
Abstract
The kinetics of (Mg, Fe)SiO3 pyroxene layer growth within silicate thin films with total thickness <1 μm was studied experimentally at 0.1 MPa total pressure, controlled fO2 and temperatures from 1,000 to 1,300°C. The starting samples were produced by pulsed laser deposition. Layer thickness before and after the experiments and layer composition as well as microstructures, grain size and shape of the interfaces were determined by Rutherford back scattering and transmission electron microscopy assisted by focused ion beam milling. Due to the miniaturization of the starting samples and the use of high resolution analytical methods the experimentally accessible temperature range for rim growth experiments was extended by about 300°C towards lower temperatures. The thickness of the layers at a given temperature increases proprotional to the square root of time, indicating a diffusion-controlled growth mechanism. The temperature dependence of rim growth yields an apparent activation energy of 426 ± 34 kJ/mol. The small grain size in the orthopyroxene rims implies a significant contribution of grain boundary diffusion to the bulk diffusion properties of the polycrystalline rims. Based on microstructural observations diffusion scenarios are discussed for which the SiO2 component behaves immobile relative to the MgO component. Volume diffusion data for Mg in orthopyroxene from the literature indicate that the measured diffusivity is probably controlled by the mobility of oxygen. The observed reaction rates are consistent with earlier results from dry high-temperature experiments on orthopyroxene rim growth. Compared to high pressure experiments at 1,000°C and low water fugacities, reaction rates are 3–4 orders of magnitude smaller. This observation is taken as direct evidence for a strong effect of small amounts of water on diffusion in silicate polycrystals. In particular SiO2 changes from an immobile component at dry conditions to an extremely mobile component even at very low water fugacities.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Diffusive mobility of chemical components controls the rates of many rock-forming processes, either affecting the mineralogical composition of rocks, or the mineral’s compositions, or the rock’s fabric, or all of them concurrently. In polycrystalline materials, like rocks, diffusion occurs simultaneously in the grain’s interiors (volume diffusion) and along grain boundaries (grain and phase boundary diffusion). The efficiency of the respective paths depends both on the ratio of volume to grain boundary diffusion coefficients as well as grain size (Joesten 1991), but also on the partitioning between the grain boundary and the crystal volume (Dohmen and Chakraborty 2003). A number of studies used the growth of polycrystalline reaction rims between two incompatible reactants to experimentally quantify the diffusivity of the respective rate-controlling components (Schmalzried 1978; Brady 1983; Fisler and Mackwell 1994; Fisler et al. 1997; Yund 1997; Liu et al. 1997; Milke et al. 2001; Watson and Price 2001; Milke and Heinrich 2002; Milke and Wirth 2003; Abart et al. 2004). They generally found that rim thickness increases with the square root of time in accordance with a diffusion-controlled growth process.
A general problem of the rim growth method is the detection of small growth rates. Using conventional techniques, rim growth has to amount to a few μm in rim thickness to be clearly detectable. These limitations in spatial resolution generally require experiments at very high temperatures—often outside the temperature range typically present in the Earth’s crust—to derive diffusivity data on a feasible laboratory time scale. This problem can be solved by the miniaturization of the setup combined with high resolution analytical methods. In silicate thin films the entire reactant and product layer sequence is condensed to a few hundred nanometers. Using pulsed laser deposition (PLD) complex oxide or silicate thin films tens to thousands of nm thick can be produced with perfectly controlled stoichiometry and flat layer geometry (Dohmen et al. 2002a). Olivine thin films doped with 29Si and 18O prepared with the same setup as used in this study were successfully used to improve Si and O diffusion data in single crystal olivine (Dohmen et al. 2002b).
In this paper, we present rim growth experiments according to the reaction olivine + quartz = orthopyroxene:
where both olivine and orthopyroxene compositions were dominated by the Mg endmembers. Rim growth was performed in olivine (Ol)-orthopyroxene (Opx) double layers on quartz (Qtz) substrates with a total layer thickness below 1 μm. For analysing rim growth on the sub-micron scale a concert of highly resolving methods were applied. Surface topography was controlled using optical microscopy, white light interference microscopy, and scanning electron microscopy (SEM). Layer thickness and chemical composition were determined by Rutherford backscattering (RBS). The preparation of electron transparent foils vertical to the sample surface was performed by the Focused Ion Beam (FIB) method (Wirth 2004). Investigation of layer microtextures and confirmation of rim thickness was done by transmission electron microscopy (TEM). Secondary Ion Mass Spectrometry (SIMS) and numerical modelling of the isotope profiles across the layers will be published elsewhere.
The use of thin film samples yields manifold advantages over conventional rim growth experiments. Synthetic thin films can be tailored, thus offering flexibility in chemical composition and providing the ideal topology for SIMS and RBS depth scanning. Under these ideal conditions very small growth rates can be accurately measured. The experimentally accessible temperature range is thus extended by several hundred degrees to lower temperatures. Thereby the temperature range of diffusion-controlled reactions in the Earth’s crust is entered, even for absolutely dry conditions. Here, we apply the advantages of the thin film method to close the gap between dry high-temperature Opx rim growth experiments (far >1,000°C) and experiments at more or less hydrous conditions at moderate temperature (≤1,000°C). In recent years several experimental studies focused on the diffusion-controlled growth of Opx reaction rims between forsterite-rich Ol and Qtz. Fisler et al. (1997) performed dry experiments at 0.1 MPa total pressure in the temperature range from 1,350 to 1,450°C and attributed their results to silicon grain boundary diffusion. Yund (1997) complemented their high-temperature data with piston–cylinder experiments at 250–1,500 MPa and 900–1,100°C and water-contents, which varied between vacuum dried and 5 wt% water added. At 1,000°C, bulk diffusivities scatter around 10−16 m2 s−1 and a definite decision for the rate-limiting component could not be drawn. From the ‘driest’ to the ‘wettest’ experiment, bulk diffusivity increases by a factor of five. The data of Milke et al. (2001) with samples sealed ‘as is’ and reacted at 1 GPa and 1,000°C well agree with the fit in Yund’s (1997) data-set. The latter authors suggested that MgO is the rate-controlling component in the high-P experiments considering volume changes of the interface reactions and isotope profiles across the orthopyroxene rims. Abart et al. (2004) re-examined the isotope profiles and added microstructural evidence to point out that, in contrast to the previous interpretation, SiO2 was more mobile than MgO. If Fisler et al.’s (1997) high-T data were extrapolated to 1,000°C irrespective of the actually rate-controlling component, the diffusivity were three orders of magnitude lower than in the high-P experiments of Yund (1997) and Milke et al. (2001). This unsatisfying discrepancy between the data-sets provided the motivation for dry thin film experiments down to 1,000°C to allow comparison with the high-pressure data without any temperature extrapolation. The results of our investigation provide new insight in the diffusive mobility of major chemical species in polycrystalline silicate aggregates at dry conditions. They highlight the importance of extremely small portions of water for transport phenomena in polycrystalline silicate aggregates and rocks.
