
Abstract
Nuclear intermediate filament networks formed by A- and B-type lamins are major components of the nucleoskeleton that are required for nuclear structure and function, with many links to human physiology. Mutations in lamins cause diverse human diseases (‘laminopathies’). At least 54 partners interact with human A-type lamins directly or indirectly. The less studied human lamins B1 and B2 have 23 and seven reported partners, respectively. These interactions are likely to be regulated at least in part by lamin post-translational modifications. This review summarizes the binding partners and post-translational modifications of human lamins and discusses their known or potential implications for lamin function.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Lamins are major components of the nucleoskeleton in multicellular animals (metazoans), not found in plants or fungi (Dittmer and Misteli 2011). Lamins tether chromatin, bind signaling proteins and support epigenetic regulation, mechanotransduction, development, transcription, replication and DNA damage repair (Dechat et al. 2008; Dittmer and Misteli 2011; Simon and Wilson 2011). How lamins contribute to such a remarkable range of activities is for the most part unknown: a new saga in biology that begins with a seemingly simple structural polymer. Lamins form highly stable filament networks near the inner membrane of the nuclear envelope and are also distributed throughout the nucleoplasm except for the nucleolus (Gerace and Huber 2012; Dittmer and Misteli 2011; Simon and Wilson 2011). Mammals express two types of lamins, the B-type (lamins B1, B2 and B3) encoded by LMNB1 and LMNB2 (Dittmer and Misteli 2011; Schumacher et al. 2006), and A-type (lamins A, C, AΔ10, C2, and AΔ50, also known as ‘progerin’) generated by alternative splicing of LMNA (Dittmer and Misteli 2011; Bokenkamp et al. 2011).
Mutations in lamins cause a variety of diseases, collectively termed laminopathies (Worman 2012; Butin-Israeli et al. 2012). So far nearly 400 different disease-causing mutations in A-type lamins have been identified, underscoring their significance to cell and tissue biology and human physiology. Diseases are now also being mapped to B-type lamins. Duplication of the LMNB1 gene can cause leukodystrophy (Padiath et al. 2006; Schuster et al. 2011; Brussino et al. 2010; Molloy et al. 2012) or leukoencephalopathy (Brussino et al. 2009), and certain mutations in LMNB2 correlate with increased susceptibility to acquired partial lipodystrophy (Hegele et al. 2006).
Both A- and B-type lamins are synthesized as precursors that are post-translationally processed prior to filament assembly. All lamins except lamin C are first farnesylated at the cysteine of the C-terminal CaaX motif (Beck et al. 1990; Farnsworth et al. 1989), then proteolytically cleaved by either Rce1 or Zmpste24, and finally carboxymethylated by Icmt1 (Nigg et al. 1992; Young et al. 2005; Maske et al. 2003; Varela et al. 2005). The lamin A precursor (pre-lamin A) is further processed by Zmpste24-dependent cleavage after Tyr-646 to generate mature lamin A (Pendas et al. 2002; Bergo et al. 2002; Barrowman et al. 2012).
Lamin proteins have a small N-terminal ‘head’ domain, a long coiled-coil ‘rod’ domain and a large C-terminal ‘tail’ that includes a globular Ig-fold domain (Dechat et al. 2008; Dittmer and Misteli 2011). Lamin assembly was successfully reconstituted in vitro only recently (Ben-Harush et al. 2009). Studies of purified lamins show that they first dimerize via their rod domain; dimers then associate head-to-tail to form linear polymers, which in turn associate laterally in groups of three or four in a staggered anti-parallel manner to form ~10-nm-diameter filaments (Ben-Harush et al. 2009; Herrmann et al. 2004; Gerace and Huber 2012). The actual organization of lamina networks in somatic cells is unknown.
A- and B-type lamins can interact directly in vitro (Ye and Worman 1995; Schirmer and Gerace 2004), but in living cells appear to preferentially form independent filament networks. High-resolution microscopy of endogenous lamins A/C and B1 (Shimi et al. 2008) as well as FRET analysis of exogenous lamins A and B1 (Delbarre et al. 2006) support the existence of separate lamin A/C or B1 homopolymers in close contact with each other. The spatial separation of lamin A and B1 homopolymers was lost in cells that also expressed lamin A bearing the Hutchinson–Gilford progeria syndrome (HGPS)-causing Δ50 deletion (‘progerin’) (Delbarre et al. 2006). Remarkably, biochemical analysis suggests lamins A and C (the first 566 residues of which are identical) also form homodimers and homopolymers preferentially in vivo, via unknown mechanisms (Kolb et al. 2011).
In the nucleus lamins reportedly bind partners (Wilson and Foisner 2010; Zastrow et al. 2004). Lamin A is the most extensively studied with at least 29 reported direct binding partners (Fig. 1, Table 1), and at least 24 proteins identified by co-immunoprecipitation from cells or other indirect methods (Table 2). Many new potential partners are being uncovered (Roux et al. 2012; Kubben et al. 2010). The B-type lamins are less studied, with 23 reported direct or indirect partners for lamin B1 (Table 3) and only seven for lamin B2 (Table 4). Lamin partners in other animals, including Drosophila JIL-1 kinase (Bao et al. 2005) and Xenopus α-importin (Adam et al. 2008), are conserved in humans and may therefore also associate with human lamins.

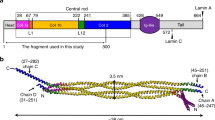
Lamin A molecule and direct binding partners. Diagram of major domains of human lamin A and mapped regions involved in binding to specific partners. The rod domain is subdivided into four coiled-coil regions (1A, 1B, 2A, 2B), which are separated by linker regions L1, L12 and L2. NLS, nuclear localization signal. Question marks indicate partners whose binding region on lamin A is unmapped
The functional association of many certain partners including LEM-domain proteins, BAF, Rb and LINC complex components has been confirmed genetically or in cells as discussed in recent reviews (Wilson and Foisner 2010; Simon and Wilson 2011; Dechat et al. 2010; Dittmer and Misteli 2011). Biochemical and biological validation will be crucial to move this field forward, since many partners identified in vitro lack proven biological relevance, and ‘associated’ proteins (e.g., those identified by co-immunoprecipitation from cells in Table 2) lack evidence that binding is direct. Nuclear lamina networks are largely insoluble under typical co-immunoprecipitation conditions, and some methods (e.g., sonication) can create small ‘chunks’ of lamina that appear soluble (e.g., not pelleted by 12,000 × g centrifugation) but might contain dozens or hundreds of different proteins that co-immunoprecipitate together. Another confounding issue is that lamins can bind DNA (Stierle et al. 2003). Hence one must rule out the possibility that ‘direct’ binding of certain partners to lamins is actually mediated by DNA in the reaction. For example, this artifact caused two proteins (Cone-rod homeobox [Crx]; HIV-1 matrix [MA]) and one polypeptide (C-terminal domain of MAN1) to be misidentified as direct partners for the dsDNA-binding protein BAF, and was corrected by NMR analysis of protein-protein binding and by re-testing under DNA-free conditions (Huang et al. 2011). A related artifact can be solved by using the DNA intercalator, ethidium bromide, to ‘bump off’ proteins (e.g., PARP1; Ku70/80) that bind DNA ends nonspecifically (Lai and Herr 1992).
A major unanswered question is how lamin associations with specific partners are regulated. To facilitate further studies, this review focuses on post-translational modifications of human lamins, including the few cases where the functional consequences of specific modifications are known.
Phosphorylation
Following the discovery that lamins are reversibly disassembled during mitosis (Gerace and Blobel 1980), early studies focused on lamin phosphorylation during mitosis. The head domain of all lamins includes an evolutionarily conserved site phosphorylated by the mitotic cyclin-dependent kinase CDK1 (Peter and Stick 2012). Peptide sequencing identified this site as Ser-22 in human lamins A/C, Ser-23 in lamin B1 and Ser-37 in lamin B2 (Fig. 2a). The first mammalian lamin B2 cDNA to be studied, thought to be the physiological form, was actually missing 20 N-terminal residues and hence dominantly disrupted nuclear lamina organization in transfected cells (Schumacher et al. 2006); please note that these 20 ‘new’ residues are included when numbering lamin B2 residues in this review. Lamin phosphorylation by CDK1 impedes assembly of head-to-tail polymers but does not disrupt lamin dimer formation (Heitlinger et al. 1991; Peter et al. 1991). CDK1 targets two regions important for head-to-tail association of lamin A dimers (Strelkov et al. 2004); phosphorylation at Ser-22, and at Ser-392, Ser-404 and Ser-406 at the opposite end of the coiled-coil domain, are required to depolymerize lamin filaments during mitosis (Heald and McKeon 1990; Peter et al. 1990; Ward and Kirschner 1990; Eggert et al. 1991; Enoch et al. 1991; Thompson and Fields 1996; Schneider et al. 1999; Fig. 2b). Phosphorylation of the A-type lamin in Drosophila, named lamin C, at Ser-37 (homologous to human lamins A/C Ser-22) increases the solubility of the lamin protein and eliminates its ability to interact with chromatin in vitro (Zaremba-Czogalla et al. 2012).

