
Abstract
We experimentally investigate the effect of water on intergranular mass transport. The growth of enstatite single rims between forsterite and quartz and forsterite-enstatite double rims between periclase and quartz was performed at 1,000°C and 1.5 GPa for incremental water–solid fractions ranging from 0 to 5 wt%. A kinetics analysis based on time series ranging from 8 min to 18 h at 1 wt% H2O demonstrates that the growth of both the single and double rims is controlled by the transport of the chemical components in the intergranular medium. Inert platinum markers reveal that MgO is always the most mobile component, and the only one, except at the highest water fractions (>~2 wt%) where some mobility of SiO2 is observed. Modelling yields that, in all of the rims from both the single and double rims, there is an increase by about six orders of magnitude between diffusivities in dry grain boundaries and in intergranular media with 5 wt% H2O of the bulk, therefore confirming that water/rock ratio is a parameter as important as temperature regarding reaction kinetics. The transition from dry to water-saturated conditions appears virtually instantaneous, occurring between 500 and 1,000 wt. ppm water–solid fraction. It corresponds to a jump of four to five orders of magnitude in diffusivities, in line with the gap between ‘dry’ and ‘wet’ enstatite single rim growth rates from various previous experimental investigations. At higher water–solid fractions, distinct increase in the diffusivities demonstrates the existence of two different intergranular transport mechanisms competing at water-saturated conditions. The first mechanism is diffusion in hydrous-saturated grain boundaries (HGBs), which dominates as long as pores are isolated. We estimate the Arrhenius law for MgO diffusion in enstatite HGBs to be \( \delta_{\text{HGB}} D_{\text{HGB}}^{\text{MgO}} = \delta_{\text{HGB}} D_{0} e^{{ - E/{\text{RT}}}} \) with E = 185 ± 20 kJ/mol and δHGB D 0 = 10−14.7±0.9 m3/s, where δHGB is the effective width of the HGBs. The second mechanism is diffusion through fluid-filled channels, which dominates when pores are interconnected. A new description of the effect of water on intergranular mass transport is proposed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Intergranular diffusion plays a key role in a variety of geological processes such as reactions between minerals, element or isotope transport and fractionation, electrical conductivity, creep and many others (see Dohmen and Milke 2010 for a review). There is general agreement that the presence of water, or rather hydrous fluid, drastically promotes element transport along grain boundaries. For example, the grain boundary diffusivity of aluminium in ‘hydrous fluid-saturated’ rocks is enhanced by seven orders of magnitude compared to ‘nearly anhydrous’ rocks (Carlson 2010), and Gasc et al. (2011) reported the same range of variations for the electrical conductivity in polycrystalline brucite from ‘dry’ to ‘wet’ conditions. Based on a consensus from literature, Farver and Yund (1995) proposed that intergranular diffusion can be classified into three categories, depending on water activity in the intergranular medium: ‘water-absent’, ‘water-unsaturated’ and ‘water-saturated’ regimes (see also Rubie 1986; Carlson 2010). However, the physico-chemical nature of the intergranular medium corresponding to these regimes remains unclear, and the transitional steps from one to another are not well specified so far. This partly results from the lack of detailed studies over the full range of water–rock fractions in a given system.
Performing rim growth experiments in model systems is one of the methods of choice to work out mechanisms and kinetics of grain boundary diffusion. Reactions between incompatible minerals require transport of chemical components between reaction interfaces to occur, and this generally takes place in the intergranular medium of polycrystalline products. In the geological context, the most studied reaction is the growth of polycrystalline single rims of enstatite between forsterite and quartz, which was investigated over a wide range of temperatures and for various pressures (Yund 1997; Fisler et al. 1997; Milke et al. 2001, 2007, 2009; Abart et al. 2004; Gardés et al. 2011; Gardés and Heinrich 2011; see also Dohmen and Milke 2010). The growth rates of enstatite fall into two groups, following distinct Arrhenius lines separated by a gap of about four orders of magnitude at 1,000°C (Fig. 1). In interpreting the results of previous experimental set-ups, Dohmen and Milke (2010) concluded that the experiments of the group with the lower growth rates were performed under ‘dry’ conditions, whereas those of higher rates were performed under ‘wet’ conditions. However, a strict definition of ‘wet’ versus ‘dry’ is missing. The crucial point is that the amount of water is not well controlled in the ‘wet’ experiments since they were performed combining, among others, pre-drying of starting materials or not, various amounts of added water and different pressure media from which water may diffuse through the capsules (Truckenbrodt and Johannes 1999). Well aware of these shortcomings, Milke et al. (2009) and Dohmen and Milke (2010) simply suggested that the two groups of rim growth rates are due to a sharp transition between two mechanisms of intergranular transport as a function of water activity. In that, ‘dry’ systems are probably better assigned as to ‘water-absent’, but a conclusive classification of the ‘wet’ systems is precluded. It is clear that further investigations with controlled and quantified water–solid fractions are required.

Rates of enstatite rim growth between forsterite and quartz according to literature data as indicated (log (Δx)2/t versus T −1, where Δx is the thickness of the enstatite rim). Growth rates are divided into two groups separated by a gap of several orders of magnitude, depending on whether the experiments were performed in ‘dry’ or ‘wet’ set-ups
The aim of this study is to investigate the mechanisms and kinetics of intergranular transport as a function of water–rock fraction using rim growth experiments in the MgO–SiO2 system. Our previous studies on the growth of reaction rims under dry conditions where controlling mechanisms are understood will serve at as starting point (Gardés et al. 2011; Gardés and Heinrich 2011). These experiments investigated dry growth of polycrystalline enstatite single rims and forsterite-enstatite double rims between single crystals of forsterite and quartz, and periclase and quartz, respectively. In the present study, we use a similar experimental approach only modified by using powder mixtures of the reactants and adding distinct amounts of water. We report results on the growth of enstatite single rims and enstatite-forsterite double rims at water–solid fractions ranging from 0 to 5 wt% at conditions of 1,000°C and 1.5 GPa and various run durations. The data are compared with those from dry set-up, intergranular diffusivities as a function of water fraction are calculated, transport mechanisms are inferred and a new description of the effect of water on intergranular mass transport is proposed.
