
Abstract
Nonsyndromic genetic deafness is highly heterogeneous in its clinical presentation, pattern of inheritance and underlying genetic causes. Mutations in TMPRSS3 gene encoding transmembrane serine protease account for <1 % of autosomal recessive nonsyndromic hearing loss (ARNSHL) in Caucasians. Targeted next generation sequencing in the index family with profound deaf parents and a son, and Sanger sequencing of selected TMPRSS3 gene regions in a cohort of thirty-five patients with suspected ARNSHL was adopted. A son and his mother in the index family were homozygous for TMPRSS3 c.208delC (p.His70Thrfs*19) variant. Father was digenic compound heterozygote for the same variant and common GJB2 c.35delG variant. Three additional patients from the ARNSHL cohort were homozygous for TMPRSS3 c.208delC. TMPRSS3 defects seem to be an important cause of ARNSHL in Slovenia resulting in uniform phenotype with profound congenital hearing loss, and satisfactory hearing and speech recognition outcome after cochlear implantation. Consequently, TMPRSS3 gene analysis should be included in the first tier of genetic investigations of ARNSHL along with GJB2 and GJB6 genes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Nonsyndromic genetic deafness is highly heterogeneous in its clinical presentation (age at onset, progression, audiological characteristics), pattern of inheritance and underlying genetic causes [1]. Mutations in several genes result in similar clinical presentation of nonsyndromic hearing loss. On the other hand, mutations in the same gene can result in various clinical presentations [2, 3]. Autosomal recessive nonsyndromic hearing loss (ARNSHL) is usually prelingual, nonprogressive and severe to profound [4]. Up to date, it was accounted to mutations in 55 genes (Hereditary Hearing Loss Homepage, http://hereditaryhearingloss.org), where the number is rapidly rising. Most frequently mutated gene in ARNSHL is GJB2, followed by SLC26A4, MYO7A, OTOF, CDH23 and TMC1 genes. In Slovenians, 26.6 % of congenitally deaf patients [5] and 11 % of progressive hearing loss patients [6] had biallelic GJB2 mutations, where other genetic causes of ARNSHL were not exploited so far. Mutations in TMPRSS3 gene encoding transmembrane serine protease are associated with two ARNSHL phenotypes, DFNB8 with childhood onset and DFNB10 where hearing loss is congenital and severe [7]. TMPRSS3 mutations are predicted to account for <1 % of ARNSHL in Caucasians [8] but are more frequent among Pakistani (1.8 %) [9], Tunisian (5 %) [10], Korean (5.9 %) [11] and especially Turkish patients (12 %) [12].
Materials and Methods
Patients
The protocol for the evaluation of hearing-impaired patients comprised a detailed family history, a medical history focusing on potential causes of acquired hearing loss (acoustic trauma, intrauterine infections, perinatal complications, meningitis, mumps, prenatal and postnatal ototoxic drug exposure), and a complete audiological history including the age of the onset of hearing loss, the rate of progression and other audiological symptoms [13]. Written informed consent/assent was obtained from the participants or parents/minors prior to the study that was conducted according to the principles in the Declaration of Helsinki. The index family with congenitally deaf parents and their son was studied initially, followed by 35 patients (23 females, aged 1.5–29 years, mean age 9.5) with ARNSHL and no mutation identified in GJB2 or GJB6 genes.
DNA isolation and quantification
Whole blood samples were used for isolation of genomic DNA with established laboratory protocols based on FlexiGene DNA isolation kit (Qiagen, Hilden, Germany). Isolated DNA samples were tested for purity and concentration using established spectroscopic protocols. The A260/A280 absorption ratio above 1.7 was the indication of successful DNA sample purification. One hundred ng/µL stock solution was prepared from each sample and 20 ng/µL working solution was used for next generation sequencing (NGS) analysis.
Library preparation and NGS analysis
The regions of interest were enriched using TruSight One library enrichment kit (Illumina, San Diego, USA) following manufacturer’s instructions. DNA samples of patients were fragmented, tagged, amplified and pooled to yield 12 pM loading library sample. NGS sequencing was performed using MiSeq desktop sequencer coupled with MiSeq Reagent kit v3 (both Illumina, San Diego, USA). 300 cycles paired-end run using Phred quality threshold >30 was executed followed by onboard primary analysis. Detected genetic variants exceeding >20× coverage threshold setting were analyzed with Variant Studio 2.2 software (Illumina). Further evaluation of variants was restricted to those located in 100 genes related to hearing loss, both syndromic and nonsyndromic, as reported in Hereditary Hearing Loss Homepage, among them, there were 52 genes related to ARNSHL. Among known variants, we eliminated those with minor allele frequency above 1 % from further evaluations.