Conservation of known phosphorylation sites in human lamins. Sequence alignment of the rod and head domains (a) or tail domains (b) of human lamins A (including precursor-specific C-terminal residues), B1 and B2 based on accession numbers NP_733821.1, NP_005564 and NP_116126, respectively. The locations of all known phosphorylation sites are indicated above; squares, circles and triangles indicate residues phosphorylated in lamins A, B1 and B2, respectively. Arrow indicates the Zmpste24 cleavage site in pre-lamin A (plamA cleavage). Underlined residues in b comprise the lamin Ig-fold domain (Dhe-Paganon et al. 2002; Krimm et al. 2002; Ruan et al. 2012)
The protein kinase C (PKC) family also regulates lamins during mitosis (Peter et al. 1990; Hocevar et al. 1993). In zebrafish, lamins are phosphorylated by PKC first (Collas 1999), suggesting PKC phosphorylation might ‘unmask’ sites for CDK1 phosphorylation (Buendia et al. 2001). Supporting this idea, mitotic PKC- and CDK1-mediated disassembly of lamin B1 is triggered by diacylglycerol (DAG) generated by either lipin in HeLa cells (Mall et al. 2012) or by PLCβ1 in mouse erythroleukemia cells (Fiume et al. 2009). Conversely, lamin filament assembly in HeLa cells during early G1 requires dephosphorylation of B-type lamins by AKAP149-PP1 (Steen et al. 2003). However, mitosis also involves dephosphorylation: in Xenopus oocytes, unidentified PKA site(s) must be dephosphorylated for lamin filaments to disassemble (Molloy and Little 1992).
Recent high-throughput proteomic studies revealed further, extensive human lamin phosphorylation during mitosis (Olsen et al. 2010; Daub et al. 2008; Malik et al. 2009; Wang et al. 2008, 2010; Fig. 2, Table 5). Many mitotic phosphorylation sites are clustered in the head domain and near the Nuclear Localization Signal (NLS) (Fig. 3). However, it is important to note that some mitotic sites are also targeted during interphase (Table 5) as discussed below. With 61 known phosphorylation sites (Fig. 3, Table 5) lamins A/C have more than twice as many known sites as lamin B1 (32 sites; Fig. 3, Table 6) or lamin B2 (28 sites; Fig. 3, Table 7). Two sites are unique to the lamin C isoform (Table 5). In general, the head and tail domains account for most phosphorylation sites, with the highest density between the rod domain and NLS (Fig. 3).

Post-translational modifications of human lamins. Lamin schematics indicating specific residues that are post-translationally modified by phosphorylation, acetylation, O-GlcNAcylation, SUMOylation, ubiquitylation, or oxidation. Arrows indicate sites cleaved by the pre-lamin A processing protease Zmpste24, or apoptotic proteases Caspase 1 (Csp1), Caspase 6 (Csp6), Granzyme A (GzmA), Granzyme B (GzmB), or CRNSP
PKC family members also regulate non-mitotic functions of lamins. PKC phosphorylation of B-type lamins in sea urchin sperm triggers lamina disassembly prior to fertilization (Collas et al. 1997). Lamin A/C Ser-525, in the Ig-fold domain, is reportedly phosphorylated only during interphase (Table 5; Eggert et al. 1993). In human dermal fibroblasts, PKC specifically modifies at least one site in lamin B2 during S-phase (Kill and Hutchison 1995). In leukemia cells, increased lamin B2 phosphorylation is proposed to extend G1 phase (Meier et al. 1997). PKC phosphorylation of chicken lamin B2 inhibits lamin B2 import into the nucleus during interphase (Hennekes et al. 1993). Finally, both PKCα (Shimizu et al. 1998) and PKCδ (Cross et al. 2000) phosphorylate B-type lamins at unknown sites during apoptosis.
Other kinases that target lamins include PKA, S6-kinase II and Akt (Tables 5 and 6). Ser-50 phosphorylation of the B-type lamin in Drosophila, named lamin Dm0, by PKA inhibits head-to-tail dimerization (Stuurman 1997); interestingly, among human lamins this modification appears to be detectably conserved only in lamin B1, on Ser-28 (Table 6; Olsen et al. 2010; Rigbolt et al. 2011). Human lamins A/C are phosphorylated by S6-kinase II at Ser-404; the functional significance of this modification is unknown (Ward and Kirschner 1990), but it has since been detected under a variety of cellular conditions (Table 5). For example, following insulin treatment, the Akt kinase phosphorylates lamins A/C at Ser-404 in HEK 293 T cells (Cenni et al. 2008). Cells that express lamin A bearing either the S404A mutation or a nearby R401C Emery–Dreifuss muscular dystrophy (EDMD)-causing mutation have disorganized lamina networks and nuclear blebbing (Cenni et al. 2008).
Phosphorylation of lamins A/C is generally reduced in myoblasts from EDMD and limb girdle muscular dystrophy (LGMD) patients (Cenni et al. 2005). The N terminus of lamins A/C is phosphorylated in cycling C2C12 myoblasts, and insulin treatment specifically increases phosphorylation of lamin A, but not lamin C (Cenni et al. 2005). Insulin stimulation also increases phosphorylation of lamins A/C in quiescent baby hamster kidney fibroblasts (Friedman and Ken 1988). Neither the kinase(s) responsible for phosphorylating lamins A/C in response to insulin signaling in myoblasts or kidney fibroblasts, nor their target sites, have been identified.
A study of lamin B2 in DLD-1 colorectal cancer cell lines using phospho-site specific antibodies revealed differential phosphorylation of five sites during the cell cycle (Kuga et al. 2010). Thr-34 and Ser-37 are phosphorylated during prophase until late anaphase. Ser-405 phosphorylation levels increase during prophase and are maintained until late G1, whereas Ser-407 is phosphorylated only during G1 and prophase, and Ser-421 is phosphorylated during the S-to-G2 transition (Kuga et al. 2010). Of these five lamin B2 phosphorylation sites, four are conserved in both lamins A/C and B1 (corresponding to lamin B2 residues Thr-34, Ser-37, Ser-405, and Ser407), and the fifth site (lamin B2 Ser-421) is conserved in lamins A/C (Fig. 2). High-throughput proteomic studies showed lamins are also phosphorylated in cells treated with EGF (Olsen et al. 2006) or MAPK inhibitors (Pan et al. 2009), and in human colon adenocarcinoma cells (Kim et al. 2005), human epithelial cancer cells (Moritz et al. 2010), and differentiating human embryonic stem cells (ESCs) (Rigbolt et al. 2011; Van Hoof et al. 2009) (Fig. 2, Tables 5–7). The criterion for including modifications in this review was access to supporting (published) evidence. Updated information about modifications and sites, both published and unpublished, can be found online (e.g., Phosphosite database at www.phosphosite.org).
Twelve phosphorylation sites are conserved in all three human lamins (Fig. 2); all but one are located in head or tail regions important for mitotic lamin depolymerization (lamins A/C residues Thr-19, Ser-22, Thr-24, Ser-390, Ser-392, Thr-394, Ser-395, Ser-398, Ser-403, Ser-404, Ser-407; Fig. 2). The other conserved phosphorylation site is lamin A/C residue Ser-303 in the coil 2B region (Fig. 2). Four additional phospho-sites are conserved between lamins A/C and B1 (lamin A residues Thr-3, Ser-18, Ser-277, Ser-652); lamins A/C and B2 share six additional sites (lamin A residues Thr-64, Ser-71, Ser-301, Ser-406, Thr-409, Ser-458), and lamins B1 and B2 share four (lamin B1 residues Ser-232, Tyr-359, Ser-401, Ser-406) (Fig. 2), potentially reflecting phospho-dependent regulation of other conserved functions. By contrast, unique phosphorylation sites are likely to reflect the differential regulation of lamins in diverse tissues (Vergnes et al. 2004; Coffinier et al. 2010; Takamori et al. 2007; Coffinier et al. 2011; Kim et al. 2011b).
Several other differences in the patterns of phosphorylation of human lamins stand out. Lamins A/C have many phosphorylation sites in coils 1A and 1B, the L1 linker in the rod domain, and the Ig-fold, whereas homologous regions in lamins B1 and B2 have few or no known sites (Fig. 3). On the other hand, lamin B1 has eight phosphorylation sites in coil 2B, whereas lamins A/C have only four and lamin B2 three (Fig. 2). Only five phosphorylated Tyr residues have been identified (lamin A Tyr-81, lamin B1 Tyr-359 and Tyr-377, lamin B2 Tyr-374 and Tyr-515); we assume more sites exist, since Tyr phosphorylation tends to be labile under the experimental conditions used in many previous studies.
