
Abstract
A common feature of inherited and sporadic ALS is accumulation of abnormal proteinaceous inclusions in motor neurons and glia. SOD1 is the major protein component accumulating in patients with SOD1 mutations, as well as in mutant SOD1 mouse models. ALS-linked mutations of SOD1 have been shown to increase its propensity to misfold and/or aggregate. Antibodies specific for monomeric or misfolded SOD1 have detected misfolded SOD1 accumulating predominantly in spinal cord motor neurons of ALS patients with SOD1 mutations. We now use seven different conformationally sensitive antibodies to misfolded human SOD1 (including novel high affinity antibodies currently in pre-clinical development) coupled with immunohistochemistry, immunofluorescence and immunoprecipitation to test for the presence of misfolded SOD1 in high quality human autopsy samples. Whereas misfolded SOD1 is readily detectable in samples from patients with SOD1 mutations, it is below detection limits for all of our measures in spinal cord and cortex tissues from patients with sporadic or non-SOD1 inherited ALS. The absence of evidence for accumulated misfolded SOD1 supports a conclusion that SOD1 misfolding is not a primary component of sporadic ALS.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Amyotrophic lateral sclerosis (ALS) is an adult-onset neurodegenerative disease characterized by the selective loss of motor neurons. While 10% of instances are dominantly inherited (known as familial ALS or FALS), the large majority have no identified genetic cause and are termed sporadic ALS (SALS). The gene encoding Cu/Zn superoxide dismutase (SOD1) was the first dominantly inherited FALS-associated gene to be identified [59]. To date more than 170 missense mutations of SOD1 have been associated with ALS, and it is the second most frequent genetic cause of FALS.
A common feature of both inherited and sporadic ALS is the accumulation of abnormal proteinaceous inclusions in motor neurons and glial cells. The major protein component of these accumulations in familial cases with SOD1 mutations [32], as well as in mutant SOD1 mouse models [8, 48, 70], is SOD1 itself. In sporadic forms of disease, protein misaccumulation is a prominent feature but the primary component of these aggregates is thought to be TDP-43 [1, 20, 46]. However, in SOD1-mediated disease TDP-43 aggregates are rarely observed [42, 43, 58, 64].
ALS-linked mutations in SOD1 have been shown to lead to an increased propensity to misfold and/or aggregate [30, 45, 55, 63, 67, 69], suggesting that SOD1 misfolding and aggregation play a preponderant role in disease pathology. In support of this, misfolded SOD1 has been detected in SOD1 FALS [18, 19, 39, 40, 56] and mutant SOD1 mouse models [25, 48,49,50, 56, 60], accumulating preferentially in spinal cord motor neurons. These findings were made possible by the use of antibodies specific for monomeric or misfolded SOD1 that do not bind to the natively folded protein.
It has been reported that certain posttranslational modifications of wild-type SOD1 (SOD1WT), including oxidation [6, 12, 16, 26, 27, 37] and demetallation [47] induce its misfolding in vitro, suggesting that even SOD1WT can acquire toxic properties like those associated with ALS-linked mutants of SOD1. In transgenic mice, ~5–20-fold higher levels of SOD1WT expression cause SOD1 aggregation and neuronal pathology in the CNS in aged mice [24, 35, 36], whereas expressing low levels of human SOD1WT does not result in ALS-like disease [9, 14, 28]. Co-expression of moderate levels of SOD1WT with the ALS-linked mutant SOD1G85R does not exacerbate ALS-like disease in mice [2, 9, 72]. However, expressing high levels of human SOD1WT in mutant SOD1G85R transgenic mice accelerates onset of disease suggesting the human SOD1WT protein itself can be toxic at high concentrations or can enhance the toxic properties of the mutant protein [15, 54, 71]. Altogether this suggests that alterations in SOD1WT protein folding induce a gain of toxic function that may implicate SOD1 in the pathology of ALS in patients without SOD1 mutations. Whether there is mechanistic similarity between SALS and SOD1 mutant-mediated disease is not established.
Several independent groups have reported that misfolded SOD1 is not detected in spinal cords [3, 7, 39, 40], lymphoblasts [49] or CSF [73] from SALS patients using conformational specific antibodies recognizing aberrantly folded SOD1. This view has been challenged by studies reporting misfolded SOD1 accumulation in motor neurons [6, 19, 22, 53] and glia [18] of spinal cords as well as in peripheral blood mononuclear cells [11] and lymphocytes [27] from SALS patients. Neural precursor cell (NPC)- [29] or patient- [57] derived astrocytes, as well as NPC-derived oligodendrocyte progenitors [17] from both SOD1 mutant or sporadic ALS patients have been reported to be toxic to co-cultured normal motor neurons. It is controversial whether this toxicity is [17, 29] or is not [57] significantly diminished upon shRNA-mediated reduction in SOD1WT, thereby leaving unsettled whether toxicity from sporadic ALS-derived astrocytes is, at least in part, through SOD1WT. Resolving this question is critical to identifying mechanistic insights into the pathogenesis of SALS and could have important implications for therapy development in ALS, including extending to SALS ongoing clinical trial using antisense oligonucleotide therapy to reduce SOD1 levels in SOD1 mutant-caused ALS [44, 61].
Here we have undertaken large-scale, unbiased analyses for accumulation of misfolded SOD1 in tissue samples from over 50 high quality human SALS autopsies using immunofluorescence/immunohistochemistry and immunoprecipitation with seven different monoclonal and polyclonal antibodies raised by independent investigators against differing epitopes exposed only in misfolded human SOD1. They include the C4F6 [25], B8H10 [25, 34], DSE2 3H1 [23, 33, 53, 68], DSE2 10E11 [23] and 131-153Ra [19] antibodies which were previously shown to be highly specific for misfolded human SOD1 (reviewed in [51]). In addition, two other conformationally sensitive monoclonal antibodies which have stronger binding to misfolded SOD1 compared to each of the five previously published antibodies were tested. Using this comprehensive approach, we find no evidence of SOD1 misfolding in the spinal cord or cortex of patients with sporadic or non-SOD1 inherited forms of ALS.
Materials and methods
Animals
SOD1G93A and their non-transgenic littermates have been previously described [31]. End-stage disease was defined when animals could not right themselves when placed on their side, in compliance with the requirements of the Animal Care and Use Committee of the University of California.
Human samples
Human control, FALS and SALS paraffin-embedded spinal cord and brain tissues for immunohistochemistry/immunofluorescence analyses and non-fixed frozen tissues embedded in OCT for immunoprecipitation were obtained through an Institutional Review Board compliant process for Human Subjects Research. Appropriate demographics and clinical information for the patient cases analyzed throughout this study are summarized in Supplementary Table 2.