Experimental and analytical methods
Single crystals of synthetic periclase (provided by CRYSTEC, Berlin) and synthetic forsterite (provided by Institut für Kristallzüchtung, Berlin) were crushed in an agate mortar with ethanol, sieved, dried and calcined at 1,000°C for several hours. Periclase and forsterite powders were sputtered three times with Pt for a few seconds using a conventional sputter coater in order to mark the initial interfaces between the reactants. Between each sputtering cycle, the powders were stirred to increase and homogenize the Pt coverage. Fragments of forsterite (>200 μm) and periclase (>100 μm) were mixed with a synthetic quartz powder (~1 μm grain size; provided by CHEMPUR) in weight proportions of about 1:2:7. Various amounts of brucite serving as source of water were added to the reactant mixture. Brucite was hydrothermally synthesized in a cold-seal pressure vessel at 600°C and 0.4 GPa for 2 days in gold capsules, using MgO powder and excess water as starting materials. X-ray powder diffraction including Rietveld refinement confirmed the formation of brucite (~95 wt%) along with traces of periclase (~5 wt%).
Pt-capsules, 3 mm in diameter, were filled with brucite and subsequently with the powder mixture before cold-sealing. By this, brucite is separated from the reactant mixture at the bottom of the capsule (Fig. 2). Charges were typically 20 mg in weight. The amount of added brucite was calculated in order to obtain the desired proportion of water, which ranged from ~0.05 to 5 wt% relative to a brucite-free sample, and weighed using a Sartorius Supermicro S4 ultramicro balance.

BSE overview of a sample with ~1 wt% H2O after 15 min at 1,000°C and 1.5 GPa (PC43). Periclase and forsterite fragments in the quartz matrix are clearly discernible. Note the initial location of brucite (Brc) at the bottom of the capsule, now reacted to periclase + water and clearly distinguishable from periclase fragments in the quartz matrix
We used the same piston-cylinder apparatus and pressure medium as described in Gardés et al. (2011). This assembly prevents water uptake or loss during the run. The pressure cell consisted of talc–Pyrex glass outer sleeves with a graphite furnace and crushable alumina as inner pressure medium. Experiments were performed at 1,000°C and 1.5 GPa, with varying water fraction and run duration. Temperature was controlled using a linear heating ramp of 30 min up to 1,000°C, followed by a plateau and a final quenching step where temperature dropped below 200°C in less than 10 s. Pressurization and depressurization were applied before and after the heating cycle. At 1.5 GPa, brucite forms periclase + water at about 900°C (Schramke et al. 1982), thus releasing water about 3 min before the plateau temperature is attained.
After the runs, the capsules were slightly abraded on two parallel sides to expose a small portion of the charge. They were embedded in epoxy under vacuum to fill porosity and again abraded until sufficient fragments of periclase and forsterite appeared on the surfaces (Fig. 2). They were then polished down to 0.3 μm using diamond pastes and mechanical-chemically polished with colloidal silica before being cleaned with ethanol and coated with carbon for electron beam analysis.
Rim thicknesses and corresponding grain sizes were measured from backscattered electron (BSE) images obtained with an electron microprobe Jeol Hyperprobe JXA-8500F equipped with a field emission gun (FEG-EMP), operated at an acceleration voltage of 15 down to 6 kV. Ion-thinned foils cut across the reaction zones were produced using a focused Ga ion beam instrument FEI FIB200TEM (Wirth 2004) and investigated in a transmission electron microscope (TEM) FEI Tecnai G2 F20 X-Twin. Rim thicknesses and grain sizes are reported as apparent on sections. Apparent grain widths were measured in direction parallel to the interfaces of the reactants. Forsterite grain widths in the double rim were measured at the vicinity of the periclase-forsterite interface. In case of the enstatite single rim, the position of the Pt-markers is defined as the distance between the markers and the middle of the rim, counted positive towards quartz. In case of the double rim, it is defined as the distance between the markers and the periclase-forsterite interface.
Results
All experiments were performed at 1,000°C and 1.5 GPa (Figs. 3, 4). Table 1 summarizes the experimental conditions, apparent layer thicknesses (Δx), apparent grain widths (a), positions of the platinum markers (d Pt), along with calculated intergranular diffusivities (see ‘Discussion’), for the enstatite single rim between forsterite and quartz, and the forsterite-enstatite double rim between periclase and quartz.

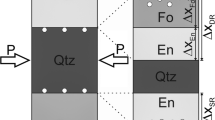
STEM (a, d) and BSE (b, c) images of enstatite single rims grown between forsterite and quartz with various water fractions. Δx En is the rim thickness, a En the apparent grain width measured parallel to the reaction interfaces and d Pt the distance of the Pt-markers from the centre of the rim (c). Pt-markers, initially sitting at the interface of the reactants, are statistically distributed around the centre of the rims (a, c, d), except in runs with the highest water fractions (>2 wt% H2O) where they are shifted towards quartz (b)

BSE (a–c) and STEM (d) images of forsterite-enstatite double rims grown between periclase and quartz with various water fractions. Δx Fo and Δx En are the respective rim thicknesses, a Fo and a En the apparent grain widths measured parallel to the reaction interfaces (in case of forsterite grains at the vicinity of the periclase-forsterite interface) and d Pt the distance of the Pt-markers from the periclase-forsterite interface (a). Pt-markers, initially sitting at the interface of the reactants, are found at or close to the periclase-forsterite interface. The periclase fragments are completely consumed in run PC39 with 5 wt% added H2O (b). Apparent thicknesses in this sample are therefore not representative of the Per + Qtz → Fo + En reaction
General features
Forsterite and quartz produced single rims of enstatite according to
with the spatial sequence (Fig. 3)
Periclase and quartz produced double rims of forsterite and enstatite according to
with the spatial sequence (Fig. 4)
The products of both reactions appear chemically homogeneous. EMP and ATEM analysis shows neither impurities nor chemical gradients within each of the rims.
Both single and double rims are polycrystalline. Enstatite grains in the single rim and forsterite grains in the double rim are texturally equilibrated (Figs. 3, 4). The size of forsterite grains systematically decreases from the periclase-forsterite interface towards the forsterite-enstatite interface (Fig. 4). By contrast, enstatite grains in the double rim are elongated normal to the reaction interfaces, thus forming a palisade microstructure. Some of these grains form needles penetrating quartz grains (Fig. 4a–c).
In most of the samples, open pores are present in the quartz matrix as well as in both the single and double rims. Voids commonly occur at the interfaces of adjacent phases, particularly at the periclase-forsterite interface in the double rim and at the enstatite-quartz interface in both the single and double rims. These voids probably result from differential dilatation during cooling and depressurization, but may alternatively denote regions with a high density of originally fluid-filled pores.
Most of the platinum markers are distributed around the centre of the single rim and at, or close to, the periclase-forsterite interface in the double rim.