Experimental methods
Sample preparation for reaction rim experiments
Two types of Quartz single crystals (Qtz1 and Qtz2) were used for our experiments. The first single crystal Qtz1 was cut into 2 mm thick slices perpendicular to the optical axes and polished with diamond powder. The second set of quartz samples were from a commercial supplier (Steeg&Reuter, Bad Homburg, Germany), which were 0.7 mm thick polished slices oriented along the [001] axis. The polished quartz samples were deposited with amorphous thin films with compositions of olivine near (Fe0.1Mg0.9)2SiO4 and orthopyroxene near (Fe0.1Mg0.9)SiO3 by Pulsed Laser Deposition as described in detail by Dohmen et al. (2002a). Some of the samples were deposited at hot conditions to crystallize films directly during deposition. The quartz samples were placed on a quartz glass plate sitting on SiC heater in a high-vaccuum chamber. The temperature of the system (deposition area on the quartz plate) was measured with a pyrometer (Typ PS28, Keller HCW GmbH, Ibbenbüren-Laggenbeck, Germany) or a NiCr–Ni thermocouple and calibrated with melting points of Al and Zn. On the basis of a sequence of heating experiments with different crucibles and samples we have estimated the temperature to be precise within ±10°C.
Table 1 lists the set of samples prepared for the reaction rim experiments along with the ablation conditions to produce the films. The thin films listed in Table 1 were produced using either a 248 nm or a 193 nm laser beam and slightly varying laser fluences (i.e. energy per surface area). The targets were dense pressed pellets of olivine (SynOldot1) and pyroxene (SynOpx4), which were synthesized by mixing the oxides SiO2, Fe2O3 and MgO and heating in gas mixing furnaces at 1,300–1,400°C for ∼48 h under an oxygen fugacity of 10−9 bar. The olivine pellet was synthesized with isotopically enriched SiO2 (50% 29SiO2 + 50% Si18O2). The oxygen fugacity in the furnace was set by a flowing gas mix of CO and CO2 in the required proportions. Preanneals of the thin film coated crystals served to crystallize the films. The preanneals as well as the reaction rim experiments were carried out in the same furnaces as the synthesis of the pellets.
We have varied the thin film structure and composition to reach a well-defined and optimized setup for the reaction rim experiments. The whole set of deposited quartz starting samples can be subdivided into four different groups: (Setup 1) Deposition of amorphous olivine layers (samples QzOl3–QzOl9) and subsequent pre-anneal at 900°C and fO2 = 10−14 bar for about 4 days. (Setup 2) Deposition of amorphous pyroxene and/or olivine layers (samples QzOl14–QzOl17 and QzOl18-20) without subsequent pre-anneal. (Setup 3) Pre-annealed pyroxene layer + pre-annealed olivine layer (samples QzOl24–QzOl30); pre-anneals performed in two steps at 1,200°C and fO2 = 10−10 bar for 160 min after deposition of the pyroxene film and 900°C and fO2 = 10−15 bar for 20 h after deposition of the olivine layer. (Setup 4) Hot deposited pyroxene + olivine layer (samples QzOl47–QzOl49 + Qz2Ol21–Qz2Ol24); produced at ∼850°C without subsequent pre-anneal.
Thin film characterization
The characterization of the samples after preanneal or reaction anneal was carried out in three steps. Surfaces were characterized with optical microscopy, white light interference microscopy (ATOS, Darmstadt, Germany) and/or scanning electron microscopy (LEO 1530 Gemini FESEM, Carl Zeiss AG, Germany) at the Institut für Geologie, Mineralogie und Geophysik, Ruhr-Universität Bochum. White light (phase shift) interference microscopy were carried out with a machine which operates in Linnick mode and provides a quantitative measure of the topography on the nm-scale, whereas SEM provides a higher resolution on the lateral scale and makes smaller structures visible, like individual grains on the nm-scale. The second step of investigation was the chemical analysis of the thin films with Rutherford-backscattering (see below) and depth profiling with secondary ion mass spectrometry (SIMS). The SIMS results and their interpretation will be published in a second paper. The last part of the sample analysis procedure was the cutting of foils for transmission electron microscopy (TEM) using the focused ion beam technique (FIB) which is essential to observe the structure of the films in a cross section.
Rutherford backscattering (RBS)
From analysis with Rutherford backscattering spectroscopy (RBS) the stoichiometry and thickness of thin films can be extracted (e.g. Jaoul et al. 1991; Dohmen et al. 2002a). The RBS measurements were carried out at the Dynamitron Tandem Accelerator facility of the Ruhr-Universität Bochum. Here, a beam of alpha particles with 2 MeV is used in combination with a silicon particle detector at a backscattering angle of 170°. The beam of the alpha particles with a current of 20 nA was collimated to a diameter of 0.5 mm. To avoid charging of the samples a thin coating with Ag at the edge of the sample was deposited with a liquid suspension.
The spectra were simulated using the software RBX (Kotai 1994). As input for the simulation in addition to the measurement conditions we have varied the stoichiometry and thickness of the films to obtain best possible matches to the observed spectra. The density was taken to be the nominal value for a crystal of the given composition.
The resolution of the spectra is relatively poor, because there is substantial overlap in signals from the substrate and the films. Moreover, recrystallization of the layers during pre-anneal and Opx-layer formation leads to a significant irregularity of the interfaces, up to ±100 nm (see below). Because of the large beam diameter the RBS analysis averages out the detailed chemical pattern. Nevertheless, the method was adequate to clearly identify the film thickness and chemistry of the starting samples and to give an idea about the reacted layer thickness. To consider the lower effective energy (depth) resolution, we used a large straggling factor between 5 and 10 for the simulations. This straggling effect is originally the broadening of the energy distribution of the alpha particles with increasing depth due to interaction with the outer electrons in the target.
Transmission electron microscopy
TEM foils were vertically extracted from the sample surface using the focused ion beam (FIB) method (Overwijk et al. 1993; Roberts et al. 2001). With FIB the preparation of electron transparent specimens—which is a cumbersome task using conventional ion thinning—becomes a routine technique that provides superior results in several respects. Milling occurs by a beam of gallium ions accelerated to 30 keV. The method allows the cutting of about 15 μm long foils with a uniform thickness of about 100 nm perpendicular to the reaction interface including the thin film and the topmost few μm of the quartz substrate. The original sample surface including the thin film forms one edge of the TEM foils. The uniform thickness of the TEM foils without any preferential thinning at the margins ensures constant electron transparency over the entire samples. Due to the vertical cutting the occurrence of beam damage or other artifacts in the sample surface are minimized. The vertical cutting also ensures that any deviations due to oblique cross sections are avoided in the layer thickness measurements. The thin film samples for this study were prepared with the FEI FIB200 instrument at the GeoForschungsZentrum (GFZ) Potsdam. For more technical details the reader is referred to Wirth (2004).
TEM investigations were carried out with a Philips CM200 TEM at GFZ-Potsdam equipped with a LaB6 filament and an energy-dispersive X-ray (EDX) analyzer with an ultrathin window. EDX profile measurements across the layer and substrate sequences were used to check the homogeneity of layer chemistry. Crystallographic information was obtained from electron diffraction patterns.
