
Abstract
Objective: the novel mutations in the LRTOMT gene was reported in a Chinese patient with nonsyndromic, congenital profound sensorineural hearing loss. Methods: one boy with hearing loss was enrolled from nonconsanguineous family in the study. Targeted genomic enrichment and massively parallel sequencing of all 415 known hearing loss genes were performed to find possible genetic etiology. Various bioinformatics tools were used to assess the pathogenicity of the variants. Interpretation of variants was performed according to the American College of Medical Genetics and Genomics (ACMG) guidelines to identify the genetic cause of hearing loss. Results: compound heterozygotes with a novel nonsense mutation (c.451C>T, p.Arg151X) and a novel missense mutation (c.358G>A, p.Gly120Ser) located in splice site were identified in the LRTOMT gene. The two mutations were segregated in both alleles of LRTOMT, present within the LRTOMT2 protein coding region. Nonsyndromic, congenital profound sensorineural hearing loss without detectable residual hearing was found in the patient, and autosomal recessive inheritance was indicated. Conclusion: a novel missense mutation located in splice site was found in the LRTOMT gene in our study. The compound heterozygous mutation increases the spectrum of LRTOMT gene mutations associated with hearing loss in the Chinese population.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
The genes related with hearing loss were studied more than thirty years, and many mutations of the genes were found. Currently, more than 108 autosomal-recessively inherited forms (ARNSHL) loci have been reported, and 75 genes for autosomal recessive nonsyndromic sensorineural HL (SNHL) have been mapped (http://hereditaryhearingloss.org). Nevertheless, only a few mutations in many autosomal genes for SNHL were reported, and these genes was described as rare hearing-loss-related gene. LRTOMT gene, as one of rare hearing-loss-related gene accounting for autosomal recessive nonsyndromic SNHL, was assigned DFNB63 locus on human chromosome 11q13.3-q13.4 in 2007 [1–3]. LRTOMT is a fusion gene that has alternative reading frames and only exists in primates [4], encoding Leucine Rich Transmembrane and O-Methyl-Transferase [5]. It was revealed that the LRTOMT gene (NM_001145309) consisted of 9 exons and was transcribed into 5 different alternative splicing transcripts, with the positional cloning of the LRTOMT gene performed from human liver cDNA [4, 6]. Further research suggests that LRTOMT gene primarily encodes two different proteins: LRTOMT1 and LRTOMT2 [4, 6]. LRTOMT2 is expressed in sensory hair cells in the inner ear and is thought to be important for auditory and vestibular functions [6]. The defects in LRTOMT2 protein resulted in degeneration of hair cells and caused SNHL [7]. Several studies have also reported that the mutations in the LRTOMT gene resulting in SNHL located in coding regions of LRTOMT2, not LRTOMT1 [8–10]. It is indicated LRTOMT-related SNHL might be more attributable to the LRTOMT2 region than to LRTOMT1.
Until now, only 17 mutations of LRTOMT gene were found in autosomal recessive nonsyndromic SNHL [2, 4, 6, 8–15]. Most of these mutations were all homozygous mutations due to consanguineous families from the Middle East and South Asia [4, 6, 8–11, 14, 16]. The highest mutation frequency in this gene is reported in Moroccan families and then in Tunisian Iranian, Turkish and Pakistani families [4, 6, 11, 16]. These mutations lead to severe-to-profound prelingual SNHL [1]. As targeted next-generation sequencing (NGS) of the known deafness genes’ exons (“gene panels”) was applied to diagnose the cause of hearing loss, more mutations will be found, because NGS of the known deafness genes’ exons is a powerful tool to reveal the disease-causing mutations in patients with hearing loss. Furthermore, more reports of pathogenic mutations are helpful for clinical gene diagnosis and genetic counselling.
In the present study, we reported a novel compound heterozygous mutation composing of a heterozygous nonsense mutation and a heterozygous missense mutation located in splice site in the LRTOMT gene identified in an eleven-month-old boy with SNHL.
MATERIALS AND METHODS
Subject and Clinical Evaluations
An eleven-month-old boy with congenital SNHL from a nonconsanguineous family with no history of HL was ascertained (pedigree is shown in Fig. 1a). Comprehensive family history, physical examination and audiological testing such as play audiometry, tympanometry, acoustic stapedial reflex, transient/distortion product otoacoustic emission (TE/DPOAEs), auditory brainstem response (ABR), auditory steady state response (ASSR) were performed. Sensorineural hearing loss diagnosis was performed in a sound-proofed room according to the clinical standards.