Immunohistochemistry
Spinal cord sections: Four micrometer tissue sections were cut from blocks of formalin-fixed paraffin embedded spinal cords from ALS patients and stained using a fully automated Ventana Discovery Ultra (Ventana Medical Systems, Tucson, AZ, USA) which allows for online deparaffinization as well as very tight control of antigen retrieval conditions. Each misfolded SOD1 antibody was carefully evaluated with regards to retrieval conditions optimizing the signal-to-noise ratio of staining and the minimal amount of retrieval that was needed for each antibody was used to prevent non-specific staining. Deparaffinization was done by heating the slides to 68 °C for 3 cycles of 4 min each in the presence of the Ventana EZ solution. Antigen retrieval was done using either a Tris–EDTA based solution (CC1) or citrate-based (CC2). Retrieval varied from 12 to 28 min (see table below). Sections were incubated sequentially with an avidin–biotin blocking system. The primary antibodies were incubated on the sections for 1 h at 37 °C followed by a biotinylated Donkey anti-rabbit or mouse secondary antibody (Jackson ImmunoResearch) for 32 min at 37 °C. Primary antibodies were visualized using the DABMap system (Ventana Medical Systems) with DAB as a chromagen. In some cases, the chromagen was followed by hematoxylin as a counterstain. Slides were rinsed, dehydrated through alcohol and xylene and coverslipped. Imaging was performed on Nanozoomer slide scanner (Hamamatsu) at the UCSD microscopy core.
Antibody | Ventana retrieval | Working dilution |
|---|---|---|
4A1 | CC1; 28 min | 1/60,000 |
A5E5 | CC1; 28 min | 1/60,000 |
B8H10 | CC1; 28 min | 1/3000 |
131-153 Ra | CC1; 12 min | 1/3000 |
10E11 | CC1; 12 min | 1/15,000 |
3H1 | CC2; 28 min | 1/15,000 |
Cortical sections: Six micrometer tissue sections were cut from blocks of formalin-fixed paraffin embedded cortices from ALS patients and deparaffinized with CitriSolv (Fisher) and hydrated with a graded alcohol series. Endogeneous peroxidase activity was quenched with 0.06% H2O2, 15 min. Antigen retrieval with high pH solution (Vector) in a pressure cooker was done for 20 min. Sections were blocked with 1% FBS (Atlanta Biologicals) for 25 min prior incubating overnight in primary monoclonal misfolded SOD1 antibodies: B8H10 (1/1000; MediMabs), 4A1 (1/10,000; kindly provided by Biogen), and A5E5 (1/10,000; kindly provided by Biogen). Secondary anti-mouse antibody (ImmPRESS reagent kit, anti-Mouse, Vector) was then incubated for 60 min at room temperature and revealed using NovaRed (Vector) for 1 min. Counterstaining with Hematoxylin (Fisher) was also done in some of the sections.
Different types of immunoreactive species detected with each conformationally sensitive antibody by IHC in patient spinal cords were scored with a semi-quantitative arbitrary scale we devised to portray the intensity of the immunoreactive signals. Globular inclusions containing misfolded SOD1-immunopositive aggregates were scored 100 to emphasize the significant and distinct immunoreactivity compared with all the other signals. Round deposits were scored 4 or 5 depending on their frequency, diffuse faint granular cytoplasmic staining was scored 3, and rare or more frequent faint cytoplasmic diffuse staining was scored 2 or 3, respectively. No immunoreactivity was scored as 0. Immunohistochemical images were independently scored by three observers who were blinded to the sample identity.
Immunofluorescence
Tissues were de-paraffinized through histology grade CitriSolv (three times for 5 min each) and a graded alcohol series (100, 95 and 70% ethanol (vol/vol) twice for 5 min each). After a 10 min permeabilization step in 1 × PBS, 0.2% TritonX100, antigen retrieval (10 mM Citrate buffer, pH 6.0, in pressure cooker at 120 °C for 20 min) was applied to the sections. This antigen retrieval step was skipped when the effects of the absence of antigen retrieval on the immunostaining were being tested. Sections were further blocked with 10% normal goat serum (vol/vol, Jackson ImmunoResearch) and incubated with misfolded SOD1 antibodies (C4F6 (1/200) and B8H10 (1/1000; MediMabs) and 3H1 (1:10,000; gift from Dr. Cashman) with or without rabbit TDP-43 (1/50; Proteintech #10782-2) prepared in antibody diluent (Dako) overnight at 4 °C. Sections were then incubated with biotinylated secondary antibodies (Jackson ImmunoResearch) followed by streptavidin-FITC (Invitrogen). To reduce autofluorescence noise, quenching with 0.1% Sudan Black in 70% EtOH for 5–10 s was applied prior to coverslip mounting with Prolong (Invitrogen). Imaging was performed on a Nikon Eclipse laser scanning confocal microscope.
Immunoprecipitation
Spinal cords from end-stage mutant SOD1G93A rats or their aged-matched littermates (5 months of age) were homogenized in cold 1 × PBS buffer plus protease inhibitors (Roche Diagnostics). The lysates were centrifuged for 10 min at 1000 g and the resulting supernatants (clarified tissue extract) were further incubated in the immunoprecipitation (IP) buffer (1 × PBS, 0.5% TritonX100 with protease inhibitors) at 4 °C for 20 h with misfolded SOD1 monoclonal B8H10 (MediMabs), DSE2 3H1 (gift from Dr. Cashman), 4A1 and A5E5 (gift from Biogen) or polyclonal 131-153Ra antibody (gift from Dr. Brännström), previously crosslinked to Dynabeads protein G (Invitrogen) with dimethyl pimelimidate (Pierce) according to the manufacturer’s instructions. The beads were magnetically isolated and washed three times with IP buffer. Samples were eluted with boiling in 2.0× sample buffer with dithiothreitol. Immunoprecipitated proteins were separated by SDS-PAGE, transferred to nitrocellulose membranes, and probed with the indicated antibodies followed by horseradish peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch). Pico or Femto ECL (Pierce) was used to detect immunoreactive bands. Primary antibodies against SOD1 (C-17; Santa Cruz Biotechnology or SOD1-100; Enzo Life Sciences), GAPDH (Abcam), were used for immunoblotting.
OCT embedded non-fixed frozen spinal cords from patients were used as starting material for immunoprecipitation. Prior to tissue homogenization the OCT was scraped out (while tissues were still kept frozen on dry ice) and spinal cords were then thawed and rinsed on cold 1 × PBS on ice. Spinal cords were rapidly weighed and homogenized as described above in 10% weight/volume of cold PBS supplemented with protease inhibitors.
Results
Multiple conformationally sensitive antibodies identify misfolded SOD1 aggregates in spinal cords of SOD1A4V patients
To test whether misfolded SOD1 accumulates in sporadic ALS, we selected six conformationally sensitive antibodies (Supplementary Table 1) for their ability to specifically detect misfolded human SOD1 conformers by immunoprecipitation. We initially compared their efficiency and specificity in spinal cords from an ALS rat model expressing ALS-linked mutation of human SOD1G93A (Fig. 1a–c) and from human patients carrying the most frequent mutation in human SOD1 in the United States (alanine substituted to valine at position 4, known as A4V) (Fig. 1a, b, d). Four different antibodies raised against misfolded SOD1 were used for immunoprecipitation starting from whole tissue extracts of freshly frozen, unfixed spinal cords from diseased SOD1G93A rats or aged-matched non-transgenic (Ntg) littermates (Fig. 1b, c). Commercially available B8H10 [25], A5E5 and 4A1 antibodies immunoprecipitated 5-, 10- and 12-fold more misfolded SOD1, respectively, compared to the previously published 3H1 DSE2 antibody [68]. While all four antibodies immunoprecipitated SOD1 from extracts of spinal cord from human patients with the SOD1A4V mutation, the A5E5 and 4A1 antibodies were more efficient compared to B8H10 and 3H1 DSE2 (Fig. 1d). The higher level of misfolded SOD1 immunoprecipitated from the rat spinal cords compared to that from ALS A4V patients correlated with the higher levels of mutant SOD1 expressed in the transgenic animals.