Lamin A residues 560–649, which have no counterpart in B-type lamins, are extensively phosphorylated and all 11 known phospho-sites in this region are eliminated by the HGPS-causing Δ50 deletion (loss of residues 608–658), pointing to lamin A misregulation as another likely consequence of this ‘accelerated aging’ mutation (Fig. 2). Among the nearly 400 disease-causing mutations in lamin A (Dittmer and Misteli 2011), remarkably only four disrupt known phosphorylation sites (Thr-10, Ser-303, Ser-395, Thr-505). However, many known phosphorylation sites are located near disease-causing mutations and might be affected indirectly. For example, lamin A Ser-458 is phosphorylated only in muscle cells from EDMD and LGMD patients who have mutations specifically in the Ig-fold domain, which can be up to seventy residues away from Ser-458 (Mitsuhashi et al. 2010).
The CDK1 phosphorylation sites on lamins are exploited by both herpes simplex virus and Epstein–Barr virus to disassemble lamins and thereby enable nascent virus particles to bud through the nuclear envelope (Lee and Chen 2010). The Epstein–Barr virus encodes its own kinase, BGLF4, which targets CDK1 sites on lamins A/C. The nuclear exit of Epstein–Barr virus is inhibited in cells that overexpress lamin A bearing five Ser-to-Ala substitutions at Ser-22, Ser-390, Ser-392, Ser-652, and Ser-657 (Lee et al. 2008). Four of these sites (all but Ser-657) can also be phosphorylated in uninfected cells (Table 5). Intriguingly, the nuclear export of large ribonucleoprotein complexes in response to Wnt signaling in Drosophila muscle cells also involves direct ‘budding’ through the nuclear envelope (Speese et al. 2012).
O-GlcNAcylation
O-GlcNAc (β-O-linked N-acetylglucosamine) is a reversible single sugar modification of Ser or Thr residues that can compete or cooperate with phosphorylation to regulate signaling, transcription and mitosis (Hart and Copeland 2010). This modification is found on both nuclear and cytoplasmic proteins (Hart and Copeland 2010). In mitotic spindles isolated from HeLa cells, lamin A was O-GlcNAcylated at Ser-612 and Thr-643 (Wang et al. 2010). Both residues are located in the unique C-terminal region of lamin A (Fig. 3). In mouse brain tissue, lamin A is O-GlcNAcylated at Ser-611 and Ser-613 (Alfaro et al. 2012); mouse Ser-613 is homologous to human lamin Ser-612. The functional consequences of lamin A O-GlcNAcylation are unknown. Lamins were first reported to be glycosylated over 20 years ago (Ferraro et al. 1989), but whether this represents O-GlcNAcylation or a different modification(s) is unknown.
Oxidation
Reactive oxygen species (ROS) are produced during oxygen metabolism and can regulate many pathways including cell senescence (Bartz and Piantadosi 2010). In primary human dermal fibroblasts, lamin A tail domain residues Cys-522, Cys-588 and Cys-591 can be oxidized, yielding both intra- and inter-molecular disulfide bridges (Pekovic et al. 2011). In cells that overexpress lamin A bearing the triple C522A/C588A/C591A mutation, nuclei are misshapen and cells enter senescence prematurely in response to oxidative stress (Pekovic et al. 2011; Sieprath et al. 2012). Premature senescence was also reported in lamin A null fibroblasts (Pekovic et al. 2011), suggesting A-type lamins are an important ‘sink’ for ROS that helps protect cells.
SUMOylation
SUMO (small ubiquitin-like modifier) proteins are covalently and reversibly attached to Lys residues on target proteins (Gareau and Lima 2010). SUMO modifications can regulate the localization, function and interactions of target proteins, and influence many pathways including nuclear import/export, transcription, apoptosis, cell cycle regulation, and protein stability (Geiss-Friedlander and Melchior 2007). The enzymes that add or remove SUMO localize mostly at the nuclear envelope or in the nucleus (Wilkinson and Henley 2010; Zhang et al. 2002; Mingot et al. 2001). However, at least one, the SUMO-specific isopeptidase SENP2, associates dynamically with nuclear pore complexes (Goeres et al. 2011) and is regulated by shuttling between the nucleus and cytoplasm (Itahana et al. 2006). Many cytosolic proteins are controlled by SUMOylation including mitochondrial proteins, plasma membrane proteins and (in yeast) septins, all of which are unlikely to shuttle into the nucleus.
Human lamins A/C are modified by SUMO2 at Lys-201, both in vitro and in vivo. This modification is important for lamin A localization and filament assembly; both activities are disrupted by K201R or by nearby cardiomyopathy-causing E203G or E203K mutations, which also decrease cell viability (Zhang and Sarge 2008). A-type lamins can also be modified by a different SUMO, SUMO1 at two positions, Lys-420 (in the NLS) and Lys-486 (in the Ig-fold) both in vitro and in vivo (Simon et al. 2013). SUMOylation of the Ig-fold residue Lys-486 is disrupted by the familial partial lipodystrophy-causing G465D and K486N mutations (Simon et al. 2013). Lys-420 is alternatively modified by SUMO3 in HEK293 cells (Galisson et al. 2011).
In contrast to lamin A/C residues Lys-201 and Lys-420, which are located at canonical SUMOylation consensus sites, Lys-486 is not. Instead, Lys-486 represents a proposed ‘conformational’ consensus SUMOylation site, recognition of which is proposed to require Gly-465 and negatively charged residues Glu-460 and Asp-461, located directly beneath Lys-486 in the Ig-fold domain structure (Krimm et al. 2002; Simon et al. 2013). Lamins A/C and lamin B1 were also identified as potential targets of SUMO4 in serum-starved HEK293 cells (Guo et al. 2005).
At any given time only a few percent, at most, of lamins are SUMOylated (Zhang and Sarge 2008; Simon et al. 2013), similar to other characterized SUMO substrates (Johnson 2004; Hay 2005). This scarcity suggests the enzymes that add and remove SUMO either have limited access to lamin A, or are tightly controlled by other regulators in the nuclei of specific cell types. The timing and extent to which lamin A is SUMOylated in human tissues affected by FPLD disease (Simon et al. 2013) or cardiomyopathy (Zhang and Sarge 2008), and the downstream consequences of modification by SUMO1, SUMO2 or SUMO3 are open questions.
Acetylation
First discovered as a modification of histones, many other proteins are now known to be acetylated, including some (e.g., tubulin) that reside in the cytoplasm (Glozak et al. 2005). Both A- and B-type lamins were reportedly acetylated in high-throughput studies of HeLa cells (Kim et al. 2006) and a human acute myeloid leukemia cell line (MV4-11 cells; Choudhary et al. 2009). Eight acetylation sites were identified in A-type lamins: six in the rod domain (Lys-97, Lys-108, Lys-114, Lys-270, Lys-311, Lys-378), one in the NLS (Lys-417) and one in the Ig-fold (Lys-450) (Fig. 3). Lamin B1 has six acetylation sites (Lys-33, Lys-123, Lys-157, Lys-181, Lys-271, Lys-483) and lamin B2 has four (Lys-47, Lys-81, Lys-393, Lys-520) (Fig. 3). All three human lamins have one known acetylation site in the Ig-fold domain. Both B-type lamins have a known acetylation site at the border between the head domain and coiled-coil rod (Fig. 3; Choudhary et al. 2009). Nothing is known about the timing or functional consequences of lamin acetylation. However, since lamins associate with LEM-domain proteins (emerin and LAP2β) and HDAC3 to tether silent chromatin (Somech et al. 2005; Guelen et al. 2008; Zullo et al. 2012; Reddy et al. 2008; Demmerle et al. 2012), we speculate lamin acetylation might influence chromatin tethering.
Ubiquitylation
Ubiquitin was the first discovered small protein modification of other proteins. Like SUMO, it is covalently attached to Lys residues on target proteins; two enzymes (E1, E2) first prepare ubiquitin for transfer, with target specificity dictated by a variety of ubiquitin ligase (E3) enzymes (Neutzner and Neutzner 2012). Ubiquitin can be attached to another ubiquitin, creating a ‘chain’ that marks the target for proteolytic degradation. By contrast, attachment of a single ubiquitin is known to influence target proteins in diverse ways and regulates many specific cellular pathways and nuclear functions (Strieter and Korasick 2012). The impact of poly- and mono-ubiquitinylation, which can have major roles in the regulation of protein function and the spatial and temporal coordination of pathways, on the functions of lamins A/C, B1 and B2 are essentially unknown.
High-throughput mass spectrometry analysis of ubiquitylated proteins in HEK293T cells (Wagner et al. 2011) and HCT-116 cells, a colon adenocarcinoma cell line (Kim et al. 2011a), revealed widespread ubiquitylation of human lamins A/C, B1 and B2. Most ubiquitylation sites are located in the rod domain (Fig. 3) and might therefore influence lamin dimerization or filament assembly. Whereas poly-ubiquitylation is assumed to influence lamin turnover, the functional consequences of lamin mono-ubiquitylation are unknown.
