 53:167–175
Camacho JPM, Cabrero J, Viseras E, Lopez-Leon MD, Navas-Castillo J, Alche JD (1991) G-banding in two species of grasshoppers and its relationship to C, N and fluorescence banding techniques. Genome 34:638–643
Craig-Cameron TA (1970) Actinomycin-D and chiasma frequency in Schistocerca gregaria (Forskal). Chromosoma 30:169–179
Craig-Cameron T, Jones GH (1970) The analysis of exchanges in tritium-labelled meiotic chromosomes. 1. Heredity (Edinb) 25:223–232
Csik L, Koller PC (1939) Relational coiling and chiasma frequency. Zeitschr Zellforsch Microsk Anat Abt B Chromosoma 1:191–196
Diaz MO, Barsacchi-Pilone G, Mahon KA, Gall JG (1981) Transcripts from both strands of a satellite DNA occur on lampbrush chromosome loops of the newt Notophthalmus. Cell 24:649–659
Dolezel J, Bartos J, Voglmayr H, Greilhuber J (2003) Nuclear DNA content and genome size of trout and human. Cytometry A 51:127–128, author reply 129
Drummond AJ, Ashton B, Cheung M, Heled J, Kearse M (2009) Geneious 4.8. Biomatters Auckland, New Zealand
Dutt K (1966) Karyotype study of a fresh water snake Cerberus rhynohops. (Abstr). In: Proc Int Symp Animal Venoms Inst. Butantan, Sao Paulo, pp 15–16
Epstein LM, Mahon KA, Gall JG (1986) Transcription of a satellite DNA in the newt. J Cell Biol 103:1137–1144
Ferrier DE, Akam M (1996) Organization of the Hox gene cluster in the grasshopper, Schistocerca gregaria. Proc Natl Acad Sci U S A 93:13024–13029
Fox DP (1966a) The effect of X-rays on the chromosomes of locust embryos: II Chromatid interchanges and the organization of the interphase nucleus. Chromosoma 20:173–194
Fox DP (1966b) The effects of X-rays on the chromosomes of locust embryos. I. The early responses. Chromosoma 19:300–316
Fox DP (1967a) The effects of X-rays on the chromosomes of locust embryos. 3. The chromatid aberration types. Chromosoma 20:386–412
Fox DP (1967b) The effects of X-rays on the chromosomes of locust embryos. IV. Dose–response and variation in sensitivity of the cell cycle for the induction of chromatid aberrations. Chromosoma 20:413–441
Fox DP (1970) A non-doubling DNA series in somatic tissues of the locusts Schistocerca gregaria (Forskal) and Locusta migratoria (Linn.). Chromosoma 29:446–461
Fox DP (1973) The control of chiasma distribution in the locust, Schistocerca gregaria (Forskal). Chromosoma 43:289–328
Fox DP, Santos JL (1985) N-bands and nucleolus expression in Schistocerca gregaria and Locusta migratoria. Heredity 54:333–341
Fox DP, Carter KC, Hewitt GM (1973) Giemsa banding and chiasma distribution in the desert locust. Heredity (Edinb) 31:272–276
Gaubatz JW, Cutler RG (1990) Mouse satellite DNA is transcribed in senescent cardiac muscle. J Biol Chem 265:17753–17758
Gordon A, Hannon GJ (2010) Fastx-toolkit. FASTQ/A short-reads pre-processing tools. Unpublished http://hannonlab.cshl.edu/fastx_toolkit
Gregory TR (2005) Synergy between sequence and size in large-scale genomics. Nat Rev Genet 6:699–708
Hagele K (1979) Characterization of heterochromatin in Schistocerca gregaria by C- and N-banding methods. Chromosoma 70:239–250
Hardie DC, Gregory TR, Hebert PD (2002) From pixels to picograms: a beginners’ guide to genome quantification by Feulgen image analysis densitometry. J Histochem Cytochem 50:735–749
Henderson SA (1962) Temperature and chiasma formation in Schistocerca gregaria. Chromosoma 13:437–463
Henderson SA (1963) Chiasma distribution at diplotene in a locust. Heredity 18:173
Henderson SA (1964) RNA synthesis during male meiosis and spermiogenesis. Chromosoma 15:345–366
Henderson SA (1988) Four effects of elevated temperature on chiasma formation in the locust Schistocerca gregaria. Heredity 60:387–401