Sanger sequencing
The presence of candidate causative variant was confirmed with Sanger sequencing. Exon 3 of the TMPSSS3 gene was PCR amplified with in-house designed primers TMPRSS3e3F (gacagggacgcaatttcaata) and TMPRSS3e3R (tacagatgggaagggtcagg), exon 6 with primers TMPRSS3e6F (ttcaaagagggggaaatagaa) and TMPRSS3e6R (ctgagggcaaggagatagga). Amplicons were sequenced using a BigDye Terminator v3.1 sequencing kit and an ABI Genetic Analyzer 3500 (both Applied Biosystems, Foster City, USA).
Results
Clinical characteristics of the patients
Family with profound hearing loss, parents and their son
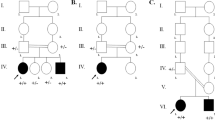
The index family comprised congenitally deaf parents and their son. All others possible causes of hearing loss were excluded [13]. The father has never received hearing aids, but mother has had hearing aids for <2 years in her early childhood without any hearing improvement and still refuses to use them. When Slovenian cochlear implant program started, both parents were adults with only sign language developed. They never complained regarding vestibular disorders. The son is their only offspring and was conceived at their age of 37 years. He was referred to an audiologist after he failed at the National newborn hearing screening program where evoked otoacoustic emissions were not present in first 2 days after birth. He underwent a clinical otorhinolaryngological examination, including ear microscopy, with a systematic search for signs of a syndromic form of hearing loss. All others possible causes of hearing loss were excluded [13]. Before each hearing test, a tympanogram was performed and middle ear causes of hearing loss were excluded. The brain stem evoked acoustic potentials (BERA) were not detected even at stimulation with 110 dB click. He underwent ophthalmological and neurological examinations. They revealed no other pathology. He was immediately fitted with bilateral hearing aids at his age of 5 months but without any benefit. As he was a candidate for cochlear implantation, computed aid tomography (CT scan) of the temporal bone was performed and showed normal medial and inner ear. The cochleography measurements were negative and the electrically evoked auditory brain responds (EABR) were not detectable. For the stimulation, the stimulating golf electrode was placed on round window and reversed electrical stimuli with intensity from 200 μA to 1 mA and duration of 200 μs were used. He received cochlear implant (CI) on his right side at age of 11 months. Now, he has pure tone audiometry hearing thresholds in speech frequencies at 25 dB and he is integrated in regular school. In all of them, GJB2, GJB6 variants and mitochondrial A1555G variant were previously excluded. Only in the father, common c.35delG variant in GJB2 gene in a heterozygous state was identified.
Patients with suspected ARNHL
After identifying TMPRSS3 variant in the index family, the same variant was also screened in other Slovenian congenitally deaf patients with suspected ARNHL. Thirty-five patients were included, all with normally hearing parents and no signs of syndromic forms of hearing loss. In all of them, GJB2, GJB6 mutations and mitochondrial A1555G mutation were previously excluded. All were congenitally deaf, confirmed with BERA measurements. They all received CI as children at average age of 3 years. All except one had good hearing with cochlear implant at the average level 35 DB; in one, the hearing with CI was immeasurable due to severe mental disability not accounted to syndromic forms of hearing loss. Clinical characteristics of patients with identified TMPRSS3 mutations were summarized in the Table 1.
Identification of the disease associated variant
In all three family members, NGS sequencing revealed TMPRSS3 variants that were subsequently confirmed with Sanger sequencing. The son and his mother carried c.208delC (p.His70Thrfs*19) variant on both alleles. It was detected with read depth 62/65 in the son and 31/32 in the mother. Father was compound heterozygous for the same c.208delC variant (read depth 31/66) and additional c.579dupA (p.Cys194Metfs*17) variant (read depth 27/66). c.208delC variant is a known frame-shift variant associated with ARNSHL. Sanger sequencing of the group of patients with suspected ARNHL identified three additional patients with c.208delC variant in homozygous state. c.579dupA variant (rs397517376) is also a frame-shift variant but is located in noncoding exon and has no known association with ARNHL. Therefore, in the father digenic compound heterozygousity of TMPRSS3, c.208delC variant and previously detected GJB2c.35delG variant was proposed.