Several residues can be either ubiquitylated or acetylated as reported for seven lamin A residues (Lys-97, Lys-108, Lys-270, Lys-311, Lys-378, Lys-417, Lys-450), four lamin B1 residues (Lys-123, Lys-157, Lys-271, Lys-483), and two lamin B2 residues (Lys-81, Lys-520). Lamin A/C Lys-201 can be either ubiquitylated or SUMO2-modified and Lys-486 can be either ubiquitylated or SUMO1-modified (Simon et al. 2013). Lamin A/C Lys-420, located in the NLS, can be either ubiquitylated, SUMO1-modified or SUMO3-modified. These competing modifications may differentially regulate lamin interactions and functions in specific tissues.
Lamins, as major structural proteins of the cell, are targeted for destruction early in apoptosis. They are directly cleaved by caspases 1 and 6 (Takahashi et al. 1996), granzymes A and B (Zhang et al. 2001), and CRNSP (Ca+2-regulated nuclear scaffold protease; Clawson et al. 1992) at sites located near many ubiquitylation sites (Fig. 3).
Conclusion and perspectives
In humans, 92 residues in lamins A/C, 52 in lamin B1, and 51 in lamin B2 are reportedly post-translationally modified, yet the only well-defined functional consequence (mitotic disassembly) was discovered more than 20 years ago. Lamins are probably also regulated by other modifications not discussed here, including ADP ribosylation (Adolph 1987). Furthermore, lamin modifications in other organisms might differ from those in human lamins both in detail (e.g., due to amino acid sequence differences; e.g., mouse lamin A/C phosphosites Ser-5, Thr-199, Thr-480, Ser-572; Eggert et al. 1993) and in substance, as diverging metazoan lineages evolved. Indeed, species-specific posttranslational modifications might explain why some lamin A mutations that cause a specific human disease, yield a different phenotype in mice (Stewart et al. 2007).
Modifications have the potential to regulate all aspects of lamin function, from filament assembly to the nuanced binding of tissue-specific partners. However, one must keep these modifications in perspective — even rare modification sites can be detected by modern mass spectrometry, giving the erroneous impression that lamins are always modified. Lamin modifications in living cells are likely to be relatively rare and transient due to the constant interplay between different modifying and de-modifying enzymes, with one known exception: mitosis, when many specific residues are phosphorylated at >80 % stoichiometry (Olsen et al. 2010; Ward and Kirschner 1990; Peter et al. 1990; Kill and Hutchison 1995). Huge gaps in knowledge about the nature, regulation and consequences of these modifications must be filled to understand how lamins function, and how specific mutations lead to disease.
References
Adam SA, Sengupta K, Goldman RD (2008) Regulation of nuclear lamin polymerization by importin alpha. J Biol Chem 283(13):8462–8468
Adolph KW (1987) ADPribosylation of nuclear proteins labeled with [3H]adenosine: changes during the HeLa cycle. Biochim Biophys Acta 909(3):222–230
Al-Haboubi T, Shumaker DK, Koser J, Wehnert M, Fahrenkrog B (2011) Distinct association of the nuclear pore protein Nup153 with A- and B-type lamins. Nucleus 2(5):500–509
Alfaro JF, Gong CX, Monroe ME, Aldrich JT, Clauss TR, Purvine SO, Wang Z, Camp DG 2nd, Shabanowitz J, Stanley P, Hart GW, Hunt DF, Yang F, Smith RD (2012) Tandem mass spectrometry identifies many mouse brain O-GlcNAcylated proteins including EGF domain-specific O-GlcNAc transferase targets. Proc Natl Acad Sci U S A 109(19):7280–7285
Bao X, Zhang W, Krencik R, Deng H, Wang Y, Girton J, Johansen J, Johansen KM (2005) The JIL-1 kinase interacts with lamin Dm0 and regulates nuclear lamina morphology of Drosophila nurse cells. J Cell Sci 118(Pt 21):5079–5087
Barrowman J, Hamblet C, Kane MS, Michaelis S (2012) Requirements for efficient proteolytic cleavage of prelamin A by ZMPSTE24. PLoS One 7(2):e32120
Barton RM, Worman HJ (1999) Prenylated prelamin A interacts with Narf, a novel nuclear protein. J Biol Chem 274(42):30008–30018
Bartz RR, Piantadosi CA (2010) Clinical review: oxygen as a signaling molecule. Crit Care 14(5):234
Beck LA, Hosick TJ, Sinensky M (1990) Isoprenylation is required for the processing of the lamin A precursor. J Cell Biol 110(5):1489–1499
Ben-Harush K, Wiesel N, Frenkiel-Krispin D, Moeller D, Soreq E, Aebi U, Herrmann H, Gruenbaum Y, Medalia O (2009) The supramolecular organization of the C. elegans nuclear lamin filament. J Mol Biol 386(5):1392–1402
Bengtsson L, Otto H (2008) LUMA interacts with emerin and influences its distribution at the inner nuclear membrane. J Cell Sci 121(4):536–548
Bergo MO, Gavino B, Ross J, Schmidt WK, Hong C, Kendall LV, Mohr A, Meta M, Genant H, Jiang Y, Wisner ER, Van Bruggen N, Carano RA, Michaelis S, Griffey SM, Young SG (2002) Zmpste24 deficiency in mice causes spontaneous bone fractures, muscle weakness, and a prelamin A processing defect. Proc Natl Acad Sci U S A 99(20):13049–13054
Beullens M, Vancauwenbergh S, Morrice N, Derua R, Ceulemans H, Waelkens E, Bollen M (2005) Substrate specificity and activity regulation of protein kinase MELK. J Biol Chem 280(48):40003–40011
Bokenkamp R, Raz V, Venema A, DeRuiter MC, van Munsteren C, Olive M, Nabel EG, Gittenberger-de Groot AC (2011) Differential temporal and spatial progerin expression during closure of the ductus arteriosus in neonates. PLoS One 6(9):e23975
Brachner A, Reipert S, Foisner R, Gotzmann J (2005) LEM2 is a novel MAN1-related inner nuclear membrane protein associated with A-type lamins. J Cell Sci 118(24):5797–5810
Brussino A, Vaula G, Cagnoli C, Mauro A, Pradotto L, Daniele D, Di Gregorio E, Barberis M, Arduino C, Squadrone S, Abete MC, Migone N, Calabrese O, Brusco A (2009) A novel family with Lamin B1 duplication associated with adult-onset leucoencephalopathy. J Neurol Neurosurg Psychiatry 80(2):237–240
Brussino A, Vaula G, Cagnoli C, Panza E, Seri M, Di Gregorio E, Scappaticci S, Camanini S, Daniele D, Bradac GB, Pinessi L, Cavalieri S, Grosso E, Migone N, Brusco A (2010) A family with autosomal dominant leukodystrophy linked to 5q23.2–q23.3 without lamin B1 mutations. Eur J Neurol 17(4):541–549
Buendia B, Courvalin JC, Collas P (2001) Dynamics of the nuclear envelope at mitosis and during apoptosis. Cell Mol Life Sci 58(12–13):1781–1789
Butin-Israeli V, Adam SA, Goldman AE, Goldman RD (2012) Nuclear lamin functions and disease. Trends Genet 28(9):464–471
Cenni V, Bertacchini J, Beretti F, Lattanzi G, Bavelloni A, Riccio M, Ruzzene M, Marin O, Arrigoni G, Parnaik V, Wehnert M, Maraldi NM, de Pol A, Cocco L, Marmiroli S (2008) Lamin A Ser404 is a nuclear target of Akt phosphorylation in C2C12 cells. J Proteome Res 7(11):4727–4735
Cenni V, Sabatelli P, Mattioli E, Marmiroli S, Capanni C, Ognibene A, Squarzoni S, Maraldi NM, Bonne G, Columbaro M, Merlini L, Lattanzi G (2005) Lamin A N-terminal phosphorylation is associated with myoblast activation: impairment in Emery–Dreifuss muscular dystrophy. J Med Genet 42(3):214–220
Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M (2009) Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325(5942):834–840
Clawson GA, Norbeck LL, Hatem CL, Rhodes C, Amiri P, McKerrow JH, Patierno SR, Fiskum G (1992) Ca(2+)-regulated serine protease associated with the nuclear scaffold. Cell Growth Differ 3(11):827–838
Coffinier C, Chang SY, Nobumori C, Tu Y, Farber EA, Toth JI, Fong LG, Young SG (2010) Abnormal development of the cerebral cortex and cerebellum in the setting of lamin B2 deficiency. Proc Natl Acad Sci U S A 107(11):5076–5081