Results
Starting samples
Grain sizes
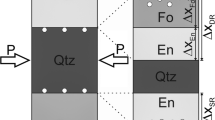
During pre-annealing of initially amorphous films both olivine and orthopyroxene crystallized to a characteristic grain size and texture. The pre-anneals of the Opx layers at 1,200°C (Setup 3) lead to fairly homogeneous grain sizes in each specimen in the 100–200 nm range (Fig. 1a). Initially amorphous olivine layers with Fo90 composition annealed at 900°C crystallize within several hours to a dense polycrystalline matrix with grain sizes ranging roughly from 50 to 200 nm (Dohmen et al. 2002a). We observed the same behavior for Setup 1 samples (only Ol layer) as well as Setup 3 samples (Fig. 1a, Ol layer on top of pre-annealed Opx-layer). The Opx crystallized at 1,200°C was not effected in any visible way by the following anneal at 900°C (Setup 3). In all these samples the crystal diameters of Opx are approximately equal to the respective layer thickness. It must be emphasized that on the bright field TEM photos crystal sizes may easily be underestimated because many grain boundaries do not show up and the real grain size becomes only visible by orientation contrasts on dark field images. The orthopyroxene crystals assume a palisade-like shape preferred texture with grain boundaries oriented almost perpendicular to the Opx–Qz reaction front. Other than Opx, Ol assumes a polygonal fabric without any preferred grain orientation or elongation.

a Bright field TEM image of a FIB section through a layered olivine–orthopyroxene thin film on quartz. This sample (QzOl-29) shows the initial state after layer deposition and pre-anneals according to Setup 3. Annealing temperature for the Opx layer was 1,200°C. The Opx layer crystallized during the pre-anneal, but no Opx rim growth did occur so far. Typical crystal sizes in the Opx layer are 150–200 nm. Single crystals are not always discernable on bright field images, but only on series of dark field images. The bright and dark coating on top of the olivine layer are carbon and platinum, respectively, applied for FIB preparation. b Section through a layered Opx–Ol thin film couple on Qtz (QzOl2–24) prepared by hot deposition of both layers at ∼850°C (Setup 4). Crystal sizes in the Opx layer are often only 10–20 nm, but crystallographically coherent areas with more than 100 nm diameter occur as well
In thin films produced by hot deposition (∼850°C, Setup 4) of first Opx and then Ol on top, both phases crystallized instantaneously from the plasma. Most Opx crystals only measure 10–20 nm, but high-resolution TEM (HRTEM) images also indicated the presence of structurally coherent areas with about 100 nm diameter. Crystal sizes in the Ol layer measure mostly 20–50 nm.
Chemical composition and thickness
In Table 1, the chemical composition of the layers as measured by RBS and the thickness of the layers as measured by RBS and TEM are listed in comparison. The resolution of the RBS spectra is relatively poor for the investigated samples with two layers of several hundred nm thickness. In particular, the measurement of the Mg and O contents in the layers has a relatively large error, because the cross section of the backscattering is proportional to the square of the number of protons in the atom. However, there are very significant features in the spectrum, caused for example from the compositional jumps of Si between the layers and the Qtz substrate, from which a very good estimate of the layer thickness can be drawn (Fig. 2). From the best fits it is evident that the Ol- and Opx-layers are in general slightly enriched in Fe compared to the nominal composition of the targets used for ablation. A small deviation from an ideal Ol- or Opx-stoichiometry cannot be excluded from the RBS analysis, but any small excess of either SiO2 or (Fe, Mg)O would not significantly influence the process as long as the majority of the crystals in the film are Opx or Ol. In fact, the TEM images show a rather homogeneous film with respect to phase assemblage. Chemical compositions of the films are also homogeneous, except for those films that were heated during deposition, as identified for sample Qz2Ol24. Here, a slight zoning in Fe in the range of a few mol% can be observed, a corresponding zoning for Mg might be present, but cannot be resolved by RBS. The reason could be exchange processes between the Ol- and Opx-layer or partitioning between crystallized and still amorphous regions in the film during deposition.

Example of a RBS depth scan fitted with the software RBX to obtain chemical composition and thickness of individual layers (sample QzOl-29, prepared by Setup 3). a The spectrum with its best fit. The four vertical lines indicate the characteristic energy of α-particles back-scattered at a given element (here Fe, Si, Mg and O), which is located at the surface of the sample. b Here the contribution of the individual layers to the bulk spectrum is illustrated. The bulk simulated spectrum shown in (a) is simply the sum of the individual contributions. For the Opx-layer and the Qtz substrate the contribution of each element is also indicated by the element symbol. The small sketch in the upper right of the figure illustrates schematically the sequence of the layers. This specific sample has been deposited in addition with a very thin graphite film for the investigation with SEM
Interfaces
After deposition, the layer interfaces are very smooth depending on the polishing quality of the quartz substrate and the evenness of the pyroxene layer surface. For instance, quantitative measurement of the surface topography of sample QzOl29 using interference microscopy gives an average roughness of about 2–3 nm (calculated by the software ATOS Mark III V3.7.1.5). The irregularity of the Ol–Opx interface of the same sample seems to be slightly larger, of the order of 10 nm, according to the bright field TEM image of a cross section of this sample (Fig. 1a).
To summarize, irrespective of the detailed experimental procedure used to produce the diffusion couples, the samples are chemically and geometrically well defined and therefore suitable as starting samples for layer growth experiments. The different layers are in direct physical contact and are separated by real interphase boundaries without any visible porosity that could be a potential barrier for element transport.
Microstructural observations of reacted samples
Grain size evolution
Figure 3 displays a bright field TEM image of an Ol–Opx layer sample after an Opx growth experiment. Initially amorphous Opx–Ol double layers (QzOl 15 and 16) crystallized during 15 min annealing at 1,100°C to grain sizes of approximately 10–50 nm. The crystallization rate seems to be fast enough in this case to avoid any chemical mixing between the two layers. After 300 min the grains measured 50–100 nm, well below the thickness of the Opx layer. These observations contrast with the fast Opx crystal growth during the pre-anneals at 1,200°C (cf. Fig. 1a). In all experiments using samples pre-annealed at 1,200°C it is obvious that Opx crystal sizes and textures after the experiments do not significantly differ from the pre-annealed starting samples (cf. Fig. 4). It is thus concluded that after initial grain growth a stable grain boundary configuration is attained that persists during the experiments.

Bright field TEM image of an initially amorphous two-layer sample with Opx–Ol stoichiometry on Qtz after a rim growth experiment at 1,100°C for 15 min (QzOl–15). The Opx and Ol layers crystallized without chemical mixing. Crystal sizes in the Opx layer are predominantly in the 10–50 nm range

Bright field TEM images of a starting sample (Setup 3) and three experiments of a time series at 1,200°C. The scale bar applies for all four images. Black outlines on both sides of the Opx layer accentuate the evolution of layer thickness and interface morphology. The Opx layer thickness increases with run duration. Variations in Opx layer thickness and perturbations in the initially even interfaces evolve in the course of rim growth
Unlike Opx the pre-annealed olivine grains coarsen significantly from an average diameter of about 50 nm to about 200 nm after the reaction anneal (e.g., Figs. 4, 5a). This coarsening does not significantly increase surface roughness (Fig. 5b), whereas the Opx/Ol interface seems to become increasingly irregular (Fig. 4).