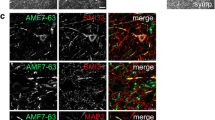
Misfolded SOD1 positive aggregates are immunodetected in spinal cords of FALS SOD1 patients using multiple conformationally sensitive antibodies against misfolded SOD1 species. a Scheme depicting the method used to immunoprecipitate misfolded SOD1 from rat or human samples. Immunoblots demonstrating b total SOD1 levels (as the 10% of input) in spinal cord from human patients or rats, immunoprecipitated misfolded SOD1 using A5E5, 4A1, 3H1 DSE2, B8H10 antibodies or mouse immunoglobulins (mIgG; as a negative control) from spinal cords of c non-transgenic (Ntg) and transgenic rats expressing mutant SOD1G93A (G93A) that develop fatal paralytic ALS-like disease or d healthy control (Ctrl65) or FALS human patient carrying the SOD1A4V mutation (A4V24). GAPDH was used as a loading control. e Immunochemical analysis of misfolded SOD1 in two FALS with SOD1A4V mutation (#15 and #24) and two non-neurological control patients (CTRL #19 and #44) using the 10E11 DSE2, B8H10, 131-153Ra, 4A1 and A5E5 antibodies. Arrows indicate the presence of globular inclusions or intense positive aggregates of misfolded SOD1. Arrowheads point to immunoreactive round deposits found randomly, more frequently outside cells. The asterisk signs indicate cytoplasmic diffuse or granular signal in neurons, respectively. Scale bar 50 µm. f Immunofluorescence analysis of misfolded SOD1 in FALS with SOD1A4V mutation (#24) and non-neurological control (#44) patients using the B8H10, 3H1 DSE2 and C4F6 antibodies (green). TDP-43 antibody (red) was used as a control marker to ascertain quality of tissue. Scale bar 50 µm. Dashed white lines outline motor neurons
To confirm the presence and to identify the location of misfolded SOD1, we utilized immunohistochemistry (IHC) and immunofluorescence (IF) using B8H10, polyclonal 131-153Ra, 4A1, A5E5, C4F6 antibodies, and an additional antibody recognizing the DSE2 epitope (10E11 and 3H1 clones) (Fig. 1e, f, Supplementary Fig. 1). Each of these antibodies detected misfolded SOD1-immunopositive aggregates reminiscent of globular inclusions in motor neuron perikarya and neurite processes of spinal cords from human patients carrying the SOD1A4V mutation. None identified any such structure in samples from a non-neurological patient control. In control patients, faint diffuse, granular, or with dense round deposits identified to be corpora amylacea in neuronal cytoplasm were observed by IHC using each of these antibodies (Fig. 1e). In all cases nuclear TDP-43 immunostaining was detected as expected in our collection of human spinal cord samples, thus validating that the quality of the sections was not compromised (Fig. 1f, Supplementary Fig. 1).
Misfolded SOD1 aggregates are not in sporadic ALS tissues using conformationally sensitive antibodies that detect misfolded SOD1 conformers
We used the conformationally sensitive antibodies to SOD1 (Supplementary Table 1) validated in Fig. 1 in immunoprecipitation (Fig. 2a–d) and IHC (Figs. 2e, f, 3, 4, 5) to test for misfolded SOD1 in a large bank of short post-mortem interval human autopsied samples (Supplementary Table 2). Misfolded SOD1 was not specifically immunodetected in spinal cords from SALS patients when compared to control patients after immunoprecipitation with 3H1 DSE2 antibody (Fig. 2a). In addition, no misfolded SOD1 was observed in spinal cords from 10 different SALS patients compared to 7 control samples when the 4A1 and A5E5 antibodies were used for immunoprecipitation, although these two antibodies were able to bind misfolded SOD1 with higher affinity compared to the other antibodies tested in this study (Fig. 1b–d). Of note, misfolded SOD1 was not detected in FALS patients carrying mutations in a gene other than SOD1, namely C9ORF72 (Fig. 2c; patients #13 and #14). A faint band with a size similar to that of SOD1 was detected at comparable levels in some control or SALS patients after immunoprecipitation with either misfolded SOD1 antibody. Importantly this band was also present in some control or SALS patients when the immunoprecipitation was performed with control IgGs, suggesting that it is non-specific (Supplementary Fig. 2).

No misfolded SOD1 is immunodetected in SALS patients using multiple conformationally sensitive antibodies against misfolded SOD1 species. Immunoblots demonstrating immunoprecipitated misfolded SOD1 using (a) 3H1, (b) 4A1, (c, d) A5E5 antibodies in spinal cords from FALS with SOD1A4V mutation (#15, 24 or 101), FALS with no mutations in SOD1 (#13 [n.i.: genotype not identified]; or #14 [C9:C9ORF72 repeat expansion]), non-neurological control (CTRL #65, 31, 42, 19, 23, 39, 83) and SALS (#11, 18, 33, 35, 36, 16, 17, 22, 32, 6) patients. Total levels of SOD1 and loading control GAPDH are shown as 10% of input. e Immunochemical analysis of misfolded SOD1 in two FALS with SOD1A4V mutation (#15 and #101), two non-neurological controls (#20 and #31) and two SALS (#17 and #64) patients using the 10E11 DSE2, B8H10, 131-153Ra, 4A1 and A5E5 antibodies. Arrows indicate the presence of globular inclusions or intense positive aggregates of misfolded SOD1. Arrowheads point to immunoreactive round deposits found randomly, more frequently outside cells. The cross or asterisk signs indicate cytoplasmic diffuse or granular signal in neurons, respectively. Scale bar 50 µm. f Semi-quantification in controls and SALS of the different types of immunoreactive species detected with each conformationally sensitive antibody by IHC in patient spinal cords was scored according to the presence of globular inclusions (found only in FALS samples with SOD1 mutations), rare or frequent round deposits (lower or upper blue squares, respectively), cytoplasmic granular staining (green triangle), sparse or frequent cytoplasmic diffuse staining (lower or upper grey triangles, respectively) or no immunoreactivity (orange diamonds). Each symbol corresponds to one patient

No misfolded SOD1 is immunodetected in SALS patients using the 10E11 DSE2 misfolded SOD1 antibody. Immunochemical analysis of misfolded SOD1 in two FALS patients with SOD1A4V mutation (#15 and #24), two with expansion repeats in C9ORF72 gene (#14 and #81), eight non-neurological controls (#19, 37, 83, 44, 23, 31, 20 and 65) and twelve SALS (#63, 60, 82, 6, 64, 68, 36, 35, 32, 17, 11, 16) using the 10E11 DSE2 antibody. Arrows indicate the presence of globular inclusions or intense positive aggregates of misfolded SOD1. Arrowheads point to immunoreactive round deposits found randomly, more frequently outside cells. The cross or asterisk signs indicate cytoplasmic diffuse or granular signal in neurons, respectively. Scale bar 100 µm