Coffinier C, Jung HJ, Nobumori C, Chang S, Tu Y, Barnes RH 2nd, Yoshinaga Y, de Jong PJ, Vergnes L, Reue K, Fong LG, Young SG (2011) Deficiencies in lamin B1 and lamin B2 cause neurodevelopmental defects and distinct nuclear shape abnormalities in neurons. Mol Biol Cell 22(23):4683–4693
Collas P (1999) Sequential PKC- and Cdc2-mediated phosphorylation events elicit zebrafish nuclear envelope disassembly. J Cell Sci 112(6):977–987
Collas P, Thompson L, Fields AP, Poccia DL, Courvalin JC (1997) Protein kinase C-mediated interphase lamin B phosphorylation and solubilization. J Biol Chem 272(34):21274–21280
Crisp M, Liu Q, Roux K, Rattner JB, Shanahan C, Burke B, Stahl PD, Hodzic D (2006) Coupling of the nucleus and cytoplasm: role of the LINC complex. J Cell Biol 172(1):41–53
Cross T, Griffiths G, Deacon E, Sallis R, Gough M, Watters D, Lord JM (2000) PKC-delta is an apoptotic lamin kinase. Oncogene 19(19):2331–2337
Daub H, Olsen JV, Bairlein M, Gnad F, Oppermann FS, Korner R, Greff Z, Keri G, Stemmann O, Mann M (2008) Kinase-selective enrichment enables quantitative phosphoproteomics of the kinome across the cell cycle. Mol Cell 31(3):438–448
Dechat T, Adam SA, Taimen P, Shimi T, Goldman RD (2010) Nuclear lamins. Cold Spring Harb Perspect Biol 2(11):a000547
Dechat T, Korbei B, Vaughan OA, Vlcek S, Hutchison CJ, Foisner R (2000) Lamina-associated polypeptide 2alpha binds intranuclear A-type lamins. J Cell Sci 113(19):3473–3484
Dechat T, Pfleghaar K, Sengupta K, Shimi T, Shumaker DK, Solimando L, Goldman RD (2008) Nuclear lamins: major factors in the structural organization and function of the nucleus and chromatin. Genes Dev 22(7):832–853
Delbarre E, Tramier M, Coppey-Moisan M, Gaillard C, Courvalin JC, Buendia B (2006) The truncated prelamin A in Hutchinson–Gilford progeria syndrome alters segregation of A-type and B-type lamin homopolymers. Hum Mol Genet 15(7):1113–1122
Demmerle J, Koch AJ, Holaska JM (2012) The nuclear envelope protein emerin binds directly to histone deacetylase 3 (HDAC3) and activates HDAC3 activity. J Biol Chem 287(26):22080–22088
Dhe-Paganon S, Werner ED, Chi YI, Shoelson SE (2002) Structure of the globular tail of nuclear lamin. J Biol Chem 277(20):17381–17384
Dittmer TA, Misteli T (2011) The lamin protein family. Genome Biol 12(5):222
Dreuillet C, Tillit J, Kress M, Ernoult-Lange M (2002) In vivo and in vitro interaction between human transcription factor MOK2 and nuclear lamin A/C. Nucleic Acids Res 30(21):4634–4642
Eggert M, Radomski N, Linder D, Tripier D, Traub P, Jost E (1993) Identification of novel phosphorylation sites in murine A-type lamins. Eur J Biochem 213(2):659–671
Eggert M, Radomski N, Tripier D, Traub P, Jost E (1991) Identification of phosphorylation sites on murine nuclear lamin C by RP-HPLC and microsequencing. FEBS Lett 292(1–2):205–209
Enoch T, Peter M, Nurse P, Nigg EA (1991) p34cdc2 acts as a lamin kinase in fission yeast. J Cell Biol 112(5):797–807
Farnsworth CC, Wolda SL, Gelb MH, Glomset JA (1989) Human lamin B contains a farnesylated cysteine residue. J Biol Chem 264(34):20422–20429
Favale NO, Sterin Speziale NB, Fernandez Tome MC (2007) Hypertonic-induced lamin A/C synthesis and distribution to nucleoplasmic speckles is mediated by TonEBP/NFAT5 transcriptional activator. Biochem Biophys Res Commun 364(3):443–449
Ferraro A, Eufemi M, Cervoni L, Marinetti R, Turano C (1989) Glycosylated forms of nuclear lamins. FEBS Lett 257(2):241–246
Fiume R, Ramazzotti G, Teti G, Chiarini F, Faenza I, Mazzotti G, Billi AM, Cocco L (2009) Involvement of nuclear PLCbeta1 in lamin B1 phosphorylation and G2/M cell cycle progression. FASEB J 23(3):957–966
Foisner R, Gerace L (1993) Integral membrane proteins of the nuclear envelope interact with lamins and chromosomes, and binding is modulated by mitotic phosphorylation. Cell 73(7):1267–1279
Foisner R, Traub P, Wiche G (1991) Protein kinase A- and protein kinase C-regulated interaction of plectin with lamin B and vimentin. Proc Natl Acad Sci U S A 88(9):3812–3816
Friedman DL, Ken R (1988) Insulin stimulates incorporation of 32Pi into nuclear lamins A and C in quiescent BHK-21 cells. J Biol Chem 263(3):1103–1106
Furukawa K, Kondo T (1998) Identification of the lamina-associated-polypeptide-2-binding domain of B-type lamin. Eur J Biochem 251(3):729–733
Galisson F, Mahrouche L, Courcelles M, Bonneil E, Meloche S, Chelbi-Alix MK, Thibault P (2011) A novel proteomics approach to identify SUMOylated proteins and their modification sites in human cells. Mol Cell Proteomics 10 (2):M110 004796.
Gareau JR, Lima CD (2010) The SUMO pathway: emerging mechanisms that shape specificity, conjugation and recognition. Nat Rev Mol Cell Biol 11(12):861–871
Geiss-Friedlander R, Melchior F (2007) Concepts in sumoylation: a decade on. Nat Rev Mol Cell Biol 8(12):947–956
Gerace L, Blobel G (1980) The nuclear envelope lamina is reversibly depolymerized during mitosis. Cell 19(1):277–287
Gerace L, Huber MD (2012) Nuclear lamina at the crossroads of the cytoplasm and nucleus. J Struct Biol 177(1):24–31
Glozak MA, Sengupta N, Zhang X, Seto E (2005) Acetylation and deacetylation of non-histone proteins. Gene 363:15–23
Goeres J, Chan PK, Mukhopadhyay D, Zhang H, Raught B, Matunis MJ (2011) The SUMO-specific isopeptidase SENP2 associates dynamically with nuclear pore complexes through interactions with karyopherins and the Nup107-160 nucleoporin subcomplex. Mol Biol Cell 22(24):4868–4882
Gonzalez JM, Navarro-Puche A, Casar B, Crespo P, Andres V (2008) Fast regulation of AP-1 activity through interaction of lamin A/C, ERK1/2, and c-Fos at the nuclear envelope. J Cell Biol 183(4):653–666
Goss VL, Hocevar BA, Thompson LJ, Stratton CA, Burns DJ, Fields AP (1994) Identification of nuclear beta II protein kinase C as a mitotic lamin kinase. J Biol Chem 269(29):19074–19080
Grimsby S, Jaensson H, Dubrovska A, Lomnytska M, Hellman U, Souchelnytskyi S (2004) Proteomics-based identification of proteins interacting with Smad3: SREBP-2 forms a complex with Smad3 and inhibits its transcriptional activity. FEBS Lett 577(1–2):93–100
Guelen L, Pagie L, Brasset E, Meuleman W, Faza MB, Talhout W, Eussen BH, de Klein A, Wessels L, de Laat W, van Steensel B (2008) Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature 453(7197):948–951
Guo D, Han J, Adam BL, Colburn NH, Wang MH, Dong Z, Eizirik DL, She JX, Wang CY (2005) Proteomic analysis of SUMO4 substrates in HEK293 cells under serum starvation-induced stress. Biochem Biophys Res Commun 337(4):1308–1318
Haas M, Jost E (1993) Functional analysis of phosphorylation sites in human lamin A controlling lamin disassembly, nuclear transport and assembly. Eur J Cell Biol 62(2):237–247
Han X, Feng X, Rattner JB, Smith H, Bose P, Suzuki K, Soliman MA, Scott MS, Burke BE, Riabowol K (2008) Tethering by lamin A stabilizes and targets the ING1 tumour suppressor. Nat Cell Biol 10(11):1333–1340
Haque F, Lloyd DJ, Smallwood DT, Dent CL, Shanahan CM, Fry AM, Trembath RC, Shackleton S (2006) SUN1 interacts with nuclear lamin A and cytoplasmic nesprins to provide a physical connection between the nuclear lamina and the cytoskeleton. Mol Cell Biol 26(10):3738–3751
Hart GW, Copeland RJ (2010) Glycomics hits the big time. Cell 143(5):672–676
Hay RT (2005) SUMO: a history of modification. Mol Cell 18(1):1–12
Heald R, McKeon F (1990) Mutations of phosphorylation sites in lamin A that prevent nuclear lamina disassembly in mitosis. Cell 61(4):579–589
Hegele RA, Cao H, Liu DM, Costain GA, Charlton-Menys V, Rodger NW, Durrington PN (2006) Sequencing of the reannotated LMNB2 gene reveals novel mutations in patients with acquired partial lipodystrophy. Am J Hum Genet 79(2):383–389