(a) Pedigree of the family. The proband is marked by an arrow. (b) Pure tone audiogram of patient. Audiogram indicate profound hearing loss in both ears. Frequency in hertz (Hz) and the hearing threshold in decibels (dB) are shown.
Clinical examinations did not reveal any other symptoms except for HL. The boy received cochlear implant at the age of 11 months. Auditory and speech performance in this case were evaluated using the categories of auditory performance (CAP) scale and speech intelligibility rating (SIR) scale. These scales are reliable for measuring the outcome of cochlear implantation. CAP consists of 8 categories, ranging from “no awareness of environment” (CAP score 0) to “use of telephone with known users” (CAP score 7) and SIR consist of 5 categories, ranging from “unintelligible speech” (SIR score 1) to “speech intelligible to all listeners” (SIR score 5).
Genomic DNA Sample Collection
Blood samples were collected from the boy and his parents in this pedigree, as well as 76 patients with sporadic hearing loss and 145 normal individuals as controls at the Department of Otolaryngology and Head-Neck Surgery, Peking University First Hospital. Genomic DNA was extracted using the Qiagen blood DNA extraction kit (Qiagen, Hilden, Germany).
Whole Exome Capture and Library Construction
Human exome capture was performed following the protocol from Illumina’s TruSeq™ Exome Enrichment Guide (Illumina, San Diego, CA, USA). Illumina’s TruSeq 62 Mb Exome Enrichment kit was used as exome enrichment probe sets, and 5 μg of genomic DNA in 80 μL of Buffer EB (Qiagen) was fragmented in a Biorupter UCD-200 (Diagenode, Belgium) to sizes of 100–500 bp. DNA concentration was estimated by OD260 measurement and quantitative real-time polymerase chain reaction (PCR) analysis. Captured DNA libraries were sequenced with the Illumina HiSeq 2000, yielding 200 (2 × 100) base pairs from the final library fragments using V2 reagent. After performing WES, the released raw data were converted to the FASTQ file. Bioinformatic analysis included GATK (Genome Analysis Toolkit) (https://gatk.broadinstitute.org/) for variant calling, BWA (Burrows-Wheeler Aligner) (http://bio-bwa.sourceforge.net/) for genome alignments and variant detection (hg19, NCBI Build 38) and Picard to mark duplicate reads were used. Variant filtering was performed based on homozygous missense, start codon change, splice site, nonsense, stop loss, and indel variants with MAF <1% in databases such as: dbSNP version 147, 1000 genomes project phase 3 database (https://www.internationalgenome.org/), NHLBI GO exome sequencing project (ESP) (https://evs.gs.washington.edu/), and exome aggregation consortium (ExAC) (http://exac.broadinstitute.org).
After the filtration, the candidate variants were confirmed using bidirectional Sanger sequencing. PCR amplification and sequencing of the variants were performed using the forward primer: 5′-TATGGGATTCCCTCCCTACC-3′ and the reverse primer: 5′-AGCAGCAACAGATCCCAAAT-3′ for exon 7 of LRTOMT, the forward primer: 5′-TGTCTTCTGCAACAGCCATC-3′ and the reverse primer: 5′-CTCACACACCATCCAGCATC-3′ for exon 8 of LRTOMT. Chromatograms were compared with reference sequence (NM_001145308), encoding a 291-residue protein (NP_001138780.1), using SeqMan software version 5.00© (DNASTAR, Madison, WI, USA). Next, the variants were investigated in the Human Gene Mutation Database (HGMD) (http://www.hgmd.cf.ac.uk/) and the literature to seek the novelty of the variant or its association with HL. Variant nomenclature was based on Human Genome Variation Society (HGVS) [17].
Prediction of Pathogenicity of Mutation
For missense mutation, SIFT, MutationTaster, and PolyPhen2 software programs were applied to predict the influence on the protein function by amino acid substitution [18]. The splice variant was evaluated by different in silico software tools such as FSPLICE (http://www.softberry.com/), NetGene2 Server (www.cbs.dtu.dk/), PANTHER (http://www.pantherdb.org/), MutationTaster (http://www.mutationtaster.org/), SIFT (https://sift.bii.a-star.edu.sg/) and CADD (https://cadd.gs.washington.edu/) to predict its deleterious effect on protein in terms of function. The American College of Medical Genetics and Genomics (ACMG) guidelines were also used to classify the variants [19].