In a given sample, the thickness of both the single and double rim may vary from one crystal fragment to another and even around the same fragment (Figs. 3, 4). These variations are probably due to sectioning effects because the inclination between the rims and the surface of the section is not determined. Variations may also result from local departures in growth or nucleation rate, as indicated by abrupt thickness changes commonly observed around fragments. Variations in rim thickness are randomly distributed, and there is no systematic gradient across the samples from the bottom to the top. This demonstrates rapid release and homogeneous distribution of water from brucite across the capsule. Despite these fluctuations, in the double rim, the forsterite rim is always thicker than the enstatite rim and the latter is always thinner than the single enstatite rim.
Evolution with time
Five experiments were performed at run durations of 8, 15 min, 1, 4 and 18 h using a constant water–solid fraction of 1 wt% (Table 1). Rim thicknesses continuously increase with time (Fig. 5). From 8 min to 18 h, the enstatite single rim increases from 1.4 ± 0.1 to 7.5 ± 0.5 μm, the enstatite rim in the double rim from 0.97 ± 0.15 to 6.4 ± 0.7 μm and the forsterite rim in the double rim from 6.5 ± 0.3 to 38.4 ± 1.5 μm. Least squares refinement of the data with power laws of the form \( \Updelta x \propto t^{m} \) yields m = 0.39 ± 0.17 for the enstatite single rim, m = 0.39 ± 0.11 for enstatite in the double rim and m = 0.37 ± 0.06 for forsterite in the double rim (Fig. 5). It is evident that rim growth does not follow a parabolic power law, i.e., \( \Updelta x \propto t^{0.5} \). Rims grow slower.

Time evolution of the thickness (Δx) of the single and double rims grown with 1 wt% added H2O. Rim growth, it is neither linear nor parabolic, but slower
Forsterite and enstatite grains continuously coarsen with time (Fig. 6). From 8 min to 18 h, grain width increases from 0.95 ± 0.13 to 2.5 ± 0.7 μm in the enstatite single rim, from 0.41 ± 0.05 to 1.4 ± 0.2 μm for enstatite grains in the double rim and from 2.4 ± 0.4 to 12.0 ± 3.3 μm for forsterite grains in the double rim. Least squares refinement of the data with power laws of the form \( a^{n} \propto t \) yields n = 4.1 ± 2.1 for the enstatite single rim, n = 4.2 ± 1.1 for enstatite in the double rim and n = 3.1 ± 0.7 for forsterite in the double rim (Fig. 6).

Time evolution of the grain width (a) in the single and double rims grown with 1 wt% added H2O
Despite significant scattering, the Pt-markers in average are located in the centre of the enstatite single rim and, in the double rim, a few μm away from the periclase-forsterite interface (Table 1). No major evolution in porosity with time was detected.
Evolution with increasing water–solid fraction
Eight experiments were performed at a run duration of 4 h with various water–solid fractions \( (\phi_{{{\text{H}}_{2} {\text{O}}}} ) \) ranging from 0 to 5 wt% (Table 1).
No reaction is detected in the dry sample (PC37) and in that with the smallest water fraction of ~0.05 wt% H2O (PC45). By contrast, the reactions are most advanced in the sample with the highest water fraction of 5 wt% H2O (PC39). Here, the periclase is completely consumed (Fig. 4b). Once periclase is no longer available, forsterite cannot be produced further according to the reaction Per + Qtz → Fo + En. Instead, it is only consumed according to the reaction Fo + Qtz → En, similar to the enstatite single rim. Rim thicknesses are therefore irrelevant, and no data are reported for sample PC39.
As a general trend, the kinetics of the reactions increases with increasing water–solid fraction. However, it is striking that in both the single and double rims, the thickness of the rims is constant in the range from 0.1 to 0.5 wt% H2O (Fig. 7). Only at higher water contents, namely from 0.5 to 5 wt% H2O, rim thicknesses increase with increasing amounts of water. In this range, least squares refinement of the data with power laws of the form Δx \( \propto (\phi_{{{\text{H}}_{2} {\text{O}}}} )^{m} \) (Fig. 7) yields m = 0.54 ± 0.20 for enstatite single rim, m = 1.37 ± 1.00 for enstatite in the double rim and m = 0.88 ± 0.19 for forsterite in the double rim. Extrapolation of the two latter laws yields that the double rim must exceed 100 μm at 5 wt% H2O or, in other words, that periclase fragments with widths up to 200 μm were completely consumed. This is precisely what is observed in sample PC39.

Thickness (Δx) of the single and double rims after 4 h runs as a function of water fraction. They are constant in the range from 0.1 to 0.5 wt% H2O and increase in the range from 0.5 to 5 wt% H2O
Coarsening of the grains in all of the rims displays a very similar behaviour. Grain widths are constant from 0.1 to 0.5 wt% and increase by about half an order of magnitude with increasing water fractions from 0.5 to 5 wt% (Fig. 8). In the latter range, least squares refinement of the data with power laws of the form a \( \propto (\phi_{{{\text{H}}_{2} {\text{O}}}} )^{m} \) yields m = 0.59 ± 0.24 for the enstatite single rim, m = 0.43 ± 0.65 for enstatite in the double rim and m = 0.74 ± 0.18 for forsterite in the double rim (Fig. 8).

Grain width (a) in the single and double rims after 4 h runs as a function of water fraction. It is constant in the range from 0.1 to 0.5 wt% H2O and increases in the range from 0.5 to 5 wt% H2O
The Pt-markers again remain in the centre of the enstatite single rim and closely adjacent to the periclase-forsterite interface in the double rim with the exception of sample PC39 (5 wt% H2O) where they appear to be shifted from the centre of the single rim towards the quartz-enstatite interface (Table 1). This is also the case at 2 wt% H2O but to a lesser extent.
The number and size of pores and voids in the entire samples and particularly in the reaction rims also increases with water fraction (Figs. 3, 4). The increase in porosity is further indicated indirectly by the increasing number of grains that were pulled out from the surface during polishing.
Discussion
Analysis of reaction kinetics
The results of Gardés et al. (2011) on growth of enstatite single rims and forsterite-enstatite double rims performed in a dry experimental set-up display many similarities with the outcome of the experiments presented here. (1) The reactions can proceed independently via the coupled diffusion of Mg2+ and O2− or coupled diffusion of Si4+ and 2 O2−, resulting in the net transport of MgO and SiO2 components, respectively. Gardés et al. (2011) demonstrated that MgO is the only mobile—and therefore controlling—component based on the fact that the Pt-markers are located in the centre of the single rim and at the periclase-forsterite interface in the double rim. Mass balance considerations predict that if some mobility of SiO2 would have occurred, the position of the markers would have been shifted to some extent towards the enstatite-quartz interface in both the single and double rims. (2) The growth of all of the rims is not parabolic but slower, demonstrating that it is controlled by the intergranular transport of MgO. Grain coarsening continuously reduces the volume fraction of the intergranular medium, i.e., diffusion pathways, thus generating a decrease in the bulk diffusivities. Gardés and Heinrich (2011) modelled these results assuming that MgO is the only mobile component and that its transport is intergranular only, taking into account the reduction in the intergranular volume fraction associated with grain coarsening. They showed that rim growth then must obey \( \Updelta x \propto t^{(1 - 1/n)/2} \), where n is the exponent of the power law for grain coarsening (\( a^{n} \propto t \)). The model was validated since this evolution is precisely that experimentally observed.