No misfolded SOD1 is immunodetected in SALS patients using the monoclonal B8H10 or polyclonal 131-153Ra misfolded SOD1 antibodies. Immunochemical analysis of misfolded SOD1 in two FALS patients with SOD1A4V mutation (#15 and #24), two with expansion repeats in C9ORF72 gene (#14 and #81), two non-neurological controls (#19and #44) and nine SALS (#16, 17, 35, 36, 41, 66, 6, 48 and 82) using the B8H10 (two left panels) or 131-153Ra (two right panels) antibodies. Arrows indicate the presence of globular inclusions or intense positive aggregates of misfolded SOD1. Arrowheads point to immunoreactive round deposits found randomly, more frequently outside cells. The cross or asterisk signs indicate cytoplasmic diffuse or granular signal in neurons, respectively. Scale bar 100 µm

No misfolded SOD1 is immunodetected in SALS patients using antibodies with increased binding to misfolded SOD1 (4A1 and A5E5). Immunochemical analysis of misfolded SOD1 in three FALS patients with SOD1A4V mutation (#15, 24 and 101), two with expansion repeats in C9ORF72 gene (#14 and #81), six non-neurological controls (#19, 31, 42, 44, 65, 20) and four SALS (#16, 22, 33 and 35) using the 4A1 (two left panels) or A5E5 (two right panels) antibodies. Arrows indicate the presence of globular inclusions or intense positive aggregates of misfolded SOD1. Arrowheads point to immunoreactive round deposits found randomly, more frequently outside cells. The cross or asterisk signs indicate cytoplasmic diffuse or granular signal in neurons, respectively. Scale bar 50 µm
Although misfolded SOD1 was undetectable in whole spinal cord extracts from SALS patients using conformationally sensitive antibodies that detect misfolded SOD1 conformers with high affinity, we reasoned that it was possible that misfolded conformers may be present at levels below detection by immunoprecipitation but enriched in a small subset of spinal cord cells such as motor neurons, which make up less than 10% of the overall number of cells within spinal cords. To test this possibility, we exploited IHC and immunofluorescence using our panel of conformationally sensitive antibodies on lumbar, thoracic and cervical regions of spinal cords in a total of 7 FALS, 14 control and 30 SALS patients (Supplementary Table 3). Consistent with our other findings, misfolded SOD1 positive inclusions primarily in neuronal perikarya and neuronal processes were consistently immunodetected in spinal cords of the three FALS patients carrying the SOD1A4V mutation with antibodies DSE2 10E11 or 3H1 (Figs. 2e, f, 3; Supplementary Table 3), B8H10 and 131-153Ra (Figs. 2e, f, 4; Supplementary Table 3), 4A1 and A5E5 (Figs. 2e, f, 5; Supplementary Table 3) and C4F6 (Supplementary Table 3). In contrast, no immunoreactive signal was found in any spinal cord region of any of the SALS patients (DSE2 antibody-Figs. 2e, f, 3), B8H10 and 131-153Ra antibodies-Figs. 2e, f, 4) and 4A1 and A5E5 antibodies-Figs. 2e, f, 5). Faint diffuse, granular immunoreactivity which appeared in neuronal cytoplasm or with dense round deposits was indistinguishable between SALS and control patients (Figs. 2e, f, 3, 4, 5; Supplementary Table 3). Additionally, immunofluorescence performed in the absence of commonly used antigen retrieval approaches also did not reveal specific signal in SALS patient samples stained with misfolded SOD1 3H1 DSE2 or C4F6, indicating antigen retrieval could not explain the absence of signal (Supplementary Fig. 3; Supplementary Table 3).
Immunoreactive species detected with each conformationally sensitive antibody by IHC in patient spinal cords (shown in Fig. 2e) were quantified using a semi-quantitative scale (Fig. 2f). Misfolded SOD1 containing globular inclusions (red circles), which had the most intense and distinctive staining were detected only in patients carrying mutations in SOD1 but not in SALS patients, FALS patients with non-SOD1 mutations, or non-neurological controls. The three classes of fainter immunoreactive species (round deposits (blue squares), granular (green triangles) or diffuse (grey triangles) cytoplasmic staining) were identified equally in SALS, non-SOD1 FALS and non-neurological control patients (Fig. 2f; Supplementary Table 3). Similarly, misfolded SOD1 was uniquely (but rarely) detected by IHC in the Betz cells of cortical layer 5 of patients with the SOD1A4V mutation but not in control or SALS patients (Fig. 6; Supplementary Table 3).

No misfolded SOD1 is immunodetected in the cortex of ALS patients using multiple antibodies. Immunochemical analysis of misfolded SOD1 in two FALS patients with SOD1A4V mutation (#15 and 24), two non-neurological controls (#65 and #83) and two SALS (#48 and #82) using the B8H10, 4A1 and A5E5 antibodies. Arrows indicate the presence of globular inclusions or positive aggregates of misfolded SOD1
Altogether, no misfolded SOD1 immunopositive signal was detected with any of the seven misfolded SOD1 antibodies (Supplementary Table 1), tested in SALS patient samples regardless of age/site of onset, disease duration (Supplementary Table 2), region of the CNS, immunodetection approach (IHC or IF) (Supplementary Table 3).
Discussion
Misfolded SOD1 has been proposed as a component of disease in sporadic ALS patients [6, 7, 18, 19, 23, 53], in large part based on detection of misfolded SOD1 in the CNS of SALS patients using conformationally sensitive antibodies for misfolded SOD1. However, this evidence has been challenged by independent reports which failed to detect misfolded SOD1 in the CNS of sporadic ALS patients using a similar approach [4, 39, 40]. In light of the importance that resolving this controversy may have on identifying new therapeutic approaches to treat ALS, we used a comprehensive, unbiased approach using immunofluorescence/immunohistochemistry and immunoprecipitation with seven independent antibodies raised against different epitopes specific to misfolded human SOD1. With these, we tested whether misfolded SOD1 is present in the CNS of a large cohort of high quality autopsied samples (including over 40 FALS and SALS patients). In multiple samples from ALS patients with SOD1 mutations, this panel of antibodies consistently detected the presence of SOD1 globular inclusions in anterior horn spinal cord motor neurons (cell body and processes) and less abundantly in Betz cells of cortex layer 5. However, using these same antibodies, we found no evidence of this pattern of misfolded SOD1 accumulation in non-SOD1 FALS, SALS, or control tissues. Therefore, our analysis, including five antibodies whose prior use [6, 7, 18, 19, 23, 53] had been interpreted to indicate that misfolded SOD1 may be present in SALS, refutes that misfolded human SOD1 accumulates in the CNS of sporadic ALS patients. We conclude, therefore, that misfolding of SOD1 is unlikely to contribute to disease pathogenesis in SALS.