Discussion
The family history is usually not as evident in ARNSHL as it is in dominant nonsyndromic hearing loss. In the investigated family, both parents and their son were congenitally deaf, and we could not reliably predict the inheritance and suggest more limited targeted genetic testing for autosomal recessively or dominantly inherited deafness. Additionally, father was known to carry GJB2 variant, which was implying possible digenic inheritance in the family. Therefore, NGS sequencing targeting all genes, related to both syndromic and nonsyndromic hearing loss was used successfully. Targeted NGS sequencing seems suitable for identification of causative mutations in hearing-loss patients due to many genes involved in its etiology and various mode of inheritance. It was previously reported as efficient in identifying mutations in 33 % [14] or 12.7 % [15] of sensorineural hearing-loss cases.
Estimated frequency of TMPRSS3 mutations in general childhood Caucasian deaf population is 0.38 % [8]. c.207delC variant was so far reported in homozygous state in one Spanish [8], one Dutch [16], 6 Pakistani and 8 Newfoundland patients, each from one family [17]. In compound heterozygous state, the mutation was reported in one Greek [8], one Dutch [16] and 2 Newfoundland patients [17]. This one nucleotide deletion is leading to frame-shift at the amino acid position 160 and premature termination after 18 unrelated amino acids. The mutation is located just after the transmembrane domain and the truncated protein is lacking LDLRA, SRCR and protease domain [8] suggesting that variant is seriously affecting the function of the truncated protein. Minor allele frequency according to Exome Sequencing Project (http://evs.gs.washington.edu/EVS/) in apparently healthy European-American population is 0.06 where it is not present in homozygous state. This is further confirming its pathogenicity.
The phenotype resulting from the DMPRSS3 variants is dependent on residual proteolitic activity of the mutated protein [18]. Combination of two severe variants on separated alleles leads to profound deafness with prelingual onset, while the combination of severe and mild variant leads to milder phenotype with postlingual onset [16]. In concordance, patients presented here with homozygous c.207delC variant had the same level of profound hearing loss.
Variant c.208delC was detected in homozygous state in two patients from the index family and in three additional ARNHL patients, altogether 5 out of 38 patients (13.1 %). Therefore, TMPRSS3 defects seem to be an important cause of ARNHL in Slovenians, even though it was previously reported to be very rare in Caucasians [8]. Consequently, TMPRSS3 gene analysis should be included in the first tier of genetic investigations of ARNSHL along with GJB2 and GJB6 genes. Identification of causative variants in hearing-loss patients is important to at least partly predict the severity of hearing loss and assist in the decision regarding cochlear implant. Patients with TMPRSS3 mutations have satisfactory speech recognition after the cochlear implantation [16]. The son from the index family received cochlear implant at the age of 13 months with an excellent outcome. During the 6 years of using the implant, he developed good verbal communication and attends regular school. The three additional patients from ARNSHL group with the same mutation were also congenitally deaf and received cochlear implants at the age of 11–30 months with comparably favorable outcome.
In conclusion, TMPRSS3 mutations seem to be an important cause of ARNSHL in Slovenia resulting in rather uniform phenotype with profound congenital hearing loss, and satisfactory hearing and speech recognition outcome after cochlear implantation.