In the present case, the location of the Pt-markers shows some scattering but they are still distributed around the centre of the single rim and close to the periclase-forsterite interface in the double rim. This indicates that the reactions are also mainly controlled by diffusional transport of MgO in the water-bearing system. The predominance of MgO mobility is corroborated by the decrease in forsterite grain size across the forsterite rim from the periclase-forsterite interface to the forsterite-enstatite interface (Fig. 4). This indicates that new forsterite grains indeed form at the forsterite-enstatite interface and, while coarsening, are subsequently shifted towards the periclase-forsterite interface by the new generations of grains. Thus, growth of forsterite occurs at the expense of enstatite and this requires MgO to diffuse to the forsterite-enstatite interface: enstatite (MgSiO3) + MgO(mobile) → forsterite (Mg2SiO4). At 5 wt% H2O and to a small extent also at 2 wt% H2O, the slight shift of the markers from the centre of the enstatite single rim towards the enstatite-quartz interface indicates that in these cases, SiO2 was also somewhat mobile and contributed moderately to the overall reaction (Figs. 3, 4; Table 1).
Rim growth with time at 1 wt% H2O in both the single and double rims is also not parabolic but slower, as observed by Gardés et al. (2011) in the dry system. A power law results that has the form \( \Updelta x \propto t^{m} \) with m = 0.39 ± 0.17 for the enstatite single rim, 0.39 ± 0.11 for enstatite in the double rim and 0.37 ± 0.06 for forsterite in the double rim. This is in perfect agreement with the model of Gardés and Heinrich (2011, see above) since the (1 − 1/n)/2 values are 0.38 ± 0.06 for the enstatite single rim, 0.38 ± 0.03 for enstatite in the double rim and 0.34 ± 0.04 for forsterite in the double rim, according to the n values of the grain coarsening laws derived above. We therefore conclude that growth of the rims in the water-bearing system is controlled by the transport of MgO in the intergranular medium. The contribution of SiO2 is certainly negligible up to 1 wt% H2O and becomes noticeable only above 2 wt% H2O.
Quantification of intergranular transport enhancement versus water–solid fraction
Growth of enstatite single rims and forsterite-enstatite double rims in the presence of water displays many similarities with that in dry set-up to safely apply the model of Gardés and Heinrich (2011) for extracting intergranular diffusivities. Additional mobility of SiO2 detected in the samples containing 2 and 5 wt% H2O is minor and neglected for the sake of clarity here. In grain boundary diffusion models, an important parameter is the effective grain boundary width δ, that is, the width of the disturbed region across the grain boundary where diffusion is facilitated relative to grain interiors. It is often estimated to be 1 nm (e.g. Joesten 1991). Recent TEM investigations suggest values up to 10 nm for the effective grain boundary width, where 1 nm corresponds rather to the structural grain boundary width (Marquardt et al. 2011). It is not possible to assign a definite value here due to possible presence of intergranular fluid films with different widths as a function of water fraction. Therefore, in the following, we consider δig, the effective width of the intergranular medium, as a variable. Consequently, we do not report intergranular diffusivities D ig but their product with effective intergranular medium widths δig D ig. These are obtained from Eqs. 22, 20b and 20a along with Eqs. 18, 15 and 5 of Gardés and Heinrich (2011)
where R is the gas constant, ΩEn and ΩFo the molar volumes of enstatite and forsterite (3.13 × 10−5 and 4.38 × 10−5 m3/mol, respectively; Gottschalk 1997), Δ r G En and Δ r G Fo the Gibbs energies of the reactions Mg2SiO4 + SiO2 → 2 MgSiO3 and MgO + MgSiO3 → Mg2SiO4 at 1,000°C and 1.5 GPa (−10.5 and −24.8 kJ/mol, respectively; Gottschalk 1997). n is the exponent of the power law for grain coarsening and was determined for the time series at 1 wt% H2O only. For simplification, we assume it is identical for all other water fractions. We take an average value for n for both enstatite and forsterite coarsening in the double rim because n is supposed to be identical for both rims in the model of Gardés and Heinrich (2011). Hence, for all water fractions, we calculate the δig D igs with n = 4.1 for enstatite grain coarsening in the single rim and n = 3.7 for both enstatite and forsterite grain coarsening in the double rim. These approximations do not introduce a significant error since Eqs. 1–3 are rather insensitive to n. Indeed, even when n would vary from 2 to 6, (1 − n)/n varies from −0.5 to −0.83 only. Results are reported in Table 1 and in Figs. 9 and 10.

MgO intergranular diffusivities—reported as their product with the effective width of the intergranular medium (δig D ig)—calculated for the various run durations. Calculations were performed according to Eqs. 1–3 derived from the model of Gardés and Heinrich (2011). The invariability of the δig D igs demonstrates the validity of the model, especially the fact that the growth of the rims is controlled by the diffusion of MgO in the intergranular medium only

MgO intergranular diffusivities—reported as their product with the effective width of the intergranular medium (δig D ig)—as a function of water fraction. Calculations were performed according to Eqs. 1–3 derived from the model of Gardés and Heinrich (2011). The δig D igs are constant in the 0.1–0.5 wt% H2O range and increase strongly in the 0.5–5 wt% H2O range. This is interpreted as resulting from the existence of two competing diffusion mechanisms in water-saturated conditions, i.e., diffusion in hydrous-saturated grain boundaries and diffusion in interconnected fluid-filled porosity
The invariability of the δig D igs at 1 wt% H2O as a function of run durations confirms the appropriateness of the model (Fig. 9). The transport is intergranular and the observed growth, slower than parabolic growth as in case of volume diffusion, is due to the reduction in the volume fraction of the intergranular medium resulting from grain coarsening.
The evolution of the δig D igs with water fraction is similar to that of rim thickness and grain coarsening. They are independent on water fraction in the 0.1–0.5 wt% range, with average values being 10−22.26±0.05 m3/s for enstatite in the single rim, 10−22.58±0.24 m3/s for enstatite in the double rim and 10−20.35±0.30 m3/s for forsterite in the double rim (Fig. 10).