Prior studies reporting the presence of misfolded SOD1 in motor neurons [6, 19, 53], axons [23] and glia [18] of SALS patients relied primarily on immunohistochemistry using a single misfolded SOD1 antibody and in three of the five reports no controls were shown [19, 23, 53]. Comparison of the data between these different studies reveals a high degree of inconsistency in the staining patterns attributed to misfolded SOD1. It is described as diffuse in the neuronal cytoplasm [6], granular in neuronal cell bodies [19], and even as deposits in the extracellular space [18, 19, 53]. The immunoreactive signal attributed to misfolded SOD1 in a SALS sample from Forsberg et al. [19] is identical to what we observe with the same antibody in multiple samples from our control individuals. Moreover, some of this putative immunoreactive signal resembles corpora amylacea structures, glycoproteinacous inclusions frequently found in the CNS tissues of elderly people [10, 38, 52], including both non-neurological control and SALS patients [4]. Such structures have been reported to immunostain for multiple proteins including ubiquitin, heme oxygenase, Mn SOD1, and alpha-synuclein [62], and do not represent disease-specific pathology.
Divergent outcomes of studies [4, 6, 7, 18, 19, 23, 39, 40, 53] for the detection of misfolded SOD1 in SALS patient samples were proposed to be due to variations in the IHC conditions used by the different groups. In particular, Bosco et al. [6] reported that antigen retrieval approaches prevent detection of immunoreactive misfolded SOD1 in the motor neuron cell bodies of SALS patients using the C4F6 antibody. With or without antigen retrieval methods, we found no evidence of misfolded SOD1 immunostaining in SALS patient samples regardless of the staining protocol used not only for the C4F6 antibody but also for the remaining six antibodies.
Nevertheless, it is certainly true that immunohistochemical analyses of autopsied human tissues following typical fixation may hide (or more rarely expose) antibody epitopes. Therefore, to verify that the absence of misfolded SOD1 in nervous systems from SALS patient may reflect fixation-mediated epitope masking, we examined a large number of unfixed human autopsied samples that were freshly collected and then subjected to immunoprecipitation with three different antibodies. Whereas misfolded SOD1 was consistently immunoprecipitated in SOD1-positive FALS, we did not detect misfolded SOD1 in immunoprecipitates from 10 SALS and 7 control patients by immunoprecipitation using the 3H1, and two additional misfolded SOD1 antibodies with higher binding to SOD1 (4A1 and A5E5). A faint immunoreactive band migrating at the same molecular weight as SOD1 was detected in some control and SALS patients but also when the immunoprecipitation was performed with control IgGs, indicating that this band is non-specific. Two prior teams had attempted to immunoprecipitate misfolded SOD1 from SALS patients, but the conclusions drawn from these studies diverged sharply [23, 39, 40]. Grad et al. [23] reported accumulation of misfolded SOD1 immunoprecipitated in four SALS and four FALS patients compared to six controls using two DSE monoclonal antibodies (including the 3H1 that we also tested by immunoprecipitation). However, the amounts of misfolded SOD1 that were immunoprecipitated were normalized to the total amounts of SOD1 present in each sample, and therefore, the starting conditions of immunoprecipitation were not comparable as higher amounts of homogenates from SALS and FALS were used to compensate for the proposed reduced levels of SOD1. Recognizing that, we believe the evidence actually strongly supports the conclusion that misfolded SOD1 is not accumulated to level above that found in non-ALS tissues.
Mutant SOD1 has been reported to be secreted in vitro [5, 21, 23, 45, 66], in mouse models [41, 65] and is detected in CSF from patients with SOD1 mutations [13]. Whether wild-type SOD1 is also released into the extracellular space is not established. Urushitani et al. [66] reported that secreted mutant SOD1 caused microgliosis and neuronal death, while SOD1WT induced suppression of microglial activation in cell culture. Grad et al. [23] proposed that SOD1WT is exported from cells that have been exposed to mutant SOD1 but did not test whether naïve cells under stress conditions would release misfolded SOD1WT. In CSF from ALS patients, misfolded SOD1 (scored by ELISA using polyclonal misfolded SOD1 antibodies) has been detected at very low levels not just in FALS and SALS samples, but also controls [73], thus arguing against a direct cytotoxic role of misfolded SOD1 specifically in sporadic ALS.
In this study using different methods to detect misfolded SOD1, multiple misfolded SOD1 antibodies raised against different epitopes, appropriate parallel controls and by controlling for staining artefacts through the comparison of different antigen retrieval methods, we demonstrate that misfolded SOD1 is not a contributing component of sporadic ALS.
References
Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y et al (2006) TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 351:602–611
Audet JN, Gowing G, Julien JP (2010) Wild-type human SOD1 overexpression does not accelerate motor neuron disease in mice expressing murine Sod1 G86R. Neurobiol Dis 40:245–250. doi:10.1016/j.nbd.2010.05.031
Ayers JI, Fromholt S, Koch M, DeBosier A, McMahon B, Xu G, Borchelt DR (2014) Experimental transmissibility of mutant SOD1 motor neuron disease. Acta Neuropathol 128:791–803. doi:10.1007/s00401-014-1342-7
Ayers JI, Xu G, Pletnikova O, Troncoso JC, Hart PJ, Borchelt DR (2014) Conformational specificity of the C4F6 SOD1 antibody; low frequency of reactivity in sporadic ALS cases. Acta Neuropathol Commun 2:55. doi:10.1186/2051-5960-2-55
Basso M, Pozzi S, Tortarolo M, Fiordaliso F, Bisighini C, Pasetto L, Spaltro G, Lidonnici D, Gensano F, Battaglia E et al (2013) Mutant copper-zinc superoxide dismutase (SOD1) induces protein secretion pathway alterations and exosome release in astrocytes: implications for disease spreading and motor neuron pathology in amyotrophic lateral sclerosis. J Biol Chem 288:15699–15711. doi:10.1074/jbc.M112.425066