Heitlinger E, Peter M, Haner M, Lustig A, Aebi U, Nigg EA (1991) Expression of chicken lamin B2 in Escherichia coli: characterization of its structure, assembly, and molecular interactions. J Cell Biol 113(3):485–495
Hennekes H, Peter M, Weber K, Nigg EA (1993) Phosphorylation on protein kinase C sites inhibits nuclear import of lamin B2. J Cell Biol 120(6):1293–1304
Herrmann H, Kreplak L, Aebi U (2004) Isolation, characterization, and in vitro assembly of intermediate filaments. Methods Cell Biol 78:3–24
Hocevar BA, Burns DJ, Fields AP (1993) Identification of protein kinase C (PKC) phosphorylation sites on human lamin B. Potential role of PKC in nuclear lamina structural dynamics. J Biol Chem 268(10):7545–7552
Holaska JM, Lee KK, Kowalski AK, Wilson KL (2003) Transcriptional repressor germ cell-less (GCL) and barrier to autointegration factor (BAF) compete for binding to emerin in vitro. J Biol Chem 278(9):6969–6975
Holaska JM, Wilson KL (2007) An emerin "proteome": purification of distinct emerin-containing complexes from HeLa cells suggests molecular basis for diverse roles including gene regulation, mRNA splicing, signaling, mechanosensing, and nuclear architecture. Biochemistry 46(30):8897–8908
Huang Y, Cai M, Clore GM, Craigie R (2011) No interaction of barrier-to-autointegration factor (BAF) with HIV-1 MA, cone-rod homeobox (Crx) or MAN1-C in absence of DNA. PLoS One 6(9):e25123
Imami K, Sugiyama N, Kyono Y, Tomita M, Ishihama Y (2008) Automated phosphoproteome analysis for cultured cancer cells by two-dimensional nanoLC-MS using a calcined titania/C18 biphasic column. Anal Sci 24(1):161–166
Isokane M, Hieda M, Hirakawa S, Shudou M, Nakashiro K, Hashimoto K, Hamakawa H, Higashiyama S (2008) Plasma-membrane-anchored growth factor pro-amphiregulin binds A-type lamin and regulates global transcription. J Cell Sci 121(21):3608–3618
Itahana Y, Yeh ET, Zhang Y (2006) Nucleocytoplasmic shuttling modulates activity and ubiquitination-dependent turnover of SUMO-specific protease 2. Mol Cell Biol 26(12):4675–4689
Ivorra C, Kubicek M, Gonzalez JM, Sanz-Gonzalez SM, Alvarez-Barrientos A, O'Connor JE, Burke B, Andres V (2006) A mechanism of AP-1 suppression through interaction of c-Fos with lamin A/C. Genes Dev 20(3):307–320
Johnson ES (2004) Protein modification by SUMO. Annu Rev Biochem 73:355–382
Kasahara K, Chida K, Tsunenaga M, Kohno Y, Ikuta T, Kuroki T (1991) Identification of lamin B2 as a substrate of protein kinase C in BALB/MK-2 mouse keratinocytes. J Biol Chem 266(30):20018–20023
Kill IR, Hutchison CJ (1995) S-phase phosphorylation of lamin B2. FEBS Lett 377(1):26–30
Kim JE, Tannenbaum SR, White FM (2005) Global phosphoproteome of HT-29 human colon adenocarcinoma cells. J Proteome Res 4(4):1339–1346
Kim SC, Sprung R, Chen Y, Xu Y, Ball H, Pei J, Cheng T, Kho Y, Xiao H, Xiao L, Grishin NV, White M, Yang XJ, Zhao Y (2006) Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol Cell 23(4):607–618
Kim W, Bennett EJ, Huttlin EL, Guo A, Li J, Possemato A, Sowa ME, Rad R, Rush J, Comb MJ, Harper JW, Gygi SP (2011a) Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol Cell 44(2):325–340
Kim Y, Sharov AA, McDole K, Cheng M, Hao H, Fan CM, Gaiano N, Ko MS, Zheng Y (2011b) Mouse B-type lamins are required for proper organogenesis but not by embryonic stem cells. Science 334(6063):1706–1710
Kolb T, Maass K, Hergt M, Aebi U, Herrmann H (2011) Lamin A and lamin C form homodimers and coexist in higher complex forms both in the nucleoplasmic fraction and in the lamina of cultured human cells. Nucleus 2(5):425–433
Krimm I, Ostlund C, Gilquin B, Couprie J, Hossenlopp P, Mornon JP, Bonne G, Courvalin JC, Worman HJ, Zinn-Justin S (2002) The Ig-like structure of the C-terminal domain of lamin A/C, mutated in muscular dystrophies, cardiomyopathy, and partial lipodystrophy. Structure 10(6):811–823
Kubben N, Voncken JW, Demmers J, Calis C, van Almen G, Pinto Y, Misteli T (2010) Identification of differential protein interactors of lamin A and progerin. Nucleus 1(6):513–525
Kuga T, Nozaki N, Matsushita K, Nomura F, Tomonaga T (2010) Phosphorylation statuses at different residues of lamin B2, B1, and A/C dynamically and independently change throughout the cell cycle. Exp Cell Res 316(14):2301–2312
Lai JS, Herr W (1992) Ethidium bromide provides a simple tool for identifying genuine DNA-independent protein associations. Proc Natl Acad Sci U S A 89(15):6958–6962
Lee CP, Chen MR (2010) Escape of herpesviruses from the nucleus. Rev Med Virol 20(4):214–230
Lee CP, Huang YH, Lin SF, Chang Y, Chang YH, Takada K, Chen MR (2008) Epstein-Barr virus BGLF4 kinase induces disassembly of the nuclear lamina to facilitate virion production. J Virol 82(23):11913–11926
Lee KK, Haraguchi T, Lee RS, Koujin T, Hiraoka Y, Wilson KL (2001) Distinct functional domains in emerin bind lamin A and DNA-bridging protein BAF. J Cell Sci 114(Pt 24):4567–4573
Libotte T, Zaim H, Abraham S, Padmakumar VC, Schneider M, Lu W, Munck M, Hutchison C, Wehnert M, Fahrenkrog B, Sauder U, Aebi U, Noegel AA, Karakesisoglou I (2005) Lamin A/C-dependent localization of Nesprin-2, a giant scaffolder at the nuclear envelope. Mol Biol Cell 16(7):3411–3424
Lloyd DJ, Trembath RC, Shackleton S (2002) A novel interaction between lamin A and SREBP1: implications for partial lipodystrophy and other laminopathies. Hum Mol Genet 11(7):769–777
Lussi YC, Hugi I, Laurell E, Kutay U, Fahrenkrog B (2011) The nucleoporin Nup88 is interacting with nuclear lamin A. Mol Biol Cell 22(7):1080–1090
Ma L, Tsai MY, Wang S, Lu B, Chen R, Iii JR, Zhu X, Zheng Y (2009) Requirement for Nudel and dynein for assembly of the lamin B spindle matrix. Nat Cell Biol 11(3):247–256
Maison C, Pyrpasopoulou A, Theodoropoulos PA, Georgatos SD (1997) The inner nuclear membrane protein LAP1 forms a native complex with B-type lamins and partitions with spindle-associated mitotic vesicles. EMBO J 16(16):4839–4850
Malhas AN, Lee CF, Vaux DJ (2009) Lamin B1 controls oxidative stress responses via Oct-1. J Cell Biol 184(1):45–55
Malik R, Lenobel R, Santamaria A, Ries A, Nigg EA, Korner R (2009) Quantitative analysis of the human spindle phosphoproteome at distinct mitotic stages. J Proteome Res 8(10):4553–4563
Mall M, Walter T, Gorjanacz M, Davidson IF, Nga Ly-Hartig TB, Ellenberg J, Mattaj IW (2012) Mitotic lamin disassembly is triggered by lipid-mediated signaling. J Cell Biol 198(6):981–990
Mancini MA, Shan B, Nickerson JA, Penman S, Lee WH (1994) The retinoblastoma gene product is a cell cycle-dependent, nuclear matrix-associated protein. Proc Natl Acad Sci U S A 91(1):418–422
Mansharamani M, Wilson KL (2005) Direct binding of nuclear membrane protein MAN1 to emerin in vitro and two modes of binding to barrier-to-autointegration factor. J Biol Chem 280(14):13863–13870
Mariappan I, Gurung R, Thanumalayan S, Parnaik VK (2007) Identification of cyclin D3 as a new interaction partner of lamin A/C. Biochem Biophys Res Commun 355(4):981–985
Martelli AM, Bortul R, Tabellini G, Faenza I, Cappellini A, Bareggi R, Manzoli L, Cocco L (2002) Molecular characterization of protein kinase C-alpha binding to lamin A. J Cell Biochem 86(2):320–330
Maske CP, Hollinshead MS, Higbee NC, Bergo MO, Young SG, Vaux DJ (2003) A carboxyl-terminal interaction of lamin B1 is dependent on the CAAX endoprotease Rce1 and carboxymethylation. J Cell Biol 162(7):1223–1232
Meier R, Muller PR, Hirt A, Leibundgut K, Ridolfi-Luthy A, Wagner HP (1997) Differential phosphorylation of lamin B2 in normal and leukemic cells. Leuk Res 21(9):841–847