The δig D igs increase strongly as a function of water fraction above 0.5 wt%. They reach 10−20.62±0.12 m3/s for enstatite in the single rim at 5 wt% H2O, 10−20.88±0.14 m3/s for enstatite in the double rim at 2 wt% H2O and 10−18.90±0.17 m3/s for forsterite in the double rim at 2 wt% H2O, i.e., about two orders of magnitude higher compared to the 0.1–0.5 wt% range (Table 1). Least squares refinement of the data in this range with power laws of the form δig D ig \( \propto (\phi_{{{\text{H}}_{2} {\text{O}}}} )^{p} \) yields p = 1.66 ± 0.53 for the enstatite single rim, p = 2.68 ± 1.96 for enstatite in the double rim and p = 2.46 ± 0.24 for forsterite in the double rim (Fig. 10).
No δig D igs are reported for 0 and 0.05 wt% H2O since no rims were detected in these samples. This, however, demonstrates a sharp jump in reaction kinetics from 0.05 to 0.1 wt% H2O. Extrapolating the Arrhenius laws of Gardés and Heinrich (2011) for the dry set-up down to 1,000°C yields δig D igs of 10−26.23 m3/s in the single enstatite rim, 10−26.24 m3/s for enstatite in the double rim and 10−25.21 m3/s for forsterite in the double rim, i.e., about four to five orders of magnitude less than derived for the 0.1–0.5 wt% H2O range.
Taking the data of Gardés and Heinrich (2011) into account, the entire increase in δig D igs from 0 to 5 wt% water–solid fraction covers about six orders of magnitude in each of the three rims. Remarkably, the increase attains four to five orders of magnitude already at 0.1 wt% H2O. Carlson (2010) observed an increase of seven orders of magnitude in Al intergranular diffusivities from nearly anhydrous to fluid-saturated conditions. Such variations definitely confirm that water/rock ratio is a parameter at least as important as temperature regarding the kinetics of mineral reactions. Consequently, it is impossible to assign a unique value for the intergranular diffusivity of a given chemical component as it may drastically vary as a function of the water–rock fraction. Moreover, as they may have different dependences on water–rock fraction, the relative intergranular diffusivities between the chemical components themselves may vary as well (e.g. Joachim et al. 2012).
The δig D igs in the forsterite rim are higher by about two orders of magnitude compared to that in enstatite in both the single and double rims, in both the 0.1–0.5 and 0.5–2 wt% H2O range (Fig. 11). This demonstrates that, even in a water-saturated rock, the intergranular diffusivity of a chemical component is strongly dependent on the adjacent phases. Particularly, when modelling reaction coronas, assuming identical mobilities in the various single or multiphase rims may introduce major errors. The ratio of the δig D igs in the double rim is slightly decreasing as a function of water fraction but this may not be significant given the large error bars (Fig. 11). In the dry set-up, the δig D ig in the forsterite rim is only about one order of magnitude higher compared to that in enstatite at 1,000°C (Gardés et al. 2011; Fig. 11). Hence, the introduction of water may also provoke significant variations in the ratio of the δig D igs of a given component between multilayered rims.

Ratio (forsterite/enstatite) of MgO intergranular diffusivities in the double rim as a function of water fraction. The overall difference of about two orders of magnitude between forsterite and enstatite demonstrates that, even if water-saturated, the intergranular diffusivity of MgO is strongly dependent on the adjacent phases
Mass transport mechanisms versus water–solid fraction
In samples PC37 and PC45 (H2O-free and with 0.05 wt% H2O), no reaction rims were detected. This suggests that grain boundaries in the sample with 0.05 wt% H2O were also free of water or hydrous species. This gains some plausibility because Kronenberg and Wolf (1990) have shown that crustal quartz under hydrous conditions may accommodate water by 0.05–0.8 wt% in the form of submicroscopic fluid inclusions (see also Johnson 2006, and references therein). If so, it is probable that this small amount of water was mainly incorporated into quartz during the fast sintering of the quartz matrix in our experiments and therefore absent in the intergranular medium. Hence, a range from zero to very low water–rock fractions may exist where transport still occurs in dry grain boundaries. The ‘dry’ group of the previous enstatite rim growth experiments (Fig. 1) typically falls in that category.
The invariability of the δig D igs in the 0.1–0.5 wt% H2O range (Fig. 10) indicates that no continuous film of water along grain boundaries is present. One would otherwise expect an increase in the film width with increasing water fraction from 0.1 to 0.5 wt% H2O resulting in increasing δig D igs, which is not observed. Moreover, the derived values of the δig D igs, for instance 10−22.26±0.05 m3/s in the enstatite single rim, are much too low to be consistent with a film of free water. Tentatively, assuming a very small film thickness of 5 nm would yield the diffusivity of 10−14 m2/s. This is still six to seven orders of magnitude lower than ionic diffusivities in water (~10−8–10−7 m2/s, Oelkers and Helgeson 1988). Finally, because SiO2 is highly soluble in water at our experimental conditions (Manning 1994), its immobility in experiments with low water contents definitely confirms that there is no thick film of water along grain boundaries. This interpretation is in line with the work of Watson and Brenan (1987) who demonstrated that most geological fluids do not exist as films along grain boundaries. It follows that water or related species are bonded in the grain boundaries and this is most likely to occur via hydroxylation. Studies of silicate–water interfaces indicate that surface silica groups are hydroxylated, thus forming silanol groups (e.g. Fenter et al. 2000; Schlegel et al. 2002; Adeagbo et al. 2008; see also Heinemann et al. 2001). It was also suggested that water is adsorbed on silicate surfaces via hydrogen bridges between the silanol groups and oxygen of water molecules (e.g. Schlegel et al. 2002). Therefore, in addition to hydroxyl groups, the presence of a layer of bonded water molecules at the interface of the grains cannot be excluded. For that reason, we do not refer to these grain boundaries simply as hydroxylated but more generally to ‘hydrous’ grain boundaries. The large enhancement of the diffusivity in such ‘hydrous’ compared to dry grain boundaries might result from the weaker hydrogen bonds that reduce the activation energy (e.g. Beach 1982). Once saturated with water-related species, the structure of these grain boundaries does not change and any additional free water is expelled into isolated pores (see Watson and Brenan 1987), hence explaining the invariability of the δig D igs in the 0.1–0.5 wt% H2O range. Tentatively, assuming an effective width of 5 nm, MgO diffusivities in hydrous-saturated grain boundaries (HGBs) result in 10−14.0 m2/s for enstatite in the single rim, 10−14.3 m2/s for enstatite in the double rim and 10−12.0 m2/s for forsterite in the double rim. This is definitely slower than diffusivities in free water. Converting the data of the 0.1–0.5 wt% H2O range for the enstatite single rim growth into ‘rim growth rates’ (Δx)2/t—which is not rigorous here since the growth is not parabolic—yields an average value of 10−15.3 m2/s. This perfectly coincides with the growth rates derived at 1,000°C from the ‘wet’ group of previous enstatite rim growth experiments (Fig. 1). We therefore suggest that the water fractions in all these experiments were in about the 0.1–0.5 wt% H2O range, and that intergranular transport occurred mainly via HGBs as defined above. They lie in a relatively narrow range because the transport properties of these grain boundaries are independent on water fraction. Taking all growth rates for enstatite of the ‘wet’ group from the literature (Fig. 1) and using a linear least squares refinement yields the apparent activation energy of 185 ± 20 kJ/mol. Assuming this energy as the activation energy of diffusion—as it should be if the growth was parabolic—and using 10−22.26 m3/s at 1,000°C from our experiments, the Arrhenius law for MgO diffusion in enstatite HGBs is \( \delta_{\text{HGB}} D_{\text{HGB}}^{\text{MgO}} = \delta_{\text{HGB}} D_{0} e^{{ - E/{\text{RT}}}} \) with E = 185 ± 20 kJ/mol and δHGB D 0 = 10−14.7±0.9 m3/s, or D 0 = 10−6.4±0.9 m2/s if δHGB is tentatively assumed to be 5 nm (Fig. 12). This activation energy is about two times lower than that in dry grain boundaries, i.e., 417 kJ/mol (Gardés and Heinrich 2011).