Bosco DA, Morfini G, Karabacak NM, Song Y, Gros-Louis F, Pasinelli P, Goolsby H, Fontaine BA, Lemay N, McKenna-Yasek D et al (2010) Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat Neurosci 13:1396–1403. doi:10.1038/nn.2660
Brotherton TE, Li Y, Cooper D, Gearing M, Julien JP, Rothstein JD, Boylan K, Glass JD (2012) Localization of a toxic form of superoxide dismutase 1 protein to pathologically affected tissues in familial ALS. Proc Natl Acad Sci U S A 109:5505–5510. doi:10.1073/pnas.1115009109
Bruijn LI, Becher MW, Lee MK, Anderson KL, Jenkins NA, Copeland NG, Sisodia SS, Rothstein JD, Borchelt DR, Price DL et al (1997) ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron 18:327–338
Bruijn LI, Houseweart MK, Kato S, Anderson KL, Anderson SD, Ohama E, Reaume AG, Scott RW, Cleveland DW (1998) Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science 281:1851–1854
Cavanagh JB (1999) Corpora-amylacea and the family of polyglucosan diseases. Brain Res Brain Res Rev 29:265–295
Cereda C, Leoni E, Milani P, Pansarasa O, Mazzini G, Guareschi S, Alvisi E, Ghiroldi A, Diamanti L, Bernuzzi S et al (2013) Altered intracellular localization of SOD1 in leukocytes from patients with sporadic amyotrophic lateral sclerosis. PLoS One 8:e75916. doi:10.1371/journal.pone.0075916
Chattopadhyay M, Nwadibia E, Strong CD, Gralla EB, Valentine JS, Whitelegge JP (2015) The disulfide bond, but not zinc or dimerization, controls initiation and seeded growth in amyotrophic lateral sclerosis-linked Cu, Zn superoxide dismutase (SOD1) fibrillation. J Biol Chem 290:30624–30636. doi:10.1074/jbc.M115.666503
Crisp MJ, Mawuenyega KG, Patterson BW, Reddy NC, Chott R, Self WK, Weihl CC, Jockel-Balsarotti J, Varadhachary AS, Bucelli RC et al (2015) In vivo kinetic approach reveals slow SOD1 turnover in the CNS. J Clin Investig 125:2772–2780. doi:10.1172/JCI80705
Dal Canto MC, Gurney ME (1995) Neuropathological changes in two lines of mice carrying a transgene for mutant human Cu, Zn SOD, and in mice overexpressing wild type human SOD: a model of familial amyotrophic lateral sclerosis (FALS). Brain Res 676:25–40
Deng HX, Shi Y, Furukawa Y, Zhai H, Fu R, Liu E, Gorrie GH, Khan MS, Hung WY, Bigio EH et al (2006) Conversion to the amyotrophic lateral sclerosis phenotype is associated with intermolecular linked insoluble aggregates of SOD1 in mitochondria. Proc Natl Acad Sci U S A 103:7142–7147. doi:10.1073/pnas.0602046103
Ezzi SA, Urushitani M, Julien JP (2007) Wild-type superoxide dismutase acquires binding and toxic properties of ALS-linked mutant forms through oxidation. J Neurochem 102:170–178. doi:10.1111/j.1471-4159.2007.04531.x
Ferraiuolo L, Meyer K, Sherwood TW, Vick J, Likhite S, Frakes A, Miranda CJ, Braun L, Heath PR, Pineda R et al (2016) Oligodendrocytes contribute to motor neuron death in ALS via SOD1-dependent mechanism. Proc Natl Acad Sci U S A 113:6496. doi:10.1073/pnas.1607496113
Forsberg K, Andersen PM, Marklund SL, Brannstrom T (2011) Glial nuclear aggregates of superoxide dismutase-1 are regularly present in patients with amyotrophic lateral sclerosis. Acta Neuropathol 121:623–634. doi:10.1007/s00401-011-0805-3
Forsberg K, Jonsson PA, Andersen PM, Bergemalm D, Graffmo KS, Hultdin M, Jacobsson J, Rosquist R, Marklund SL, Brannstrom T (2010) Novel antibodies reveal inclusions containing non-native SOD1 in sporadic ALS patients. PLoS One 5:e11552. doi:10.1371/journal.pone.0011552
Giordana MT, Piccinini M, Grifoni S, De Marco G, Vercellino M, Magistrello M, Pellerino A, Buccinna B, Lupino E, Rinaudo MT (2010) TDP-43 redistribution is an early event in sporadic amyotrophic lateral sclerosis. Brain Pathol 20:351–360. doi:10.1111/j.1750-3639.2009.00284.x
Gomes C, Keller S, Altevogt P, Costa J (2007) Evidence for secretion of Cu, Zn superoxide dismutase via exosomes from a cell model of amyotrophic lateral sclerosis. Neurosci Lett 428:43–46. doi:10.1016/j.neulet.2007.09.024
Grad LI, Pokrishevsky E, Silverman JM, Cashman NR (2014) Exosome-dependent and independent mechanisms are involved in prion-like transmission of propagated Cu/Zn superoxide dismutase misfolding. Prion 8:331–335. doi:10.4161/19336896.2014.983398
Grad LI, Yerbury JJ, Turner BJ, Guest WC, Pokrishevsky E, O’Neill MA, Yanai A, Silverman JM, Zeineddine R, Corcoran L et al (2014) Intercellular propagated misfolding of wild-type Cu/Zn superoxide dismutase occurs via exosome-dependent and -independent mechanisms. Proc Natl Acad Sci U S A 111:3620–3625. doi:10.1073/pnas.1312245111
Graffmo KS, Forsberg K, Bergh J, Birve A, Zetterstrom P, Andersen PM, Marklund SL, Brannstrom T (2013) Expression of wild-type human superoxide dismutase-1 in mice causes amyotrophic lateral sclerosis. Hum Mol Genet 22:51–60. doi:10.1093/hmg/dds399
Gros-Louis F, Soucy G, Lariviere R, Julien JP (2010) Intracerebroventricular infusion of monoclonal antibody or its derived Fab fragment against misfolded forms of SOD1 mutant delays mortality in a mouse model of ALS. J Neurochem 113:1188–1199. doi:10.1111/j.1471-4159.2010.06683.x
Gruzman A, Wood WL, Alpert E, Prasad MD, Miller RG, Rothstein JD, Bowser R, Hamilton R, Wood TD, Cleveland DW et al (2007) Common molecular signature in SOD1 for both sporadic and familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A 104:12524–12529. doi:10.1073/pnas.0705044104
Guareschi S, Cova E, Cereda C, Ceroni M, Donetti E, Bosco DA, Trotti D, Pasinelli P (2012) An over-oxidized form of superoxide dismutase found in sporadic amyotrophic lateral sclerosis with bulbar onset shares a toxic mechanism with mutant SOD1. Proc Natl Acad Sci U S A 109:5074–5079. doi:10.1073/pnas.1115402109
Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX et al (1994) Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science 264:1772–1775
Haidet-Phillips AM, Hester ME, Miranda CJ, Meyer K, Braun L, Frakes A, Song S, Likhite S, Murtha MJ, Foust KD et al (2011) Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat Biotechnol 29:824–828. doi:10.1038/nbt.1957