a SEM image of the olivine layer surface of an annealed sample (QzOl–24). b Interference microscopy image of the same sample. The grey scale indicates the topography. Along the black solid line a height profile is shown on the right side of the image
In hot-deposited samples fast grain growth of Opx and Ol occured in the first minutes of experiments at 1,200°C or higher, leading to crystal sizes and textures identical to the pre-annealed specimens (for Opx), independently of run temperature and duration, thus suggesting that the final crystal diameters are controlled by layer thickness. The time needed until the final grain size is reached remains unknown, because a mature texture did already form in the shortest experiments during 10 min run time. In thin layers with individual crystals in the range of tens of nanometers minimization of interface energies is possibly attained by rotating crystals to low-energy mutual orientations (Harris et al. 1998; Moldovan et al. 2001). Small grains therefore might form clusters that assist in the recrystallization to an equilibrium structure made up of larger grains. Similar processes may contribute to the fast crystallization of the initially small crystals to the coherent palisade-like crystals extending over the entire Opx layer widths.
Once formed, texture and grain size remain almost unchanged except for the increase in thickness of the Opx layers. However, as Opx proceeds to grow the interface roughness of both the Opx–Qtz and Opx–Ol layers increase notably. Locally, pyroxene bulges into the quartz substrate leading to protrusions up to 300 nm, whereas in other areas the interface remains smooth on the 10–20 nm scale (Fig. 6).

An extreme case of interface roughness at the Opx–Qtz growth front from sample QzOl–27 (1,200°C for 360 h). In the thick parts the Opx layer apparently has used up the Ol source layer. Reasons for this locally aberrant behaviour are unknown. Arrows point to strain contrast in quartz. The brighter circle below the centre of the image is not a structure in the sample, but a hole in the carbon film on which the studied sample is placed in the TEM
Polysynthetic twinning of enstatite
TEM investigation reveals that the pyroxene consists of twinned lamellar intergrowths of orthoenstatite and clinoenstatite (for convenience we use Opx as a summary term in the entire paper). Such microstructures are explained by formation of orthoenstatite plates due to (100) twinning of clinoenstatite on the unit cell scale and stacking ortho- and clinoenstatite lamellae in two twin orientations (Buseck et al. 1980). Alternating ortho- and clinoenstatite lamellae have a thickness below 100 Å as can be discerned from dark field images (Fig. 7a). Frequent irregularities in fringe contrasts and spacings are indicative of high stacking disorder. Such lamellar microstructures are present in the quenched pyroxenes over the entire temperature range of this study. They are consistent with pyroxene microstructures formed upon instantaneous transformation from the high-temperature polymorph protoenstatite to metastable clinoenstatite and stable orthoenstatite during quenching (Buseck et al. 1980). Although stability relations and transition temperatures between the enstatite polymorphs are debatable (Jackson et al. 2004 and references therein), these observations indicate that all pyroxenes were protoenstatite during the reaction anneals. Consequently, the transition from protoenstatite to the low-temperature modifications must occur below 1,000°C. The grain boundary structure and hence diffusion properties in protoenstatite potentially differ from those in orthoenstatite. However, since the displacive proto- to orthoenstatite transition essentially involves just a slight rotation of SiO4-tetrahedra in the chains, its effect on grain boundary diffusivity is probably of minor importance.

a HRTEM image of an Opx crystal at the Opx–Qtz interface viewed approximately along the b-axis. The white and black stripes are clinopyroxene (cell width ∼9 Å in a-direction) or orthopyroxene (cell width ∼18 Å in a-direction) lamellae, both of them in two twin orientations. b TEM bright field image of an Ol–Opx thin film on Qtz substrate (QzOl–30; 1,005°C; 4,200 min). Quartz shows pronounced strain contrast. All lobes are emerging from strain contrast centres in the Opx–Qtz interface
Elastic deformation of quartz
After the Opx layer has increased in width during the anneals, chains of strain contrast centers are present in quartz along the Opx reaction front (Fig. 7b). The strained areas show up on TEM bright field images as bent contrasts typically reaching about 200 nm, but in places up to 2 μm into the Qtz substrate crystal. Their appearance is equivalent to strain contrasts observed in quartz in areas where local volume increase was induced due to beam damage (Martin et al. 1996) or to diffusion-controlled crystal growth (Milke and Wirth 2003). The most pronounced examples emerge where Opx protrudes into the Qtz surface, but small strain halos also occur in regions where the interface roughness after reaction does not exceed the original morphology of the Qtz surface (cf. Figs. 6, 7b). Strikingly, strain contrasts are present in all samples that have undergone growth of the Opx layer whereas they are completely absent in the pre-annealed starting samples.
Reaction-induced stress at the Opx–Qtz interface might have profound implications on local reactions. If purely diffusion controlled, a planar growth front under isobaric conditions should move in a straight way preserving its smoothness. However, if local stress occured, this would locally add a mechanical contribution to the chemical potentials of the diffusing components and thereby change the driving force for diffusion across the reaction rim. In the case of the reaction Ol + Qtz → Opx the reaction volume is negative and ΔG of the reaction as well as the chemical potential gradients for the diffusing components increase with pressure. Any local growth-induced stress should therefore locally increase diffusion rates resulting in a positive feed back for any protrusion that starts to form at the Opx–Qtz growth front. It is thus expected that small undulations initially present at the Opx–Qtz interface tend to build up, leading to increased irregularity of the growth fronts as layer thickness increases (cf. Fig. 4). In the context of our experiments the positive feed back between reaction-induced stress and growth rates sets a limit for rim growth to be feasibly treated as planar layer growth. Generally, the increasing deviations from planar rim geometry indicate that reaction-induced stress significantly contributes to the driving force of diffusion-controlled solid media reactions. Reaction-induced stress may lead to inhomogeneity and anisotropy during diffusion-controlled rim growth.
Reaction rates
Our investigations clearly show that the olivine film and the quartz crystal are continuously consumed to form orthopyroxene. Ideally, if the reaction is transport controlled the geometry of the system should not change its character—a uniform layer should grow in a uniform way. However, the time series at 1,200°C (Fig. 4a–d) shows that the interphase boundaries became more irregular with time. Above, we have already discussed that phenomena on the nano-scale, like grain-scale stress fields, are an important parameter influencing the reaction rate. Therefore, the system deviates from the ideal case. Irrespective of these growing irregularities the bulk amount of Opx increases with time, which can be shown by assigning the Opx-rim an average thickness. The total reaction progress is then equivalent to the difference between the final and the initial Opx-rim thickness: \( \Delta x_{{{\text{Opx}}}} = d_{{{\text{Opx}}}} - d^{^\circ }_{{{\text{Opx}}}} . \) For the determination of the average rim thickness, we measured the rim thickness of the Opx- and Ol-layer at up to 50 different positions on TEM images and calculated the arithmetic average (Table 2). Regions in the TEM image where strong stresses are visible and correspondingly anomalously thick Opx-layers (e.g. sample QzOl27; Fig. 7) occur were not considered for this analysis. The parabolic dependence of \( (\Delta x_{{{\text{Opx}}}} ) \) versus time of the experiment, \( \Delta t, \) observed for the time series at 1,200°C, is characteristic for a diffusion-controlled mechanism of the bulk reaction (Fig. 8). The slope, k, is defined in the following as the reaction rate constant. In comparison we show a linear fit to the data, which would be indicative for an interface-controlled reaction, where dissolution or precipitation of a chemical species is the rate determining step. However, intermediate reaction types are possible as well. The kinetics of such a combined mechanism may still result in a parabolic rate law (Lasaga 1986). We calculated a reaction rate constant for all experiments (Table 2) from \( {\left( {\Delta x_{{{\text{Opx}}}} } \right)}^{2} /\Delta t \) assuming that the parabolic rate law is valid at all temperatures even when we have only one experiment at a given temperature and the validity was not explicitly tested (Fig. 9). These data show an Arrhenean behavior over the entire investigated temperature regime between 1,000 and 1,300°C and yield an apparent activation energy of 416 ± 34 kJ/mol, which is within the error limits the same activation energy as observed by Fisler et al (1997). This linear dependence supports the assumption of the parabolic rate law at other temperatures than 1,200°C. The extrapolation of our data to higher temperatures give very similar reaction rates, slightly smaller by a factor of 2 than those of Fisler et al. (1997). Both data sets in combination show that under completely dry conditions and one atmosphere the rate-determining step for the formation of Opx does not change over the entire temperature range between 1,000 and 1,450°C. This is an important conclusion, because on the one hand it shows that our nano-scale approach gives consistent results compared to a micro-scale approach at similar conditions. On the other hand, a combined mechanism can be excluded because in this case one could expect a change in the temperature dependence at some point, since diffusion and interface processes are expected to have different activation energies.