RESULTS
Clinical Evaluations
The proband was an eleven-month-old boy who showed bilateral congenital profound NSHL, according to the audiological evaluations (Fig. 1b). No symptom of syndromic HL was found in the patient. The proband was born to a nonconsanguineous couple after a full-term natural delivery. No genetic disease other than HL was evident in the pedigree. The CT scan results of temporal bone were normal in the patient.
Two years after cochlear implantation, auditory and speech performance were evaluated and good outcome of cochlear implantation with CAP score of 6 (understanding conversation without lip reading) and SIR score of 5 (speech is intelligible to the listener) was found.
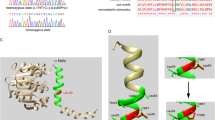
Whole-exome sequencing (WES) was applied and two variants in LRTOMT gene met the criteria for further analyses. As a result of WES, a heterozygous nonsense mutation of c.451C>T (p.Arg151X) and a heterozygous missense mutation of c.358G>A (p.Gly120Ser) located in splice site in the LRTOMT gene were found (Figs. 2a, 2b). The c.451C>T mutation leads to an early stop codon, resulting in a truncated protein with 150 residues (versus 291 residues in the intact protein). Arg151 and truncated part are modified in the mutated protein are located in a highly conserved residue of LRTOMT2 in multiple-species alignment. The c.358G>A mutation we discovered leads to Gly120Ser amino acid substitution in LRTOMT2, meanwhile, the mutation located in splice site of exon 7, resulted in the essential different protein, eliminating the catechol-O-methyltransferase domain and the part of the transmembrane domain. This c.358G>A mutation was assessed as being deleterious by Mutation Taster as well as several other prediction tools such as SIFT, PROVEAN, PANTHER, FSPLICE, NetGene2 Server and CADD. The variants were absent from dbSNP version 147, 1000 genomes project phase 3, NHLBI GO ESP, ExAC, Iranome, HGMD and Clinvar databases. It has been conserved among several species including Rhesus, Mouse, Dog, Elephant, Chicken, X_tropicalis Zebrafish and Lamprey (Fig. 2c). The two mutations were not found in the literature.

(a) One of two isoforms encoded by the LRTOMT gene: LRTOMT2. In this transcript of LRTOMT (NM_001145308), LRTOMT2, starts from exon 5 and ends in exon 9. A compound heterozygous mutation includes c.358G>A mutation located in exon 7 and c.451C>T mutation located in exon 8. (b) The electropherogram of the mutations in the patient, c.358G>A heterozygously from his father (b1) and c.451C>T heterozygously from his mother (b2). (c) The modified region (p.G120S amino acid substitution showm in the red box) is located in a highly conserved region among species.
The variant co-segregated with the disease in the family: heterozygous in parents, but compound heterozygous in the patient who was the only child of the family (Fig. 1a). None of the two variants were identified in the 76 patients with sporadic hearing loss and 145 Chinese normal hearing controls. According to the ACMG guideline, the two variants’ evidences are described below, and the two variants are classified as pathogenic variants (1 very strong, 2 moderate and 1 supporting criteria): c.451C>T (p.Arg151X) variant and c.358G>A (p.Gly120Ser) located in splice site.
—It is a nonsense variant or a splice variant (PVS1).
—It is located in a mutational hot spot and/or critical and well-established functional domain (PM1).
—The variant is absent from controls (or at extremely low frequency if recessive) in Exome Sequencing Project, 1000 Genomes Project and Exome Aggregation Consortium (PM2).
—Multiple lines of computational evidence supported the deleterious effect of the variant on the gene or gene product (conservation, splicing impact, etc.) (PP3).
DISCUSSION
The most mutations of LRTOMT gene resulting in hearing loss were homozygous due to consanguineous marriage, only two patients with compound heterozygous mutation were reported, one is Chinese (see Table 1). It is indicated that the carried rate of the LRTOMT gene mutation was extremely low in Chinese population. In the present study, we identified a novel compound heterozygous mutation in the LRTOMT gene in a boy with congenital profound hearing loss that was presumably autosomal recessive inherited. This is the second case reported to be affected by the compound heterozygous mutation in Chinese patients. One nonsense mutation c.451C>T (p.R151X) was identified in exon 8 of LRTOMT (NM_001145309). The premature stop codon is predicted to result in a truncated protein with impaired function or no protein at all, due to nonsense mediated mRNA decay, according to Alamut 2.3 software. The other mutation of the compound heterozygous mutation was c.358G>A variation which located in the end of exon 7 and resulted in both missense mutation (p.G120S) and splice mutation. The mutations are believed to be pathogenic because of nonsense mutation or splice mutation, and because neither change has been observed in a series of normal controls.