References
Morton CW, Nance E (2006) Newborn hearing screening––a silent revolution. N Engl J Med 354:2151–2164
Kenna MA, Feldman HA, Neault MW, Frangulov A, Wu BL, Fligor B, Rehm HL (2010) Audiologic phenotype and progression in GJB2 (Connexin 26) hearing loss. Arch Otolaryngol Head Neck Surg 136:81–87
Beck C, Pérez-Álvarez JC, Sigruener A, Haubner F, Seidler T, Aslanidis C et al (2014) Identification and genotype/phenotype correlation of mutations in a large German cohort with hearing loss. Eur Arch Otorhinolaryngol. doi:10.1007/s00405-014-3157-5
Petersen MB, Willems PJ (2006) Non-syndromic, autosomal–recessive deafness. Clin Genet 69:371–392
Battellino S, Rudolf G, Zargi M, Podkrajsek KT, Peterlin B (2011) Connexin 26 (GJB2) and connexin 30 del(GJB6-D13S1830) mutations in Slovenians with prelingual non-syndromic deafness. Int Adv Otol 7:372–378
Battelino S, Repič Lampret B, Zargi M, Podkrajšek KT (2012) Novel connexin 30 and connexin 26 mutational spectrum in patients with progressive sensorineural hearing loss. J Laryngol Otol 126:763–769
Scott HS, Kudoh J, Wattenhofer M, Shibuya K, Berry A, Chrast R et al (2001) Insertion of beta-satellite repeats identifies a transmembrane protease causing both congenital and childhood onset autosomal recessive deafness. Nat Genet 27:59–63
Wattenhofer M, Di Iorio MV, Rabionet R, Dougherty L, Pampanos A, Schwede T et al (2002) Mutations in the TMPRSS3 gene are a rare cause of childhood nonsyndromic deafness in Caucasian patients. J Mol Med (Berl) 80:124–131
Walsh T, Abu Rayan A, Abu Sa’ed J, Shahin H, Shepshelovich J, Lee MK et al (2006) Genomic analysis of a heterogeneous Mendelian phenotype: multiple novel alleles for inherited hearing loss in the Palestinian population. Hum Genomics 2:203–211
Masmoudi S, Antonarakis SE, Schwede T, Ghorbel AM, Gratri M, Pappasavas MP et al (2001) Novel missense mutations of TMPRSS3 in two consanguineous Tunisian families with non-syndromic autosomal recessive deafness. Hum Mutat 18:101–108
Chung J, Park SM, Chang SO, Chung T, Lee KY, Kim AR et al (2014) A novel mutation of TMPRSS3 related to milder auditory phenotype in Korean postlingual deafness: a possible future implication for a personalized auditory rehabilitation. J Mol Med (Berl) 92:651–663
Wattenhofer M, Sahin-Calapoglu N, Andreasen D, Kalay E, Caylan R, Braillard B et al (2005) A novel TMPRSS3 missense mutation in a DFNB8/10 family prevents proteolytic activation of the protein. Hum Genet 117:528–535
Fakim SA, Naderpoor M, Shahidi N, Basharhashemi F, Nejati N, Sakha SH, Alizadeh M (2010) Study of prevalence and causes of hearing loss in high risk neonates admitted to neonatal ward and neonatal intensive care unit. Int Adv Otol 6:365–370
Vozzi D, Morgan A, Vuckovic D, D’Eustacchio A, Abdulhadi K, Rubinato E et al (2014) Hereditary hearing loss: a 96 gene targeted sequencing protocol reveals novel alleles in a series of Italian and Qatari patients. Gene 542:209–216
Gu X, Guo L, Ji H, Sun S, Chai R, Wang L, Li H (2014) Genetic testing for sporadic hearing loss using targeted massively parallel sequencing identifies 10 novel mutations. Clin Genet. doi:10.1111/cge.12431
Weegerink NJ, Schraders M, Oostrik J, Huygen PL, Strom TM, Granneman S (2011) Genotype–phenotype correlation in DFNB8/10 families with TMPRSS3 mutations. J Assoc Res Otolaryngol 12:7537–7566
Ahmed ZM, Li XC, Powell SD, Riazuddin S, Young TL, Ramzan K (2004) Characterization of a new full length TMPRSS3 isoform and identification of mutant alleles responsible for nonsyndromic recessive deafness in Newfoundland and Pakistan. BMC Med Genet 5:24
Lee YJ, Park D, Kim SY, Park WJ (2003) Pathogenic mutations but not polymorphisms in congenital and childhood onset autosomal recessive deafness disrupt the proteolytic activity of TMPRSS3. J Med Genet 40:629–631
Acknowledgments
This work was supported in part by the Slovenian Research Agency grants P3-0343, J3-6798, J3-6800.
Conflict of interest
None declared.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Battelino, S., Klancar, G., Kovac, J. et al. TMPRSS3 mutations in autosomal recessive nonsyndromic hearing loss. Eur Arch Otorhinolaryngol 273, 1151–1154 (2016). https://doi.org/10.1007/s00405-015-3671-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00405-015-3671-0