Increase in the square of the Opx rim thickness, \( (\Delta x_{{{\text{Opx}}}} )^{2} , \) versus time of the experiment, \( \Delta t. \) The temperature series yield straight lines in the \( (\Delta x_{{{\text{Opx}}}} )^{2} \) vs. \( \Delta t \) plot. Their slope is the reaction rate constant, k. In comparison a fit is shown assuming a linear dependence of \( \Delta x_{{{\text{Opx}}}} \) on t (in this plot a parabolic time dependence)

Logarithm of rim growth rate (as reaction rate constant, k) as a function of reciprocal temperature. The slope yields an activation energy of 416 ± 34 kJ/mol. Data from other Opx rim growth studies (Fisler et al. 1997; Yund 1997; Milke et al. 2001) with Fo90 olivine as starting material are shown for comparison. The error bars for each data point are calculated from the standard deviations of each individual rim measurement (Table 2 )
Most importantly, compared to the high pressure data at 1,000°C (Yund 1997; Milke et al. 2001) the present low-pressure data are almost four orders of magnitude smaller. A reasonable explanation for this phenomenon is given below after a detailed discussion of possible mechanisms.
Interpretation
Rate-controlling reaction step
The overall reaction olivine + quartz = orthopyroxene can be split in two half-reactions at the opposite sides of the Opx layer (Milke et al. 2001; Abart et al. 2004):
where 0 ≤ ν ≤ 1. In the extreme case of ν = 1 there would be exclusively diffusion of SiO2, and Opx growth would be restricted to the Ol–Opx interface. In the case of ν = 0 there would only be diffusion of MgO, and Opx growth would occur at identical rates at both the Ol–Opx and the Qtz–Opx interfaces. In the P, T range of our experiments, the volume change of partial reaction (2) in the latter situation is negative (−28%), whereas that of partial reaction (3) is markedly positive (+38%). In the particular case of ν = 0.28, ΔV of reaction (3) would be zero and the replacement of Qtz by Opx could occur stress-free.
In reactions under high confining pressure, rim forming reactions tend to minimize reaction-induced stress by constant volume replacement of the source phase of the most immobile component (Abart et al. 2004). In this case, the initially present Opx layer would stay fixed with respect to an external reference frame. In contrast, under low confining pressure, as is the case in our thin film experiments, volume restrictions are eased. Volume changes, for example, a positive reaction volume at the Qtz–Opx interface, could simply shift away the initial Opx layer with respect to an external reference frame. Therefore, within stoichiometrical limits there are no volume restrictions for Opx growth at either side of the original Opx layer. Several scenarios could comply with the observed scenario. If MgO were much more mobile than SiO2, Opx would form at equal rates at both the Qtz–Opx and Ol–Opx interfaces and the total rim growth rates would reflect MgO diffusivity. If the diffusive mobility of MgO and SiO2 were in the same order of magnitude, significant growth at undefined rates would occur on both sides of the initial Opx layer and rim growth rates would be a function of both MgO and SiO2 diffusivity. In case, local reaction-induced stress at the Qtz–Opx interface would inhibit the progress of reaction (2), any stress-releasing mechanism could control the overall layer growth rates. One possible stress-releasing mechanism would be SiO2 diffusion towards the Ol–Opx interface in which case SiO2 diffusion through the Opx layer would become rate-controlling. In the absence of any textural interfaces within the Opx layer there are no univocal criteria to differentiate between these scenarios simply by (electron) optical means.
The strain contrasts at the Qtz–Opx interface help to reduce the range of feasible scenarios. They resemble the strain contrasts observed by Martin et al. (1996) around amorphisized areas in quartz and by Milke and Wirth (2003) at propagating wollastonite reaction fronts around quartz grains, who both ascribed them to the compressive stress associated with the local reactions.
Apparently, any growth-induced stress component normal to the Qtz–Opx interface could simply be released by lifting the initial Opx layer. In contrast, a sideward stress component could not be dissipated this way. Effective exertion of reaction-induced stress on the Qtz surface thus would require some morphology of the Qtz–Opx interface, where the initial interface roughness of the starting samples possibly might suffice. The prominent strain in Qtz especially around Opx protrusions in the reaction front (cf. Fig. 7) supports this argument. The most striking observation, however, is the frequent presence of strain contrasts in reacted samples where orthopyroxene growth had occured at the Opx–Qtz interface and on the other hand its complete absence in the pre-annealed (at 1,200°C) and hot-deposited (at 850°C) starting samples, although during pre-annealing or hot layer deposition they had undergone heating and quenching treatment similar or identical to the reacted samples (cf. Figs. 1a, b, 4). It can therefore be excluded that the observed intracrystalline strain is the result of thermomechanical stress due to quenching. This conclusion is further backed by the presence of strain contrasts in reacted samples in regions with relatively flat interface morphology where the interface roughness after reaction is practically equal to the starting samples. The presence of strain contrast in Qtz in such flat interface regions after growth of the Opx layer indicates that the origin of the strain contrast centers is independent from the interface morphology, but is related to the growth history of the Opx rim. Thus, this general discrepancy between reacted and non-reacted layers provides evidence that the observed strain contrast requires the existence of growth fronts and therefore bears witness of reaction-induced stress. It follows from this reasoning that reaction (2) indeed induced a positive volume change, thus indicating that ν ≤ 0.39. Consequently, MgO must be more mobile than SiO2. The discussion of the rate-limiting diffusing component therefore can be restricted to scenarios with SiO2 being rather immobile.