Mingot JM, Kostka S, Kraft R, Hartmann E, Gorlich D (2001) Importin 13: a novel mediator of nuclear import and export. EMBO J 20(14):3685–3694
Mislow JM, Holaska JM, Kim MS, Lee KK, Segura-Totten M, Wilson KL, McNally EM (2002) Nesprin-1alpha self-associates and binds directly to emerin and lamin A in vitro. FEBS Lett 525(1–3):135–140
Mitsuhashi H, Hayashi YK, Matsuda C, Noguchi S, Wakatsuki S, Araki T, Nishino I (2010) Specific phosphorylation of Ser458 of A-type lamins in LMNA-associated myopathy patients. J Cell Sci 123(Pt 22):3893–3900
Molloy A, Cotter O, van Spaendonk R, Sistermans E, Sweeney B (2012) A patient with a rare leukodystrophy related to lamin B1 duplication. Ir Med J 105(6):186–187
Molloy S, Little M (1992) p34cdc2 kinase-mediated release of lamins from nuclear ghosts is inhibited by cAMP-dependent protein kinase. Exp Cell Res 201(2):494–499
Moritz A, Li Y, Guo A, Villen J, Wang Y, MacNeill J, Kornhauser J, Sprott K, Zhou J, Possemato A, Ren JM, Hornbeck P, Cantley LC, Gygi SP, Rush J, Comb MJ (2010) Akt-RSK-S6 kinase signaling networks activated by oncogenic receptor tyrosine kinases. Sci Signal 3(136):ra64
Neutzner M, Neutzner A (2012) Enzymes of ubiquitination and deubiquitination. Essays Biochem 52:37–50
Nigg EA, Kitten GT, Vorburger K (1992) Targeting lamin proteins to the nuclear envelope: the role of CaaX box modifications. Biochem Soc Trans 20(2):500–504
Olsen JV, Blagoev B, Gnad F, Macek B, Kumar C, Mortensen P, Mann M (2006) Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 127(3):635–648
Olsen JV, Vermeulen M, Santamaria A, Kumar C, Miller ML, Jensen LJ, Gnad F, Cox J, Jensen TS, Nigg EA, Brunak S, Mann M (2010) Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci Signal 3(104):ra3
Ozaki T, Saijo M, Murakami K, Enomoto H, Taya Y, Sakiyama S (1994) Complex formation between lamin A and the retinoblastoma gene product: identification of the domain on lamin A required for its interaction. Oncogene 9(9):2649–2653
Padiath QS, Saigoh K, Schiffmann R, Asahara H, Yamada T, Koeppen A, Hogan K, Ptacek LJ, Fu YH (2006) Lamin B1 duplications cause autosomal dominant leukodystrophy. Nat Genet 38(10):1114–1123
Pan C, Olsen JV, Daub H, Mann M (2009) Global effects of kinase inhibitors on signaling networks revealed by quantitative phosphoproteomics. Mol Cell Proteomics 8(12):2796–2808
Pegoraro G, Kubben N, Wickert U, Gohler H, Hoffmann K, Misteli T (2009) Ageing-related chromatin defects through loss of the NURD complex. Nat Cell Biol 11(10):1261–1267
Pekovic V, Gibbs-Seymour I, Markiewicz E, Alzoghaibi F, Benham AM, Edwards R, Wenhert M, von Zglinicki T, Hutchison CJ (2011) Conserved cysteine residues in the mammalian lamin A tail are essential for cellular responses to ROS generation. Aging Cell 10(6):1067–1079
Pendas AM, Zhou Z, Cadinanos J, Freije JM, Wang J, Hultenby K, Astudillo A, Wernerson A, Rodriguez F, Tryggvason K, Lopez-Otin C (2002) Defective prelamin A processing and muscular and adipocyte alterations in Zmpste24 metalloproteinase-deficient mice. Nat Genet 31(1):94–99
Peter A, Stick R (2012) Evolution of the lamin protein family: what introns can tell. Nucleus 3(1)
Peter M, Heitlinger E, Haner M, Aebi U, Nigg EA (1991) Disassembly of in vitro formed lamin head-to-tail polymers by CDC2 kinase. EMBO J 10(6):1535–1544
Peter M, Nakagawa J, Doree M, Labbe JC, Nigg EA (1990) In vitro disassembly of the nuclear lamina and M phase-specific phosphorylation of lamins by cdc2 kinase. Cell 61(4):591–602
Rao L, Modha D, White E (1997) The E1B 19 K protein associates with lamins in vivo and its proper localization is required for inhibition of apoptosis. Oncogene 15(13):1587–1597
Reddy KL, Zullo JM, Bertolino E, Singh H (2008) Transcriptional repression mediated by repositioning of genes to the nuclear lamina. Nature 452(7184):243–247
Rigbolt KT, Prokhorova TA, Akimov V, Henningsen J, Johansen PT, Kratchmarova I, Kassem M, Mann M, Olsen JV, Blagoev B (2011) System-wide temporal characterization of the proteome and phosphoproteome of human embryonic stem cell differentiation. Sci Signal 4(164):rs3
Roux KJ, Kim DI, Raida M, Burke B (2012) A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J Cell Biol 196(6):801–810
Ruan J, Xu C, Bian C, Lam R, Wang JP, Kania J, Min J, Zang J (2012) Crystal structures of the coil 2B fragment and the globular tail domain of human lamin B1. FEBS Lett 586(4):314–318
Sakaki M, Koike H, Takahashi N, Sasagawa N, Tomioka S, Arahata K, Ishiura S (2001) Interaction between emerin and nuclear lamins. J Biochem 129(2):321–327
Schirmer EC, Gerace L (2004) The stability of the nuclear lamina polymer changes with the composition of lamin subtypes according to their individual binding strengths. J Biol Chem 279(41):42811–42817
Schneider U, Mini T, Jeno P, Fisher PA, Stuurman N (1999) Phosphorylation of the major Drosophila lamin in vivo: site identification during both M-phase (meiosis) and interphase by electrospray ionization tandem mass spectrometry. Biochemistry 38(14):4620–4632
Schumacher J, Reichenzeller M, Kempf T, Schnolzer M, Herrmann H (2006) Identification of a novel, highly variable amino-terminal amino acid sequence element in the nuclear intermediate filament protein lamin B(2) from higher vertebrates. FEBS Lett 580(26):6211–6216
Schuster J, Sundblom J, Thuresson AC, Hassin-Baer S, Klopstock T, Dichgans M, Cohen OS, Raininko R, Melberg A, Dahl N (2011) Genomic duplications mediate overexpression of lamin B1 in adult-onset autosomal dominant leukodystrophy (ADLD) with autonomic symptoms. Neurogenetics 12(1):65–72
Shimi T, Pfleghaar K, Kojima S, Pack CG, Solovei I, Goldman AE, Adam SA, Shumaker DK, Kinjo M, Cremer T, Goldman RD (2008) The A- and B-type nuclear lamin networks: microdomains involved in chromatin organization and transcription. Genes Dev 22(24):3409–3421
Shimizu T, Cao CX, Shao RG, Pommier Y (1998) Lamin B phosphorylation by protein kinase calpha and proteolysis during apoptosis in human leukemia HL60 cells. J Biol Chem 273(15):8669–8674
Shumaker DK, Solimando L, Sengupta K, Shimi T, Adam SA, Grunwald A, Strelkov SV, Aebi U, Cardoso MC, Goldman RD (2008) The highly conserved nuclear lamin Ig-fold binds to PCNA: its role in DNA replication. J Cell Biol 181(2):269–280
Sieprath T, Darwiche R, De Vos WH (2012) Lamins as mediators of oxidative stress. Biochem Biophys Res Commun 421(4):635–639
Simon DN, Domaradzki T, Hofmann WA, Wilson KL (2013) Lamin A tail modification by SUMO1 is disrupted by familial partial lipodystrophy-causing mutations. Mol Biol Cell 24(3):342–50
Simon DN, Wilson KL (2011) The nucleoskeleton as a genome-associated dynamic 'network of networks'. Nat Rev Mol Cell Biol 12(11):695–708
Simon DN, Zastrow MS, Wilson KL (2010) Direct actin binding to A- and B-type lamin tails and actin filament bundling by the lamin A tail. Nucleus 1(3):264–272
Smith TC, Fang Z, Luna EJ (2010) Novel interactors and a role for supervillin in early cytokinesis. Cytoskeleton (Hoboken) 67(6):346–364
Somech R, Shaklai S, Geller O, Amariglio N, Simon AJ, Rechavi G, Gal-Yam EN (2005) The nuclear-envelope protein and transcriptional repressor LAP2beta interacts with HDAC3 at the nuclear periphery, and induces histone H4 deacetylation. J Cell Sci 118(Pt 17):4017–4025
Speese SD, Ashley J, Jokhi V, Nunnari J, Barria R, Li Y, Ataman B, Koon A, Chang YT, Li Q, Moore MJ, Budnik V (2012) Nuclear envelope budding enables large ribonucleoprotein particle export during synaptic Wnt signaling. Cell 149(4):832–846