Hough MA, Grossmann JG, Antonyuk SV, Strange RW, Doucette PA, Rodriguez JA, Whitson LJ, Hart PJ, Hayward LJ, Valentine JS et al (2004) Dimer destabilization in superoxide dismutase may result in disease-causing properties: structures of motor neuron disease mutants. Proc Natl Acad Sci U S A 101:5976–5981. doi:10.1073/pnas.0305143101
Howland DS, Liu J, She Y, Goad B, Maragakis NJ, Kim B, Erickson J, Kulik J, DeVito L, Psaltis G et al (2002) Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS). Proc Natl Acad Sci U S A 99:1604–1609. doi:10.1073/pnas.032539299
Ince PG, Highley JR, Kirby J, Wharton SB, Takahashi H, Strong MJ, Shaw PJ (2011) Molecular pathology and genetic advances in amyotrophic lateral sclerosis: an emerging molecular pathway and the significance of glial pathology. Acta Neuropathol 122:657–671. doi:10.1007/s00401-011-0913-0
Israelson A, Arbel N, Da Cruz S, Ilieva H, Yamanaka K, Shoshan-Barmatz V, Cleveland DW (2010) Misfolded mutant SOD1 directly inhibits VDAC1 conductance in a mouse model of inherited ALS. Neuron 67:575–587. doi:10.1016/j.neuron.2010.07.019
Israelson A, Ditsworth D, Sun S, Song S, Liang J, Hruska-Plochan M, McAlonis-Downes M, Abu-Hamad S, Zoltsman G, Shani T et al (2015) Macrophage migration inhibitory factor as a chaperone inhibiting accumulation of misfolded SOD1. Neuron 86:218–232. doi:10.1016/j.neuron.2015.02.034
Jaarsma D, Haasdijk ED, Grashorn JA, Hawkins R, van Duijn W, Verspaget HW, London J, Holstege JC (2000) Human Cu/Zn superoxide dismutase (SOD1) overexpression in mice causes mitochondrial vacuolization, axonal degeneration, and premature motoneuron death and accelerates motoneuron disease in mice expressing a familial amyotrophic lateral sclerosis mutant SOD1. Neurobiol Dis 7:623–643
Jonsson PA, Graffmo KS, Brannstrom T, Nilsson P, Andersen PM, Marklund SL (2006) Motor neuron disease in mice expressing the wild type-like D90A mutant superoxide dismutase-1. J Neuropathol Exp Neurol 65:1126–1136. doi:10.1097/01.jnen.0000248545.36046.3c
Karch CM, Prudencio M, Winkler DD, Hart PJ, Borchelt DR (2009) Role of mutant SOD1 disulfide oxidation and aggregation in the pathogenesis of familial ALS. Proc Natl Acad Sci U S A 106:7774–7779. doi:10.1073/pnas.0902505106
Keller JN (2006) Age-related neuropathology, cognitive decline, and Alzheimer’s disease. Ageing Res Rev 5:1–13. doi:10.1016/j.arr.2005.06.002
Kerman A, Liu HN, Croul S, Bilbao J, Rogaeva E, Zinman L, Robertson J, Chakrabartty A (2010) Amyotrophic lateral sclerosis is a non-amyloid disease in which extensive misfolding of SOD1 is unique to the familial form. Acta Neuropathol 119:335–344. doi:10.1007/s00401-010-0646-5
Liu HN, Sanelli T, Horne P, Pioro EP, Strong MJ, Rogaeva E, Bilbao J, Zinman L, Robertson J (2009) Lack of evidence of monomer/misfolded superoxide dismutase-1 in sporadic amyotrophic lateral sclerosis. Ann Neurol 66:75–80. doi:10.1002/ana.21704
Liu HN, Tjostheim S, Dasilva K, Taylor D, Zhao B, Rakhit R, Brown M, Chakrabartty A, McLaurin J, Robertson J (2012) Targeting of monomer/misfolded SOD1 as a therapeutic strategy for amyotrophic lateral sclerosis. J Neurosci 32:8791–8799. doi:10.1523/JNEUROSCI.5053-11.2012
Mackenzie IR, Bigio EH, Ince PG, Geser F, Neumann M, Cairns NJ, Kwong LK, Forman MS, Ravits J, Stewart H et al (2007) Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann Neurol 61:427–434. doi:10.1002/ana.21147
Maekawa S, Leigh PN, King A, Jones E, Steele JC, Bodi I, Shaw CE, Hortobagyi T, Al-Sarraj S (2009) TDP-43 is consistently co-localized with ubiquitinated inclusions in sporadic and Guam amyotrophic lateral sclerosis but not in familial amyotrophic lateral sclerosis with and without SOD1 mutations. Neuropathol Off J Jpn Soc Neuropathol 29:672–683. doi:10.1111/j.1440-1789.2009.01029.x
Miller TM, Pestronk A, David W, Rothstein J, Simpson E, Appel SH, Andres PL, Mahoney K, Allred P, Alexander K et al (2013) An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: a phase 1, randomised, first-in-man study. Lancet Neurol 12:435–442. doi:10.1016/S1474-4422(13)70061-9
Munch C, O’Brien J, Bertolotti A (2011) Prion-like propagation of mutant superoxide dismutase-1 misfolding in neuronal cells. Proc Natl Acad Sci U S A 108:3548–3553. doi:10.1073/pnas.1017275108
Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM et al (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314:130–133
Oztug Durer ZA, Cohlberg JA, Dinh P, Padua S, Ehrenclou K, Downes S, Tan JK, Nakano Y, Bowman CJ, Hoskins JL et al (2009) Loss of metal ions, disulfide reduction and mutations related to familial ALS promote formation of amyloid-like aggregates from superoxide dismutase. PLoS One 4:e5004. doi:10.1371/journal.pone.0005004
Parone PA, Da Cruz S, Han JS, McAlonis-Downes M, Vetto AP, Lee SK, Tseng E, Cleveland DW (2013) Enhancing mitochondrial calcium buffering capacity reduces aggregation of misfolded SOD1 and motor neuron cell death without extending survival in mouse models of inherited amyotrophic lateral sclerosis. J Neurosci 33:4657–4671. doi:10.1523/JNEUROSCI.1119-12.2013
Pickles S, Destroismaisons L, Peyrard SL, Cadot S, Rouleau GA, Brown RH Jr, Julien JP, Arbour N, Vande Velde C (2013) Mitochondrial damage revealed by immunoselection for ALS-linked misfolded SOD1. Hum Mol Genet 22:3947–3959. doi:10.1093/hmg/ddt249
Pickles S, Semmler S, Broom HR, Destroismaisons L, Legroux L, Arbour N, Meiering E, Cashman NR, Vande Velde C (2016) ALS-linked misfolded SOD1 species have divergent impacts on mitochondria. Acta Neuropathol Commun 4:43. doi:10.1186/s40478-016-0313-8
Pickles S, Vande Velde C (2012) Misfolded SOD1 and ALS: zeroing in on mitochondria. Amyotroph Later Scler 13:333–340. doi:10.3109/17482968.2012.648645
Pirici I, Margaritescu C, Mogoanta L, Petrescu F, Simionescu CE, Popescu ES, Cecoltan S, Pirici D (2014) Corpora amylacea in the brain form highly branched three-dimensional lattices. Rom J Morphol Embryol 55:1071–1077
Pokrishevsky E, Grad LI, Yousefi M, Wang J, Mackenzie IR, Cashman NR (2012) Aberrant localization of FUS and TDP43 is associated with misfolding of SOD1 in amyotrophic lateral sclerosis. PLoS One 7:e35050. doi:10.1371/journal.pone.0035050