Extraction of diffusion coefficients from reaction rates
In the preceding section the relative net mobility of MgO and SiO2 through Opx reaction rims has been discussed on the basis of microscopic observation and theoretical consideration, from which MgO has been identified as the more mobile component. In principle, the flux of all ions Fe2+ (Fe3+), Mg2+, Si4+ and O2− in the reaction system has to be treated explicitly, but these fluxes are coupled by electrostatic interactions and the constraint of phase stability of Opx. The net mobility of MgO is the combination of the individual mobilities of Mg2+ + O2−, which generally do not have to obey a one to one relation. Only in the limiting case of almost immobile Si, as proposed here, the bulk fluxes of O2− and Fe2+ + Mg2+ are equal to conserve the phase stability of Opx (e.g. Fisler et al. 1997). The framework of irreversible thermodynamics has been used by Fisler and Mackwell (1994) to derive equations for the relation between the rim thickness and the bulk diffusion coefficients of the various ions in the rim for the reaction 2 FeO + SiO2 = Fe2SiO4. Fisler et al. (1997) followed this approach for the reaction Ol + Qtz = Opx and derived the following equations for the two cases considered here assuming that Si is approximately immobile and a steady-state flux in the rim has been attained:
From these equations one can calculate the bulk diffusion coefficients in the rim for the less mobile of O and Fe, Mg. The term \( \Delta G_{{{\text{Opx}}}} \) is the driving force for the chemical flux, that is identical to the \( \Delta G \) of the Opx forming reaction investigated (Reaction 1). The bulk diffusion coefficient of an ion i, \( \overline{D} _{{\text{i}}} , \) depends on both the mean grain boundary diffusion coefficient, \( \overline{D} ^{{{\text{GB}}}}_{{\text{i}}} , \) and the volume diffusion coefficient, \( D^{{\text{V}}}_{{\text{i}}} , \) as follows (e.g., see Joesten 1991):
where effects of tortuosity have been neglected because of the palisade like Opx grains. The grain boundary and the volume diffusion coefficients are weighted by the ratio of grain size of pyroxene, d, and the grain boundary width, δ. Considering the small crystal size in the Opx thin films of about 200 nm and assuming a mean effective grain boundary width of 2 nm in accordance with many previous workers (Kaur et al. 1995, p. 147f.), the weighing factor δ/d amounts to 1%. An additional assumption for Eq. 6 is that the partitioning of the diffusing species between the grain boundary and the volume is equal. A fractionation of the species between these two media can be considered by a segregation coefficient, K (e.g., see Herzig and Mishin 1998) which is incorporated into the weighing factor: \( K\frac{{\pi \delta }} {{2d}}. \)
Discussion
Mg diffusion versus O diffusion
Bulk diffusion coefficients of either Mg or O in pyroxene rims calculated using Eq. 4 yield effectively the same activation energy as the reaction rates (416 ± 34 kJ/mol, Fig. 10), because the temperature dependence of the factor ΔG/RT in Eq. 4 is almost negligible within the investigated temperature regime (varies between 0.550 and 0.545, calculated using the data of Berman 1988). From Eq. 6, it can be directly seen that measured volume diffusion coefficients constitute a lower limit for the bulk diffusion coefficient of the polycrystalline Opx rims. For this reason Mg volume diffusion and intracrystalline FeMg interdiffusion coefficients from the literature have been added to Fig. 10. Extrapolation of the literature data from Schwandt et al. (1998) and the FeMg interdiffusion data of Ganguly and Tazzoli (1994) determined for single crystals to higher temperatures predict slightly larger bulk diffusion coefficients even without considering contribution of grain boundary diffusion. Notably FeMg interdiffusion in Opx single crystals determined from naturally formed zonations at an estimated peak temperature of 1,130°C by Klügel (2001) plot close to our data. Intuitively, this accordance between diffusion coefficients in single and polycrystalline Opx would not be expected. For the given Opx texture in our experiments, if the grain boundary diffusion coefficient, \( \overline{D} ^{{{\text{GB}}}}_{{\text{i}}} , \) for the rate determining species would exceed the volume diffusion coefficient, \( D^{{\text{V}}}_{{\text{i}}} \) by about two orders of magnitude, both volume and grain boundary diffusion could in equal measure contribute to bulk diffusion. If D i,GB > 100 D i,Vol, as is the case for forsterite with D Mg,GB ≈ 1,000 D Mg,Vol (Chakraborty et al. 1994; Farver et al. 1994) and many other oxide and silicate phases (Joesten 1991), bulk diffusion in the polycrystalline Opx rims should be significantly faster than in Opx single crystals. Thus, diffusion of Mg as the rate determining step for diffusion of the mobile component MgO can be almost ruled out, since even Mg volume diffusion coefficients are larger than or at least approximately the same as that obtained from the reaction rates of Opx rims. To overcome this complication, one might consider that the extrapolation of the single crystal diffusion data (Ganguly and Tazzoli 1994; Schwandt et al. 1998) over several 100°C is prone to large uncertainty and might exaggerate cation volume diffusion coefficients. However, in this case the activation energy for grain boundary diffusion would consequently have to be higher than for intracrystalline diffusion, which seems improbable. For several diffusion scenarios in (Mg, Fe)-silicates the situation \( D_{{{\text{Si}}}} \ll D_{{\text{O}}} \ll D_{{{\text{Fe,Mg}}}} \) is well established (e.g. compilation of data in Dimanov and Sautter 2000), however, for isostatic grain boundary diffusion the situation is undecided, and at least in the presence of traces of water under high pressure may be quite different (Abart et al. 2004).

Based on the above arguments, O diffusion is the only candidate left as the rate limiting species. Whether grain boundary diffusion dominates or volume diffusion of oxygen significantly contributes to the bulk diffusion coefficient cannot be decided by the present results, but isotopic profiles across the rims give a further constraint for this discussion. A detailed report on the isotopic profiles and their simulation is in preparation (Dohmen et al., in preparation), but as a short communication at this point it has to be noted that in accordance with our interpretations Si isotopes seem to be rather immobile compared to the oxygen isotopes. The oxygen isotopes are distributed across the complete newly formed rim suggesting that diffusion of oxygen is efficient through the volume of the Opx crystals. Another indicator that volume diffusion of oxygen might contribute or even dominate the bulk diffusion through the Opx rim is the high activation energy of more than 400 kJ/mol, which has not been reproduced so far by any direct measurement of grain boundary diffusion coefficients in silicates.
Finally, we conclude that under dry conditions Opx rim growth rates are limited by diffusive transport of (Fe, Mg)O, whereby oxygen is most probably the rate-controlling species. It must be stated, however, that the bulk diffusion of Fe, Mg is probably not orders of magnitude larger than bulk diffusion of O. It cannot be excluded that Mg grain boundary diffusion also contributes significantly to the effective grain boundary diffusion coefficient controlling Opx layer growth. Moreover, given the uncertainty in the extrapolation of Mg volume diffusion it cannot by any means ruled out that both crystal interiors and grain boundaries provide efficient pathways and provide significant contributions to Opx growth.