The p.G120S and p.R151 substitutions in LRTOMT2 are predicted to alter the COMT domain of LRTOMT2, leading to the enzymatic defect associated to the c.358G>A and c.451C>T mutation, but the role of LRTOMT isoforms in the hearing process remains elusive, rendering the cause of deafness unclear. An animal model of LRTOMT would be a valuable tool to evaluating the pathophysiology of these mutations. Indeed in mouse, Tomt (residues 34–291) shows 91% amino acid identity and 92% similarity to human LRTOMT2. Tomt was detected in the cytoplasm of inner and outer hair cells and their supporting cells in the cochlea, as well as in vestibular hair cells and their supporting cells in adult mouse [6].
Loss of function in LRTOMT2 protein resulted in profound sensorineural hearing loss in the present case. The most of the previous studies reported audiologic profiles for each individual showing congenital severe to profound hearing loss regardless of missense, nonsense, frameshift, or splice mutation [4, 6, 8, 9, 11, 15]. Nevertheless, Ichinose et al. reported a case with frameshift mutation, c.565_566delT in LRTOMT gene had moderate progressive hearing loss [12]. The authors supposed c.565_566delT located in the region near the 3′-end and C-terminus of the LRTOMT2 region might cause the partial translation of the mutated allele and an incomplete LRTOMT2 protein with residual activity [12]. So, the genotype of LRTOMT gene modulated the phenotype of hearing loss from moderate to profound. In the experiment, mice carrying a mutation of the orthologous gene (COMT2) suffer from vestibular dysfunction, profound deafness and progressive degeneration of the organ of Corti [6]. However, no vestibular dysfunction was found in the patients with LRTOMT mutation.
In the present study, the patient received cochlear implantation, and documented good cochlear implantation outcome. Because the LRTOMT2 expressed in sensory hair cells in the inner ear, the effects of LRTOMT mutations are considered to be on the inner ear and the function of the auditory nerve is spared, it indicated the patients with hearing loss resulting from LRTOMT gene mutation would be good outcomes. The similar outcomes were reported yet in patients with LRTOMT gene mutations in other studies [14, 15].
So far, the reported mutations in the LRTOMT gene were rare and sporadic except for one mutation c.242G>A (p.R81Q) which has been found in deaf patients from Morocco at a frequency of 8.75%, which makes this gene the second most frequently involved deafness gene in Morocco, after GJB2 [16]. The incidence of the mutation in the LRTOMT gene in Asian and other Africa countries is unknown.
In summary, we describe a unique variation which is both missense mutation (c.358G>A, p.G120S) and splice mutation in one allele of LRTOMT. This results in elimination of the catalytic domain of LRTOMT2, which is most likely disease causing. In conclusion, our results and other studies suggest that mutations in the LRTOMT gene result in alterations in the LRTOMT2 (COMT2) protein and might be involved in moderate-to-profound NSHL. The LRTOMT gene should be studied in a larger population of families in Asian for a more thorough understanding of its role in causing HL.
REFERENCES
Kalay, E., Uzumcu, A., Krieger, E.R., et al., MYO15A (DFNB3) mutations in Turkish hearing loss families and functional modeling of a novel motor domain mutation, Am. J. Med. Genet., Part A, 2007, vol. 143A, no. 20, pp. 2382–2389.
Khan, S.Y., Riazuddin, S., Tariq, M., et al., Autosomal recessive nonsyndromic deafness locus DFNB63 at chromosome 11q13.2-q13.3, Hum. Genet., 2007, vol. 120, no. 6, pp. 789–793.
Tlili, A., Masmoudi, S., Dhouib, H., et al., Localization of a novel autosomal recessive non-syndromic hearing impairment locus DFNB63 to chromosome 11q13.3-q13.4, Ann. Hum. Genet., 2007, vol. 71, no. 2, pp. 271–275.
Ahmed, Z.M., Masmoudi, S., Kalay, E., et al., Mutations of LRTOMT, a fusion gene with alternative reading frames, cause nonsyndromic deafness in humans, Nat. Genet., 2008, vol. 40, no. 11, pp. 1335–1340.
Zhu, B.T., Catechol-O-methyltransferase (COMT)-mediated methylation metabolism of endogenous bioactive catechols and modulation by endobiotics and xenobiotics: importance in pathophysiology and pathogenesis, Curr. Drug Metab., 2002, vol. 3, no. 3, pp. 321–349.