Prudencio M, Durazo A, Whitelegge JP, Borchelt DR (2010) An examination of wild-type SOD1 in modulating the toxicity and aggregation of ALS-associated mutant SOD1. Hum Mol Genet 19:4774–4789. doi:10.1093/hmg/ddq408
Rakhit R, Crow JP, Lepock JR, Kondejewski LH, Cashman NR, Chakrabartty A (2004) Monomeric Cu, Zn-superoxide dismutase is a common misfolding intermediate in the oxidation models of sporadic and familial amyotrophic lateral sclerosis. J Biol Chem 279:15499–15504. doi:10.1074/jbc.M313295200
Rakhit R, Robertson J, Vande Velde C, Horne P, Ruth DM, Griffin J, Cleveland DW, Cashman NR, Chakrabartty A (2007) An immunological epitope selective for pathological monomer-misfolded SOD1 in ALS. Nat Med 13:754–759. doi:10.1038/nm1559
Re DB, Le Verche V, Yu C, Amoroso MW, Politi KA, Phani S, Ikiz B, Hoffmann L, Koolen M, Nagata T et al (2014) Necroptosis drives motor neuron death in models of both sporadic and familial ALS. Neuron 81:1001–1008. doi:10.1016/j.neuron.2014.01.011
Robertson J, Sanelli T, Xiao S, Yang W, Horne P, Hammond R, Pioro EP, Strong MJ (2007) Lack of TDP-43 abnormalities in mutant SOD1 transgenic mice shows disparity with ALS. Neurosci Lett 420:128–132. doi:10.1016/j.neulet.2007.03.066
Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O’Regan JP, Deng HX et al (1993) Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362:59–62
Saxena S, Roselli F, Singh K, Leptien K, Julien JP, Gros-Louis F, Caroni P (2013) Neuroprotection through excitability and mTOR required in ALS motoneurons to delay disease and extend survival. Neuron 80:80–96. doi:10.1016/j.neuron.2013.07.027
Smith RA, Miller TM, Yamanaka K, Monia BP, Condon TP, Hung G, Lobsiger CS, Ward CM, McAlonis-Downes M, Wei H et al (2006) Antisense oligonucleotide therapy for neurodegenerative disease. J Clin Investig 116:2290–2296
Song W, Zukor H, Liberman A, Kaduri S, Arvanitakis Z, Bennett DA, Schipper HM (2014) Astroglial heme oxygenase-1 and the origin of corpora amylacea in aging and degenerating neural tissues. Exp Neurol 254:78–89. doi:10.1016/j.expneurol.2014.01.006
Stathopulos PB, Rumfeldt JA, Scholz GA, Irani RA, Frey HE, Hallewell RA, Lepock JR, Meiering EM (2003) Cu/Zn superoxide dismutase mutants associated with amyotrophic lateral sclerosis show enhanced formation of aggregates in vitro. Proc Natl Acad Sci U S A 100:7021–7026. doi:10.1073/pnas.1237797100
Tan CF, Eguchi H, Tagawa A, Onodera O, Iwasaki T, Tsujino A, Nishizawa M, Kakita A, Takahashi H (2007) TDP-43 immunoreactivity in neuronal inclusions in familial amyotrophic lateral sclerosis with or without SOD1 gene mutation. Acta Neuropathol 113:535–542. doi:10.1007/s00401-007-0206-9
Turner BJ, Atkin JD, Farg MA, Zang DW, Rembach A, Lopes EC, Patch JD, Hill AF, Cheema SS (2005) Impaired extracellular secretion of mutant superoxide dismutase 1 associates with neurotoxicity in familial amyotrophic lateral sclerosis. J Neurosci 25:108–117. doi:10.1523/JNEUROSCI.4253-04.2005
Urushitani M, Ezzi SA, Julien JP (2007) Therapeutic effects of immunization with mutant superoxide dismutase in mice models of amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A 104:2495–2500. doi:10.1073/pnas.0606201104
Valentine JS, Hart PJ (2003) Misfolded CuZnSOD and amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A 100:3617–3622. doi:10.1073/pnas.0730423100
Vande Velde C, Miller TM, Cashman NR, Cleveland DW (2008) Selective association of misfolded ALS-linked mutant SOD1 with the cytoplasmic face of mitochondria. Proc Natl Acad Sci U S A 105:4022–4027. doi:10.1073/pnas.0712209105
Wang J, Xu G, Borchelt DR (2002) High molecular weight complexes of mutant superoxide dismutase 1: age-dependent and tissue-specific accumulation. Neurobiol Dis 9:139–148. doi:10.1006/nbdi.2001.0471
Wang J, Xu G, Gonzales V, Coonfield M, Fromholt D, Copeland NG, Jenkins NA, Borchelt DR (2002) Fibrillar inclusions and motor neuron degeneration in transgenic mice expressing superoxide dismutase 1 with a disrupted copper-binding site. Neurobiol Dis 10:128–138
Wang L, Deng HX, Grisotti G, Zhai H, Siddique T, Roos RP (2009) Wild-type SOD1 overexpression accelerates disease onset of a G85R SOD1 mouse. Hum Mol Genet 18:1642–1651. doi:10.1093/hmg/ddp085
Xu G, Ayers JI, Roberts BL, Brown H, Fromholt S, Green C, Borchelt DR (2015) Direct and indirect mechanisms for wild-type SOD1 to enhance the toxicity of mutant SOD1 in bigenic transgenic mice. Hum Mol Genet 24:1019–1035. doi:10.1093/hmg/ddu517
Zetterstrom P, Graffmo KS, Andersen PM, Brannstrom T, Marklund SL (2013) Composition of soluble misfolded superoxide dismutase-1 in murine models of amyotrophic lateral sclerosis. Neuromol Med 15:147–158. doi:10.1007/s12017-012-8204-z
Acknowledgements
We thank Drs. Neil Cashman, Leslie Grad and Megan O’Neill (University of British Columbia, Vancouver) for providing us with the DSE2 3H1 and 10E11 misfolded SOD1 and for allowing SDC to visit his lab to compare immunoprecipitation methods in human patient samples from JR’s and NC’s banks. We thank Drs. Alexander McCampbell, Fang Qian and Paul Weinreb at Biogen (Cambridge, MA) for providing us with the 4A1 and A5E5 antibodies; and Drs. Thomas Brännström and Stefan Marklund (Umea University, Sweden) for their polyclonal 131-153Ra misfolded SOD1 antibody. This work was supported by the ALS Association (Grant ALSA 2006 to DWC) and NIH Grant R01-NS27036 to DWC. SDC and DWC received salary support from the Ludwig Institute for Cancer Research and JR from ALSA and NINDS.
Author information
Authors and Affiliations
Contributions
SDC, PAP, DWC and JR designed the experiments and analyzed the data. SDC, PAP, AB, SS, SKL, JS, DP, DS performed the experiments. SDC, DWC and JR wrote the manuscript. MMD provided reagents.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest.
Ethical approval
All procedures performed in studies involving human participants and animals were in accordance with the ethical standards of the institution at which the studies were conducted.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Da Cruz, S., Bui, A., Saberi, S. et al. Misfolded SOD1 is not a primary component of sporadic ALS. Acta Neuropathol 134, 97–111 (2017). https://doi.org/10.1007/s00401-017-1688-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-017-1688-8