High-pressure versus low-pressure experiments
The thin film experiments permit a direct comparison of diffusivity under “really dry” conditions at 0.1 MPa and “traces of water” conditions at high pressure. Experiments at 700–1,400 MPa by Yund (1997) and Milke et al. (2001) yield at 1,000°C D bulk around 10−16 m2 s−1, about three or four orders of magnitude faster than in the dry thin film experiments. In the high-P experiments there was only an increase by a factor 5 in reaction rates from samples vacuum-dried at 250°C to samples with 5 wt% water added (Yund 1997). However, this temperature was not really suitable to dehydrate the silicate samples (Dimanov et al. 1999). Moreover, at 1,000°C the Pt tubes used in these experiments must be considered open for the diffusion of hydrogen (Truckenbrodt et al. 1997) or even water (Truckenbrodt and Johannes 1999). Therefore, hydrogen-related defects in Opx or, even more importantly in the Opx grain boundaries could have formed due to diffusion into the capsules under run conditions. The pressure media immediately surrounding the capsules used by Yund (1997) and Milke et al. (2001) were graphite, pyrophyllite and fluorite, all of which are open for hydrogen diffusion. In all experiments pyrophyllite was part of the pressure assemblies. Therefore, the water fugacities in the capsules were at least in part controlled by the water-bearing pressure media during the experiments.
At present no data on oxygen or cation diffusion in Opx polycrystals exist at either dry or hydrous conditions. Apparently the unconstrained but low water fugacities in the high-P experiments were sufficient to enhance the bulk diffusivity in the polycrystalline Opx rims by 3–4 orders of magnitude compared to the dry conditions in the thin film experiments. Especially, the diffusion of the Si species might be enhanced in polycrystals in the presence of traces of water. Abart et al. (2004) presented microstructural and isotope evidence that Opx rims between Ol and Qtz with traces of water in high-P experiments occurs by constant volume replacement at the Opx–Ol interface. This situation requires that SiO2 is more mobile than MgO (ν ≥ 0.39 in Eqs. 2, 3). Thus, not only is bulk diffusivity enhanced in the presence of traces of water at high pressure, but in particular the effective diffusivity of the SiO2 component, such that SiO2 turns from an immobile to a mobile component. Identical observations were made for the growth of wollastonite rims between quartz and calcite in either dry CO2 in hydrothermal experiments (100 MPa; 850–950°C) or in piston–cylinder experiments (1 GPa; 1,000–1,100°C), where SiO2 was less mobile than CaO in the former set of experiments, but more mobile in the latter (Milke and Heinrich 2002).
One may speculate by which means water enhances the mobility of ions in polycrystalline Opx. Water dissolved in the grain boundaries may alter the geometry of partially ordered grain boundary structures and thereby enhance both grain boundary diffusion coefficients and effective grain boundary widths. The presence of hydrolysed silicon bonds is a well established concept to explain, for example, the water weakening effect on ductile behaviour of silicates (e.g. Griggs 1967; Tullis and Yund 1989; Brodholt and Refson 2000). Strong changes of the effective mobility of chemical species due to an altered physical nature of the grain boundary (e.g. dry system versus wet system) have long been predicted (Rubie 1986; Dohmen and Chakraborty 2003), which view is supported by many studies on diffusion controlled creep of silicates (e.g. Dimanov et al. 1999, 2000; Rybacki and Dresen 2000).
The difference in water fugacity between the “wet” high-P experiments and the dry 0.1 MPa experiments could have far reaching implications for petrogenetic processes in the crust and mantle, because it may mimic the difference between in situ “wet” and “dry” rocks. Even in the absence of a free grain boundary fluid, OH-bearing minerals should buffer fH2O at a level near the “traces of water” experiments, whereas extremely low fH2O should only be prevalent in the absence of hydrous phases (Kohn 1999). In nature, oxygen isotope resetting among metamorphic minerals depends critically on fH2O in nominally dry rocks (Kohn 1999). The drastic effect of small fH2O differences to cation mobility in polycrystalline aggregates implies that traces of water may also control the efficiency of processes depending on cation diffusion in the lower crust and the mantle. This is an obvious fact for experimental studies on diffusion creep under dry and slightly water-bearing conditions (e.g. Mei and Kohlstedt 2000; Hier-Majumder et al. 2005; Rybacki et al. 2006), but so far remained obscure in direct measurements of grain boundary diffusion in geomaterials or diffusion-controlled rim growth.
Nominally anhydrous minerals from mantle xenoliths like olivine and orthopyroxene contain water in a range from a few up to more than 1,000 wt-ppm (e.g. Ingrin and Skogby 2000; Rauch and Keppler 2002; Mosenfelder et al. 2006). Moreover, metasomatized mantle regions contain OH-bearing minerals like amphibole or phlogopite. Thus, mantle rocks span a range in water saturation close to the range from really dry experiments to those in the presence of traces of water. Consequently it is inferred that processes occurring at grain boundaries in mantle rocks may also respond vigorously to subtle changes in fH2O.
References
Abart R, Kunze K, Milke R, Sperb R, Heinrich W (2004) Silicon and oxygen self diffusion in enstatite polycrystals: the Milke et al (2001) rim growth experiments revisited. Contrib Mineral Petrol 147:633–646
Berman RG (1988) Internally consistent thermodynamic data for minerals in the system Na2O-K2O-CaO-MgO-FeO-Fe2O3-Al2O3-SiO2-TiO2-H2O-CO2. J Petrol 29:445–522
Brady JB (1983) Intergranular diffusion in metamorphic rocks. Am J Sci 283A:181–200
Brodholt JP, Refson K (2000) An ab initio study of hydrogen in forsterite and possible mechanism for hydrolytic weakening. J Geophys Res Solid Earth 105:18977–18982
Buseck PR, Nord GL, Veblen DR (1980) Subsolidus phenomena in pyroxenes. In: Prewitt CT (ed) Pyroxenes, reviews in mineralogy, vol 7. Mineralogical Society of America, Washington, pp 117–211
Chakraborty S, Farver JR, Yund RA, Rubie DC (1994) Mg tracer diffusion in synthetic forsterite and San Carlos olivine as a function of P, T, and fO2. Phys Chem Minerals 21:489–500
Dimanov A, Dresen G, Xiao X, Wirth R (1999) Grain boundary diffusion creep of synthetic anorthite aggregates: the effect of water. J Geophys Res Solid Earth 104(B5):10483–10497
Dimanov A, Sautter V (2000) “Average” interdiffusion of (Fe,Mn)-Mg in natural diopside. Eur J Miner 12:749–760
Dimanov A, Wirth R, Dresen G (2000) The effect of melt distribution on the rheology of plagioclase rocks. Tectonophysics 328:307–327
Dohmen R, Becker H-W, Meissner E, Etzel T, Chakraborty S (2002a) Production of silicate thin films using pulsed laser deposition (PLD) and applications to studies in mineral kinetics. Eur J Miner 14:1155–1168