Du, X., Schwander, M., Moresco, E.M., et al., A catechol-O-methyltransferase that is essential for auditory function in mice and humans, Proc. Natl. Acad. Sci. U.S.A., 2008, vol. 105, no. 38, pp. 14609–14014.
Bazazzadegan, N., Nikzat, N., Fattahi, Z., et al., The spectrum of GJB2 mutations in the Iranian population with non-syndromic hearing loss–a twelve year study, Int. J. Pediatr. Otorhinolaryngol., 2012, vol. 76, no. 8, pp. 1164–1174.
Babanejad, M., Fattahi, Z., Bazazzadegan, N., et al., A comprehensive study to determine heterogeneity of autosomal recessive nonsyndromic hearing loss in Iran, Am. J. Med. Genet., Part A, 2012, vol. 158A, no. 10, pp. 2485–2492.
Vanwesemael, M., Schrauwen, I., Ceuppens, R., et al., A 1 bp deletion in the dual reading frame deafness gene LRTOMT causes a frameshift from the first into the second reading frame, Am. J. Med. Genet., Part A, 2011, vol. 155A, no. 8, pp. 2021–2013.
Riahi, Z., Bonnet, C., Zainine, R., et al., Whole exome sequencing identifies new causative mutations in Tunisian families with non-syndromic deafness, PLoS One, 2014, vol. 9, no. 6. e99797.
Wang, R., Han, S., Khan, A., and Zhang, X., Molecular analysis of twelve Pakistani families with nonsyndromic or syndromic hearing loss, Genet. Test. Mol. Biomarkers, 2017, vol. 21, no. 5, pp. 316–321.
Ichinose, A., Moteki, H., Hattori, M., et al., Novel mutations in LRTOMT associated with moderate progressive hearing loss in autosomal recessive inheritance, Ann. Otol. Rhinol. Laryngol., 2015, vol. 124, suppl. 1, pp. 142S–147S.
Markova, S., Safka Brozkova, D., Meszarosova, A., et al., Mutations in eight small DFNB genes are not a frequent cause of non-syndromic hereditary hearing loss in Czech patients, Int. J. Pediatr. Otorhinolaryngol., 2016, vol. 86, pp. 27–33.
Sarmadi, A., Nasrniya, S., Soleimani Farsani, M., et al., A novel pathogenic variant in the LRTOMT gene causes autosomal recessive non-syndromic hearing loss in an Iranian family, BMC Med. Genet., 2020, vol. 21, no. 1, p. 127.
Wu, C.C., Lin, Y.H., Liu, T.C., et al., Identifying children with poor cochlear implantation outcomes using massively parallel sequencing, Medicine (Baltimore), 2015, vol. 94, no. 27. e1073
Charif, M., Bounaceur, S., Abidi, O., et al., The c.242G>A mutation in LRTOMT gene is responsible for a high prevalence of deafness in the Moroccan population, Mol. Biol. Rep., 2012, vol. 39, no. 12, pp. 11011–11016.
den Dunnen, J.T., Dalgleish, R., Maglott, D.R., et al., HGVS recommendations for the description of sequence variants: 2016 update, Hum. Mutat., 2016, vol. 37, no. 6, pp. 564–569.
Adzhubei, I.A., Schmidt, S., Peshkin, L., et al., A method and server for predicting damaging missense mutations, Nat. Methods, 2010, vol. 7, no. 4, pp. 248–249.
Richards, S., Aziz, N., Bale, S., et al., Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology, Genet. Med., 2015, vol. 17, no. 5, pp. 405–424.
ACKNOWLEDGMENTS
We sincerely thank all of the individuals in the family for their participation in this study.
Funding
This work was financially supported by the National Natural Science Foundation of China (grant nos. 81271083, 81470691, 82071070) and Beijing Natural Science Foundation (grant no. 7172216), the Beijing Municipal Science and Technology Commission (grant no. Z191100006619027).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest. The authors declare that they have no conflict of interests.
Statement of compliance with standards of research involving humans as subjects. The study on this Chinese family with hearing loss was approved by the Medical Ethics Committee of Peking University First Hospital. All participants signed the informed consents. The boy’s written consent was obtained from his parents.
Rights and permissions
About this article

Cite this article
Wang, Y., Ma, Y., Qin, Y. et al. Novel Mutations in LRTOMT Associated with Congenital Profound Sensorineural Hearing Loss in a Chinese Patient. Russ J Genet 57, 1322–1327 (2021). https://doi.org/10.1134/S1022795421110144
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1022795421110144