Hori T, Suzuki Y, Solovei I, Saitoh Y, Hutchison N, Ikeda JE, Macgregor H, Mizuno S (1996) Characterization of DNA sequences constituting the terminal heterochromatin of the chicken Z chromosome. Chromosome Res 4:411–426
Jain HK, Singh U (1967) Actinomycin D induced chromosome breakage and suppression of meiosis in the locust, Schistocerca gregaria. Chromosoma 21:463–471
Jamrich M, Warrior R, Steele R, Gall JG (1983) Transcription of repetitive sequences on Xenopus lampbrush chromosomes. Proc Natl Acad Sci U S A 80:3364–3367
John B, Henderson SA (1962) Asynapsis and polyploidy in Schistocerca paranensis. Chromosoma 13:111–147
John B, Hewitt GM (1966) Karyotype stability and DNA variability in the Acrididae. Chromosoma 20:155–172
John B, Naylor B (1961) Anomalous chromosome behaviour in germ line of Schistocerca gregaria. Heredity 16:187–198
Jones GH (1977) A test for early termination of chiasmata in diplotene spermatocytes of Schistocerca gregaria. Chromosoma 63:287–294
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760
Lorite P, Renault S, Rouleux-Bonnin F, Bigot S, Periquet G, Palomeque T (2002) Genomic organization and transcription of satellite DNA in the ant Aphaenogaster subterranea (Hymenoptera, Formicidae). Genome 45:609–616
Lorite P, Torres MI, Palomeque T (2013) Characterization of two unrelated satellite DNA families in the Colorado potato beetle Leptinotarsa decemlineata (Coleoptera, Chrysomelidae). Bull Entomol Res 103:538–546
McClintock B (1934) The relationship of a particular chromosomal element to the development of nucleoli in Zea mays. Z Zellforsch 21:294–328
Meyne J, Hirai H, Imai HT (1995) FISH analysis of the telomere sequences of bulldog ants (Myrmecia: Formicidae). Chromosoma 104:14–18
Munoz-Pajares AJ, Martinez-Rodriguez L, Teruel M, Cabrero J, Camacho JP, Perfectti F (2011) A single, recent origin of the accessory B chromosome of the grasshopper Eyprepocnemis plorans. Genetics 187:853–863
Novak P, Neumann P, Macas J (2010) Graph-based clustering and characterization of repetitive sequences in next-generation sequencing data. BMC Bioinformatics 11:378
Novak P, Neumann P, Pech J, Steinhaisl J, Macas J (2013) RepeatExplorer: a galaxy-based web server for genome-wide characterization of eukaryotic repetitive elements from next-generation sequence reads. Bioinformatics 29:792–793
Oliver MJ, Petrov D, Ackerly D, Falkowski P, Schofield OM (2007) The mode and tempo of genome size evolution in eukaryotes. Genome Res 17:594–601
Otto SP, Whitton J (2000) Polyploid incidence and evolution. Annu Rev Genet 34:401–437
Palomeque T, Lorite P (2008) Satellite DNA in insects: a review. Heredity (Edinb) 100:564–573
Pezer Z, Ugarkovic D (2012) Satellite DNA-associated siRNAs as mediators of heat shock response in insects. RNA Biol 9:587–595
Rasch EM, Rasch RW (1981) Cytophotometric determination of genome size for two species of cave crickets (Orthoptera, Rhaphidophoridae). J Histochem Cytochem 29:885
Renault S, Rouleux-Bonnin F, Periquet G, Bigot Y (1999) Satellite DNA transcription in Diadromus pulchellus (Hymenoptera). Insect Biochem Mol Biol 29:103–111
Rouleux-Bonnin F, Renault S, Bigot Y, Periquet G (1996) Transcription of four satellite DNA subfamilies in Diprion pini (Hymenoptera, Symphyta, Diprionidae). Eur J Biochem 238:752–759
Rozen S, Skaletsky H (1999) Primer3 on the WWW for general users and for biologist progremmers. In: Bioinformatics methods and protocols. Humana Press, pp 365–386
Rufas JS, Gosalvez J (1982) Development of silver stained structures during spermatogenesis of Schistocerca gregaria (Forsk.) (Orthoptera:Acrididae). Caryologia 35:261–267