Dohmen R, Chakraborty S, Becker H-W (2002b) Si and O diffusion in olivine and implications for characterizing plastic flow in the mantle. Geophys Res Lett 29(21):2030
Dohmen R, Chakraborty S (2003) Mechanism and kinetics of element and isotopic exchange mediated by a fluid phase with implications for geothermometry, geospeedometry and isotopic closure. Am Mineral 88:1251–1270
Farver JR, Yund RA, Rubie DC (1994) Magnesium grain boundary diffusion in forsterite aggregates at 1000–1300°C and 0.1–10 GPa. J Geophys Res 99:19809–19819
Fisler DK, Mackwell SJ (1994) Kinetics of diffusion-controlled growth of fayalite. Phys Chem Miner 21:156–165
Fisler DK, Mackwell SJ, Petsch S (1997) Grain boundary diffusion in enstatite. Phys Chem Miner 24:264–273
Ganguly J, Tazzoli V (1994) Fe2+-Mg interdiffusion in orthopyroxene—Retrieval from the data on intracrystalline exchange reaction. Am Mineral 79:930–937
Griggs D (1967) Hydrolytic weakening of quartz and other silicates. Geophys J R Astron Soc 14:19–31
Harris KE, Singh VV, King AH (1998) Grain rotation in thin films of gold. Acta mater 46:2623–2633
Herzig C, Mishin Y (1998) Grain boundary diffusion in metals. In: Kärger J, Heitjans P, Haberlandt R (eds) Diffusion in condensed matter. Vieweg, Braunschweig/Wiesbaden, Germany, pp 90–115
Hier-Majumder S, Mei SH, Kohlstedt DL (2005) Water-weakening of clinopyroxenite in diffusion creep. J Geophys Res Solid Earth 110(B7):B07406
Ingrin J, Skogby H (2000) Hydrogen in nominally anhydrous upper-mantle minerals: concentration levels and implications. Eur J Miner 12:543–570
Jackson JM, Sinogeikin SV, Carpenter MA, Bass JD (2004) Novel phase transition in orthoenstatite. Am Mineral 89:239–244
Jaoul O, Sautter V, Abel F (1991) Nuclear microanalysis: a powerful tool for measuring low atomic diffusivity with mineralogical applications in diffusion, atomic ordering, and mass transport. Sel Top Geochem 8:198–220
Joesten R (1991) Grain-boundary diffusion kinetics in silicate and oxide minerals. In: Ganguly J (ed) Diffusion, atomic ordering, and mass transport. Springer, New York, pp 345–395
Kaur I, Mishin Y, Gust W (1995) Fundamentals of grain and interphase boundary diffusion. Wiley, Chichester, p 512
Klügel A (2001) Prolonged reactions between harzburgite xenoliths and silica-undersaturated melt: implications for dissolution and Fe-Mg interdiffusion rates of orthopyroxene. Contrib Mineral Petrol 141:1–14
Kohn MJ (1999) Why most “dry” rocks should cool “wet”. Am Mineral 84:570–580
Kotai E (1994) Computational methods for analysis and simulation of RBS and ERDA spectra. Nucl Instrum Methods B 85:588–596
Lasaga AC (1986) Metamorphic reaction-rate laws and development of isograds. Mineral Mag 50:359–373
Liu M, Peterson J, Yund RA (1997) Diffusion-controlled growth of albite and pyroxene reaction rims. Contrib Mineral Petrol 126:217–223
Martin B, Flörke OW, Kainka E, Wirth R (1996) Electron irradiation damage in quartz, SiO2. Phys Chem Miner 23:409–417
Mei S, Kohlstedt DL (2000) Influence of water on plastic deformation of olivine aggregates, 1. Diffusion creep regime. J Geophys Res Solid Earth 105(B9):21457–21469
Milke R, Wiedenbeck M, Heinrich W (2001) Grain boundary diffusion of Si, Mg, and O in enstatite reaction rims: a SIMS study using isotopically doped reactands. Contrib Mineral Petrol 142:15–26
Milke R, Heinrich W (2002) Diffusion-controlled growth of wollastonite rims between quartz and calcite: comparison between nature and experiment. J Metamorph Geol 20:467–480
Milke R, Wirth R (2003) The formation of columnar fiber texture in wollastonite rims by reaction-induced stress and implications for diffusion-controlled corona growth. Phys Chem Miner 30:230–242
Moldovan D, Wolf D, Phillipot SR (2001) Theory of diffusion-acommodated grain rotation in columnar polycrystalline microstructures. Acta mater 49:3521–3532
Mosenfelder JL, Deligne NI, Asimow PD, Rossman GR (2006) Hydrogen incorporation in olivine from 2 to 12 GPa. Am Mineral 91:285–294
Overwijk MHF, van den Heuvel FC, Bulle-Lieuwma CWT (1993) Novel scheme for the preparation of transmission electron microscopy specimens with a focused ion beam. J Vac Sci Technol 11:202
Rauch M, Keppler H (2002) Water solubility in orthopyroxene. Contrib Mineral Petrol 143:525–536
Roberts S, McCaffrey J, Gianuzzi L, Stevie F, Zaluzec N (2001) Advanced techniques in TEM specimen preparation. In: Zhang X-F, Zhang Z (eds) Progress in transmission electron microscopy, vol 1. Springer Series in Surface Sciences, 38, pp 336–342
Rubie DC (1986) The catalysis of mineral reactions by water and restrictions on the presence of aqueous fluids during metamorphism. Miner Mag 50:399–415
Rybacki E, Dresen G (2000) Dislocation and diffusion creep of synthetic anorthite aggregates. J Geophys Res Solid Earth 105(B11):26017–26036
Rybacki E, Gottschalk M, Wirth R, Dresen G (2006) Influence of water fugacity and activation volume on the flow properties of fine-grained anorthite aggregates. J Geophys Res Solid Earth 111(B3):B03203
Schmalzried H (1978) Reactivity and point-defects of double oxides with emphasis on simple silicates. Phys Chem Miner 2:279–294
Schwandt CS, Cygan RT, Westrich HR (1998) Magnesium self-diffusion in orthoenstatite. Contrib Mineral Petrol 130:390–396
Truckenbrodt J, Ziegenbein D, Johannes W (1997) Redox conditions in piston-cylinder apparatus: the different behavior of boron nitride and unfired pyrophyllite assemblies. Am Mineral 82:337–344
Truckenbrodt J, Johannes W (1999) H2O loss during piston-cylinder experiments. Am Mineral 84:1333–1335
Tullis J, Yund RA (1989) Hydrolytic weakening of quartz aggregates—The effects of water and pressure on recovery. Geophys Res Lett 16:1343–1346
Watson EB, Price JD (2001) Kinetics of the reaction MgO + Al2O3 → MgAl2O4 and Mg-Al interdiffusion in spinel at 1200–2000°C and 1.0–4.0 GPa. Geochem Cosmochim Acta 66:2123–2138
Wirth R (2004) Focused ion beam (FIB): a novel technology for advanced application of micro- and nanoanalysis in geosciences and applied mineralogy. Eur J Mineral 16:863–876
Yund RA (1997) Rates of grain boundary diffusion through enstatite and forsterite reaction rims. Contrib Mineral Petrol 126:224–236
Acknowledgements
We thank A. Dimanov for his very constructive review and J. Renner for his remarks that improved the paper. The PLD system has been funded by the Deutsche Forschungsgemeinschaft (DFG, Grant CH166/7) and we like to thank Prof. Dr. Chakraborty for the permission to use his machine. Ralf Dohmen also likes to thank the DFG for the funding of his position and this research project within the grant DO777/1. Dr. Rolf D. Neuser is thanked for SEM measurements.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by: J. Hoefs.
Rights and permissions
About this article
Cite this article
Milke, R., Dohmen, R., Becker, HW. et al. Growth kinetics of enstatite reaction rims studied on nano-scale, Part I: Methodology, microscopic observations and the role of water. Contrib Mineral Petrol 154, 519–533 (2007). https://doi.org/10.1007/s00410-007-0207-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00410-007-0207-7