Comparison of MgO diffusion in dry and hydrous-saturated enstatite grain boundaries. The activation energy of diffusion in hydrous-saturated grain boundaries was estimated according to previous ‘wet’ enstatite rim growth experiments (see Fig. 1). It is about two times lower than the activation energy in dry grain boundaries
The transition between diffusion in dry grain boundaries and hydrous-saturated grain boundaries (HGBs) is very sharp and occurs between 0.05 and 0.1 wt% water–solid fraction in our experiments. The transition is strikingly drastic because it corresponds to a gain in diffusivity of four to five orders of magnitude (Fig. 10). This explains the absence of intermediate values and the magnitude of the gap between the growth rates of the ‘dry’ and ‘wet’ enstatite rim growth experiments (Fig. 1). In their review of these experiments, Dohmen and Milke (2010) indeed suggested the transition between the two regimes to be very sharp, but the threshold at which it actually occurs, i.e., 500–1,000 wt. ppm, is much higher than the 10–100 wt ppm Milke et al. (2009) previously estimated.
The increase in the δig D igs at higher water–solid fractions demonstrates the existence of an additional regime (Fig. 10). Once the grain boundaries are saturated with H2O-related species, the excess water is expelled to pores. As long as pores are isolated at grain corners, their contribution as pathways for mass transport is insignificant. However, once they become interconnected, fast pathways for short-circuit diffusion in HGBs are provided. Because SiO2 dissolves in the fluid-filled pores (Manning 1994), minor mobility of SiO2 detected at 2 wt% H2O and moderate mobility at 5 wt% H2O clearly evidences transport of chemical components in interconnected fluid-filled porosity in this regime. This is consistent with the fact that longer-scaled interconnectivity increases with water–rock fraction and that interconnected fluid-filled porosity in rock-forming minerals forms a network of channels along grain edges (Watson and Brenan 1987). This regime starts to predominate at 0.5 wt% added H2O (Fig. 10), which is in very good agreement with the experiments of Watson (1991) on olivine aggregates where a strong dependence of Fe intergranular diffusivity on water–solid fraction was also observed from 0.5 wt% H2O onwards. Our calculated δig D ig value for the enstatite single rim grown with 5 wt% H2O is 10−20.62±0.12 m3/s. Assuming a δig of 10 nm, which represents the mean diameter of the channels in this regime, yields a diffusivity of about 10−13 m2/s. This value, which is lower if larger diameters are assumed, is still five orders of magnitude lower than ionic diffusion in water (~10−8–10−7 m2/s, Oelkers and Helgeson 1988). Hence, diffusion in interconnected fluid-filled porosity is much slower than in bulk water as already inferred by Farver and Yund (1995) and Carlson (2010). Indeed, the strong dependence of the fluids’ transport properties on the properties of adjacent solids is corroborated by the difference in the δig D igs between the forsterite and the enstatite rims, spanning about two orders of magnitude (Fig. 11). In this regime, the δig D igs are proportional to \( (\phi_{{{\text{H}}_{2} {\text{O}}}} )^{p} \) with p being about 2 in all the rims (Fig. 10). If porosity is seen as a network of channels with diameter δig, then \( \phi_{{{\text{H}}_{2} {\text{O}}}} \) should be proportional to \( \delta_{\text{ig}}^{2} \). It follows that the δig D igs are proportional to \( \delta_{\text{ig}}^{4} \), or that the D igs are proportional to \( \delta_{\text{ig}}^{3} \). This reasoning, although very rough, would suggest that the increase in the δig D igs above 0.5 wt% H2O stems not only from the increase in porosity but also from an increase in diffusivity. This is reasonable since, with channels expanding, one would expect surface effects to be lowered and the diffusivity to converge to that in bulk water.
The effect of water on intergranular mass transport revisited
In his seminal analysis, Rubie (1986) proposed a discrimination of the effect of water on intergranular diffusion into three categories based on water activity, subsequently reviewed by Farver and Yund (1995) and Carlson (2010). The description of Farver and Yund (1995) is summarized in the following.
(1) ‘Water-absent’: the intergranular medium is free of water and hydrous species. Diffusion occurs in dry grain boundaries. (2) ‘Water-unsaturated’: water or hydrous species are structurally incorporated or bonded in the grain boundaries. The grain boundary diffusion coefficient may vary as a function of their concentration. No free fluid phase is present. (3) ‘Water-saturated’: grain boundaries are saturated with H2O or hydrous species and a free fluid phase is present. Rubie (1986) proposed that this fluid was present as thick film along grain boundaries. Farver and Yund (1995) subsequently argued that water more likely was distributed in channels along grain edges according to the experimental work of Watson and Brenan (1987).