Sridharan DM, McMahon LW, Lambert MW (2006) alphaII-Spectrin interacts with five groups of functionally important proteins in the nucleus. Cell Biol Int 30(11):866–878
Steen RL, Beullens M, Landsverk HB, Bollen M, Collas P (2003) AKAP149 is a novel PP1 specifier required to maintain nuclear envelope integrity in G1 phase. J Cell Sci 116(11):2237–2246
Steen RL, Collas P (2001) Mistargeting of B-type lamins at the end of mitosis: implications on cell survival and regulation of lamins A/C expression. J Cell Biol 153(3):621–626
Stewart CL, Kozlov S, Fong LG, Young SG (2007) Mouse models of the laminopathies. Exp Cell Res 313(10):2144–2156
Stierle V, Couprie J, Ostlund C, Krimm I, Zinn-Justin S, Hossenlopp P, Worman HJ, Courvalin JC, Duband-Goulet I (2003) The carboxyl-terminal region common to lamins A and C contains a DNA binding domain. Biochemistry 42(17):4819–4828
Strelkov SV, Schumacher J, Burkhard P, Aebi U, Herrmann H (2004) Crystal structure of the human lamin A coil 2B dimer: implications for the head-to-tail association of nuclear lamins. J Mol Biol 343(4):1067–1080
Strieter ER, Korasick DA (2012) Unraveling the complexity of ubiquitin signaling. ACS Chem Biol 7(1):52–63
Stuurman N (1997) Identification of a conserved phosphorylation site modulating nuclear lamin polymerization. FEBS Lett 401(2–3):171–174
Tabellini G, Bortul R, Aluigi M, Billi AM, Bareggi R, Grill V, Narducci P, Martelli AM (2002) Binding of elements of protein kinase C-alpha regulatory domain to lamin B1. Cell Signal 14(10):819–827
Takahashi A, Alnemri ES, Lazebnik YA, Fernandes-Alnemri T, Litwack G, Moir RD, Goldman RD, Poirier GG, Kaufmann SH, Earnshaw WC (1996) Cleavage of lamin A by Mch2 alpha but not CPP32: multiple interleukin 1 beta-converting enzyme-related proteases with distinct substrate recognition properties are active in apoptosis. Proc Natl Acad Sci U S A 93(16):8395–8400
Takamori Y, Tamura Y, Kataoka Y, Cui Y, Seo S, Kanazawa T, Kurokawa K, Yamada H (2007) Differential expression of nuclear lamin, the major component of nuclear lamina, during neurogenesis in two germinal regions of adult rat brain. Eur J Neurosci 25(6):1653–1662
Tang K, Finley RL Jr, Nie D, Honn KV (2000) Identification of 12-lipoxygenase interaction with cellular proteins by yeast two-hybrid screening. Biochemistry 39(12):3185–3191
Taniura H, Glass C, Gerace L (1995) A chromatin binding site in the tail domain of nuclear lamins that interacts with core histones. J Cell Biol 131(1):33–44
Thompson LJ, Fields AP (1996) betaII protein kinase C is required for the G2/M phase transition of cell cycle. J Biol Chem 271(25):15045–15053
Van Berlo JH, Voncken JW, Kubben N, Broers JL, Duisters R, van Leeuwen RE, Crijns HJ, Ramaekers FC, Hutchison CJ, Pinto YM (2005) A-type lamins are essential for TGF-beta1 induced PP2A to dephosphorylate transcription factors. Hum Mol Genet 14(19):2839–2849
Van Hoof D, Munoz J, Braam SR, Pinkse MW, Linding R, Heck AJ, Mummery CL, Krijgsveld J (2009) Phosphorylation dynamics during early differentiation of human embryonic stem cells. Cell Stem Cell 5(2):214–226
Varela I, Cadinanos J, Pendas AM, Gutierrez-Fernandez A, Folgueras AR, Sanchez LM, Zhou Z, Rodriguez FJ, Stewart CL, Vega JA, Tryggvason K, Freije JM, Lopez-Otin C (2005) Accelerated ageing in mice deficient in Zmpste24 protease is linked to p53 signalling activation. Nature 437(7058):564–568
Vaughan A, Alvarez-Reyes M, Bridger JM, Broers JL, Ramaekers FC, Wehnert M, Morris GE, Whitfield WGF, Hutchison CJ (2001) Both emerin and lamin C depend on lamin A for localization at the nuclear envelope. J Cell Sci 114(14):2577–2590
Vergnes L, Peterfy M, Bergo MO, Young SG, Reue K (2004) Lamin B1 is required for mouse development and nuclear integrity. Proc Natl Acad Sci U S A 101(28):10428–10433
Vlcek S, Foisner R, Wilson KL (2004) Lco1 is a novel widely expressed lamin-binding protein in the nuclear interior. Exp Cell Res 298(2):499–511
Wagner SA, Beli P, Weinert BT, Nielsen ML, Cox J, Mann M, Choudhary C (2011) A proteome-wide, quantitative survey of in vivo ubiquitylation sites reveals widespread regulatory roles. Mol Cell Proteomics 10 (10):M111 013284.
Wang B, Malik R, Nigg EA, Korner R (2008) Evaluation of the low-specificity protease elastase for large-scale phosphoproteome analysis. Anal Chem 80(24):9526–9533
Wang Z, Udeshi ND, Slawson C, Compton PD, Sakabe K, Cheung WD, Shabanowitz J, Hunt DF, Hart GW (2010) Extensive crosstalk between O-GlcNAcylation and phosphorylation regulates cytokinesis. Sci Signal 3(104):ra2
Ward GE, Kirschner MW (1990) Identification of cell cycle-regulated phosphorylation sites on nuclear lamin C. Cell 61(4):561–577
Wilkinson KA, Henley JM (2010) Mechanisms, regulation and consequences of protein SUMOylation. Biochem J 428(2):133–145
Wilson KL, Foisner R (2010) Lamin-binding proteins. Cold Spring Harb Perspect Biol 2(4):a000554
Worman HJ (2012) Nuclear lamins and laminopathies. J Pathol 226(2):316–325
Ye Q, Worman HJ (1994) Primary structure analysis and lamin B and DNA binding of human LBR, an integral protein of the nuclear envelope inner membrane. J Biol Chem 269(15):11306–11311
Ye Q, Worman HJ (1995) Protein–protein interactions between human nuclear lamins expressed in yeast. Exp Cell Res 219(1):292–298
Young SG, Fong LG, Michaelis S (2005) Prelamin A, Zmpste24, misshapen cell nuclei, and progeria—new evidence suggesting that protein farnesylation could be important for disease pathogenesis. J Lipid Res 46(12):2531–2558
Zaremba-Czogalla M, Piekarowicz K, Wachowicz K, Koziol K, Dubinska-Magiera M, Rzepecki R (2012) The different function of single phosphorylation sites of Drosophila melanogaster lamin Dm and lamin C. PLoS One 7(2):e32649
Zastrow MS, Flaherty DB, Benian GM, Wilson KL (2006) Nuclear titin interacts with A- and B-type lamins in vitro and in vivo. J Cell Sci 119(2):239–249
Zastrow MS, Vlcek S, Wilson KL (2004) Proteins that bind A-type lamins: integrating isolated clues. J Cell Sci 117(Pt 7):979–987
Zhang D, Beresford PJ, Greenberg AH, Lieberman J (2001) Granzymes A and B directly cleave lamins and disrupt the nuclear lamina during granule-mediated cytolysis. Proc Natl Acad Sci U S A 98(10):5746–5751
Zhang H, Saitoh H, Matunis MJ (2002) Enzymes of the SUMO modification pathway localize to filaments of the nuclear pore complex. Mol Cell Biol 22(18):6498–6508
Zhang YQ, Sarge KD (2008) Sumoylation regulates lamin A function and is lost in lamin A mutants associated with familial cardiomyopathies. J Cell Biol 182(1):35–39
Zhong N, Radu G, Ju W, Brown WT (2005) Novel progerin-interactive partner proteins hnRNP E1, EGF, Mel 18, and UBC9 interact with lamin A/C. Biochem Biophys Res Commun 338(2):855–861
Zullo JM, Demarco IA, Pique-Regi R, Gaffney DJ, Epstein CB, Spooner CJ, Luperchio TR, Bernstein BE, Pritchard JK, Reddy KL, Singh H (2012) DNA sequence-dependent compartmentalization and silencing of chromatin at the nuclear lamina. Cell 149(7):1474–1487
Acknowledgments
We thank Michael J. Matunis, Gerald W. Hart and Jason M. Berk for insightful discussions and comments on the manuscript. We apologize to authors whose work could not be cited here due to space limitations. We gratefully acknowledge support from the NIH (RO1 048646 to K.L.W.).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Erich Nigg
Rights and permissions
About this article
Cite this article
Simon, D.N., Wilson, K.L. Partners and post-translational modifications of nuclear lamins. Chromosoma 122, 13–31 (2013). https://doi.org/10.1007/s00412-013-0399-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00412-013-0399-8