Ruiz-Ruano FJ, Ruiz-Estevez M, Rodriguez-Perez J, Lopez-Pino JL, Cabrero J, Camacho JP (2011) DNA amount of X and B chromosomes in the grasshoppers Eyprepocnemis plorans and Locusta migratoria. Cytogenet Genome Res 134:120–126
Solovei IV, Joffe BI, Gaginskaya ER, Macgregor HC (1996) Transcription of lampbrush chromosomes of a centromerically localized highly repeated DNA in pigeon (Columba) relates to sequence arrangement. Chromosome Res 4:588–603
Timothy D, Littlewood J, Olson P (2000) Small subunit rDNA and the platyhelminthes: signal, noise, conflict and compromise. In: Littlewood DTJ, Bray RA (eds) Interrelationships of the platyhelminthes. CRC Press, UK, pp 381–330, 380
Treangen TJ, Salzberg SL (2012) Repetitive DNA and next-generation sequencing: computational chalenges and solutions. Nat Rev Genet 13:36–46
Varadaraj K, Skinner DM (1994) Cytoplasmic localization of transcripts of a complex G + C-rich crab satellite DNA. Chromosoma 103:423–431
Varley JM, Macgregor HC, Nardi I, Andrews C, Erba HP (1980) Cytological evidence of transcription of highly repeated DNA sequences during the lampbrush stage in Triturus cristatus carnifex. Chromosoma 80:289–307
Vaughan HE, Heslop-Harrison JS, Hewitt GM (1999) The localization of mitochondrial sequences to chromosomal DNA in orthopterans. Genome 42:874–880
Wang S, Lorenzen MD, Beeman RW, Brown SJ (2008) Analysis of repetitive DNA distribution patterns in the Tribolium castaneum genome. Genome Biol 9:R61
Wang X, Fang X, Yang P, Jiang X, Jiang F, Zhao D, Li B, Cui F, Wei J, Ma C, Wang Y, He J, Luo Y, Wang Z, Guo X, Guo W, Zhang Y, Yang M, Hao S, Chen B, Ma Z, Yu D, Xiong Z, Zhu Y, Fan D, Han L, Wang B, Chen Y, Wang J, Yang L, Zhao W, Feng Y, Chen G, Lian J, Li Q, Huang Z, Yao X, Lv N, Zhang G, Li Y, Zhu B, Kang L (2014) The locust genome provides insight into swarm formation and long-distance flight. Nat Commun 5:2957
Westerman M (1967) The effect of X-irradiation on male meiosis in Schistocerca gregaria (Forskal). Chromosoma 22:401–416
Westerman M (1968) The effect of x-irradiation on male meiosis in Schistocerca gregaria (Forskal). II. The induction of chromosome mutations. Chromosoma 24:17–36
Westerman M, Barton NH, Hewitt GM (1987) Differences in DNA content between two chromosomal races of the grasshopper Podisma pedestris. Heredity 58:221–228
White MJD (1934) The influence of temperature on chiasma frequency. J Genet 29:203–216
Wilmore PJ, Brown AK (1975) Molecular properties of orthopteran DNA. Chromosoma 51:337–345
Wolf KW (1996) The structure of condensed chromosomes in mitosis and meiosis of insects. Int J Insect Morphol Embryol 25:37–62
Wolf KW, Sumner AT (1996) Scanning electron microscopy of heterochromatin in chromosome spreads of male germ cells in Schistocerca gregaria (Acrididae, Orthoptera) after trypsinization. Biotech Histochem 71:237–244
Wolf KW, Turner BM (1996) The pattern of histone H4 acetylation on the X chromosome during spermatogenesis of the desert locust Schistocerca gregaria. Genome 39:854–865
Acknowledgments
This study was supported by grants from the Spanish Ministerio de Ciencia y Tecnología (CGL2009-11917 and BFU2010-16438) and Plan Andaluz de Investigacion (CVI-6649 and BIO-220), and was partially performed by FEDER funds. M. Bakkali was supported by a Ramón y Cajal fellowship from the Spanish Ministerio de Ciencia e Innovación.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 3481 kb)
Rights and permissions
About this article

Cite this article
Camacho, J.P.M., Ruiz-Ruano, F.J., Martín-Blázquez, R. et al. A step to the gigantic genome of the desert locust: chromosome sizes and repeated DNAs. Chromosoma 124, 263–275 (2015). https://doi.org/10.1007/s00412-014-0499-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00412-014-0499-0