Our observations on the effect of water on the intergranular transport of chemical components are not completely described by these categories. (1) ‘Water-unsaturated’ conditions were not observed, neither by us, nor in previous investigations of enstatite rim growth. The transition from dry to hydrous-saturated grain boundaries is very sharp, occurring between 0.05 and 0.1 wt% water–solid fraction in our experiments, probably because only a very small water fraction is required to saturate grain boundaries with H2O-related species. For that reason, the ‘water-unsaturated’ regime with gradually increasing diffusivities is probably virtually non-existent in many systems. (2) Two transport mechanisms compete in water-saturated conditions. These are diffusion along hydrous-saturated grain boundaries, which dominates as long as fluid-filled pores remain isolated, and diffusion through fluid-filled channels, which dominates when pores are interconnected because of the higher diffusivities in aqueous fluid. In this latter regime, there is a continuous enhancement of mass transport because diffusivity as well as porosity and interconnectivity increase with water fraction. In other words, transport properties in polycrystalline materials are dependent on the water/rock ratio even at water-saturated conditions, contrary to what this terminology would suggest. The classification of Carlson (2010) is also divided into three categories, basically similar to those of Farver and Yund (1995): ‘nearly anhydrous’, ‘hydrous-but-fluid-undersaturated’ and ‘fluid-saturated’ systems. Actually, Carlson (2010) pointed out that transport in water-saturated rocks where pores are isolated must be distinguished from that where pores do form an interconnected network. In fact, this division directly corresponds to our two different regimes at water-saturated conditions. Carlson (2010) associated the former case to ‘hydrous-but-fluid-undersaturated’ systems, which is justified from a physical point of view but is not reasonable in terms of thermodynamics. Hence, the description of intergranular mass transport as a function of water activity is insufficient.
We propose a new description based on the macroscopic and the microscopic nature of the intergranular medium (Fig. 13). Due to the fact that two regimes compete at water-saturated conditions, it comprises four categories sorted by increasing transport kinetics, which are presented in the following.

Proposed classification of intergranular mass transport as a function of water–rock fraction. (1) Diffusion in anhydrous grain boundaries, which holds as long as water can be accommodated in the volume of the minerals. (2) Diffusion in hydrous-undersaturated grain boundaries, which is a transition regime that is most likely rarely observed as occurring in a very narrow range in our systems. (3) Diffusion in hydrous-saturated grain boundaries, which is the first regime occurring at water-saturated conditions and corresponds to a jump of several orders of magnitude in reaction kinetics. (4) Diffusion in interconnected fluid-filled porosity, where there is a continuous enhancement of mass transport due to the concomitant increases in porosity, interconnectivity and diffusivity with water–rock fraction
-
1.
Diffusion in anhydrous grain boundaries: there is no water or hydrous species in the intergranular medium. Diffusion occurs along dry grain boundaries. This regime holds as long as water can be accommodated in the crystal structure of the minerals, either as dissolved hydrous species or as molecules in inclusions. In that case, mass transport, and processes controlled by mass transport, can be considered as dry even if the water–rock fraction departs from zero.
-
2.
Diffusion in hydrous-undersaturated grain boundaries: the surface sites which can accommodate H2O-related species, as hydroxyl groups, are partly occupied. This transitional regime, where diffusivities are expected to vary as a function of water–rock fraction, is likely to be rarely observed as only a minute fraction of water is required to saturate grain boundaries. How much water is required for this remains an open question and also may differ from one system to the other.
-
3.
Diffusion in hydrous-saturated grain boundaries: the surface of the grain boundaries are saturated with H2O-related species and excess free water is expelled to isolated pores. Diffusivity in these grain boundaries is much higher than in dry grain boundaries and is independent on water–rock fraction. This is the first regime occurring in water-saturated conditions. Its extent, in term of water–rock fraction, depends on the ability of pores to interconnect. It may not exist in systems where pores always form an interconnected network. This additional regime comprises the main distinction between our analysis and those of the previous works.
-
4.
Diffusion in interconnected fluid-filled porosity: this regime occurs at the highest water–rock fractions. Mass transport occurs in interconnected pores filled with aqueous fluid, most likely distributed as channels along grain edges. The transport properties of the fluid differ from but tend towards those of bulk water as channels expand. Therefore, there is a continuous enhancement of mass transport as a function of water fraction in rocks, even when fluid-saturated. Consequently, at a given higher water fraction, it might well be that reactions would not be transport-controlled anymore but taken over by the dissolution and precipitation processes at reaction interfaces.
Conclusion
Experimental investigation and modelling of the growth of enstatite single rims and forsterite-enstatite double rims with incremental water–solid fractions brings new insights into the effect of water on intergranular mass transport. The increase in diffusivities by six orders of magnitude between dry grain boundaries and intergranular media of samples containing 5 wt% H2O confirms that water–rock fraction is a parameter at least as important as temperature with respect to mass transport-controlled processes. The diffusivities extracted from the forsterite and the enstatite rims differs by two orders of magnitude. This demonstrates that, even in a water-saturated rock, intergranular mass transport may be strongly dependent on the adjacent solid-phase framework and implies, in case of reaction coronas, that the diffusivity of a given chemical component may differ from one rim to the other.
The evolution of intergranular transport properties as a function of water–solid fraction is better described by categories based on the explicit macroscopic and/or microscopic nature of the intergranular medium rather than by water activity. We propose a classification where intergranular mass transport is divided in four regimes, sorted by increasing kinetics: (1) diffusion in anhydrous grain boundaries, (2) diffusion in hydrous-undersaturated grain boundaries, (3) diffusion in hydrous-saturated grain boundaries and (4) diffusion in interconnected fluid-filled porosity.
Regime (1) holds as long as water can be accommodated in the volume of the minerals. Diffusivities in hydrous-saturated grain boundaries (regime 3) are independent on water–solid fraction and much higher than those in dry grain boundaries (regime 1), i.e., four to five orders of magnitude in our systems. We estimate the Arrhenius law for MgO diffusion in enstatite HGBs to be \( \delta_{\text{HGB}} D_{\text{HGB}}^{\text{MgO}} = \delta_{\text{HGB}} D_{0} e^{{ - E/{\text{RT}}}} \) with E = 185 ± 20 kJ/mol and δHGB D 0 = 10−14.7±0.9 m3/s. No intermediate diffusivities were detected. Thus, the transition regime (2) may be irrelevant as virtually instantaneous. Regimes (3) and (4) correspond to competing mechanisms, but both in water-saturated condition. Regime (3) dominates as long as excess water is distributed into isolated pores. It is replaced by regime (4) when pores form an interconnected network of channels. In this regime, the transport properties of the fluid differ from but tend towards those of bulk water as porosity increases, provoking a continuous enhancement of mass transport as a function of water–rock fraction.
References
Abart R, Kunze K, Milke R, Sperb R, Heinrich W (2004) Silicon and oxygen self diffusion in enstatite polycrystals: the Milke et al. (2001) rim growth experiments revisited. Contrib Mineral Petrol 147:633–646
Adeagbo WA, Doltsinis NL, Klevakina K, Renner J (2008) Transport processes at α-quartz-water interfaces: insights from first-principles molecular dynamics simulations. Chem Phys Chem 9:994–1002
Beach A (1982) Deformation mechanisms in some cover thrust sheets from the external French Alps. J Struct Geol 4:137–149
Carlson WD (2010) Dependence of reaction kinetics on H2O activity as inferred from rates of intergranular diffusion of aluminium. J Metamorph Geol 28:735–752
Dohmen R, Milke R (2010) Diffusion in polycrystalline materials: grain boundaries, mathematical models, and experimental data. Rev Miner Geochem 72:921–970
Farver JR, Yund RA (1995) Grain boundary diffusion of oxygen, potassium and calcium in natural and hot-pressed feldspar aggregates. Contrib Mineral Petrol 118:340–355
Fenter P, Teng H, Geissbühler P, Hanchar JM, Nagy KL, Sturchio NC (2000) Atomic-scale structure of the orthoclase (001)-water interface measured with high-resolution X-ray reflectivity. Geochim Cosmochim Acta 64:3663–3673
Fisler DK, Mackwell SJ, Petsch S (1997) Grain boundary diffusion in enstatite. Phys Chem Miner 24:264–273
Gardés E, Heinrich W (2011) Growth of multilayered polycrystalline reaction rims in the MgO–SiO2 system, part II: modelling. Contrib Miner Petrol 162:37–49
Gardés E, Wunder B, Wirth R, Heinrich W (2011) Growth of multilayered polycrystalline reaction rims in the MgO–SiO2 system, part I: experiments. Contrib Miner Petrol 161:1–12
Gasc J, Brunet F, Bagdassarov N, Morales-Flόrez V (2011) Electrical conductivity of polycrystalline Mg(OH)2 at 2 GPa: effect of grain boundary hydration–dehydration. Phys Chem Miner 38:543–556
Gottschalk M (1997) Internally consistent thermodynamic data for rock-forming minerals in the system SiO2–TiO2–Al2O3–FeO3–CaO–MgO–FeO–K2O–Na2O–H2O–CO2. Eur J Miner 9:175–223
Heinemann S, Wirth R, Dresen G (2001) Synthesis of feldspar biocrystals by direct bonding. Phys Chem Miner 28:685–692
Joachim B, Gardés E, Velickov B, Abart R, Heinrich W (2012) Experimental growth of diopside + merwinite reaction rims: the effect of water on microstructure development. Am Miner 97:220–230
Joesten R (1991) Grain-boundary diffusion kinetics in silicate and oxide minerals. In: Ganguly J (ed) Advances in physical geochemistry, vol 8, pp 345–395
Johnson EA (2006) Water in nominally anhydrous crustal minerals: speciation, concentration, and geologic significance. Rev Miner Geochem 62:117–154
Kronenberg AK, Wolf GH (1990) Fourier transform infrared spectroscopy determinations of intergranular water content in quartz-bearing rocks: implications for hydrolytic weakening in the laboratory and within the earth. Tectonophysics 172:255–271
Manning CE (1994) The solubility of quartz in H2O in the lower crust and upper mantle. Geochim Cosmochim Acta 58:4831–4839
Marquardt K, Ramasse QM, Kisielowski C, Wirth R (2011) Diffusion in yttrium aluminium garnet at the nm-scale—insight into the effective grain boundary width. Am Miner 96:1521–1529
Milke R, Wiedenbeck M, Heinrich W (2001) Grain boundary diffusion of Si, Mg, and O in enstatite reaction rims: a SIMS study using isotopically doped reactants. Contrib Miner Petrol 142:15–26
Milke R, Dohmen R, Becker HW, Wirth R (2007) Growth kinetics of enstatite reaction rims studied on nano-scale, part I: methodology, microscopic observations and the role of water. Contrib Miner Petrol 154:519–533
Milke R, Kolzer K, Koch-Müller M, Wunder B (2009) Orthopyroxene rim growth between olivine and quartz at low temperatures (750–950°C) and low water concentration. Miner Petrol 97:223–232
Oelkers EH, Helgeson HC (1988) Calculation of the thermodynamic and transport properties of aqueous species at high pressures and temperatures: aqueous tracer diffusion coefficients of ions to 1000°C and 5 kb. Geochim Cosmochim Acta 52:63–85
Rubie DC (1986) The catalysis of mineral reactions by water and restrictions on the presence of aqueous fluid during metamorphism. Miner Mag 50:399–415
Schlegel ML, Nagy KL, Fenter P, Sturchio NC (2002) Structures of quartz (100)- and (101)-water interfaces determined by x-ray reflectivity and atomic force microscopy of natural growth surfaces. Geochim Cosmochim Acta 66:3037–3054
Schramke JA, Kerrick MK, Blencoe JG (1982) Experimental determination of the brucite = periclase + water equilibrium with a new volumetric technique. Am Miner 67:269–276
Truckenbrodt J, Johannes W (1999) H2O loss during piston-cylinder experiments. Am Miner 84:1333–1335
Watson EB (1991) Diffusion in fluid-bearing and slightly-melted rocks: experimental and numerical approaches illustrated by iron transport in dunite. Contrib Miner Petrol 107:417–434
Watson EB, Brenan JM (1987) Fluids in the lithosphere. 1. Experimentally-determined characteristics of CO2–H2O fluids and their implications for fluid transport, host-rock physical properties, and fluid inclusion formation. Earth Planet Sci Lett 85:497–515
Wirth R (2004) Focused ion beam (FIB): a novel technology for advanced application of micro- and nanoanalysis in geosciences and applied mineralogy. Eur J Mineral 16:863–876
Yund RA (1997) Rates of grain boundary diffusion through enstatite and forsterite reaction rims. Contrib Mineral Petrol 126:224–236
Acknowledgments
This study greatly benefited from TEM investigations on pilot experiments and fruitful discussions with R. Wirth. We deeply thank him. We also thank D. Rhede for his invaluable assistance with microprobe. We are grateful to A. Schreiber for producing FIB foils, and G. Neusser and N. Norberg for their help with sample preparation. This paper benefited from the helpful comments of W. D. Carlson and an anonymous reviewer. This work was funded by the Deutsche Forschungsgemeinschaft within the framework of FOR 741, project HE 2015/9-1.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by J. Hoefs.
Rights and permissions
About this article
Cite this article
Gardés, E., Wunder, B., Marquardt, K. et al. The effect of water on intergranular mass transport: new insights from diffusion-controlled reaction rims in the MgO–SiO2 system. Contrib Mineral Petrol 164, 1–16 (2012). https://doi.org/10.1007/s00410-012-0721-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00410-012-0721-0