
Abstract
Amyotrophic lateral sclerosis (ALS) is a conformational disease in which misfolding and aggregation of proteins such as SOD1 (familial ALS) and TDP-43 (sporadic ALS) are central features. The conformations adopted by such proteins within motor neurons in affected patients are not well known. We have developed a novel conformation-specific antibody (USOD) targeted against SOD1 residues 42–48 that specifically recognizes SOD1 in which the beta barrel is unfolded. Use of this antibody, in conjunction with the previously described SEDI antibody that recognizes the SOD1 dimer interface, allows a detailed investigation of the in vivo conformation of SOD1 at the residue-specific level. USOD and SEDI immunohistochemistry of spinal cord sections from ALS cases resulting from SOD1 mutations (A4V and ΔG27/P28) shows that inclusions within remaining motor neurons contain SOD1 with both an unfolded beta barrel and a disrupted dimer interface. Misfolded SOD1 can also be immunoprecipitated from spinal cord extracts of these cases using USOD. However, in ten cases of sporadic ALS, misfolded SOD1 is not detected by either immunohistochemistry or immunoprecipitation. Using the amyloid-specific dyes, Congo Red and Thioflavin S, we find that SOD1-positive inclusions in familial ALS, as well as TDP-43- and ubiquitin-positive inclusions in sporadic ALS, contain non-amyloid protein deposits. We conclude that SOD1 misfolding is not a feature of sporadic ALS, and that both SOD1-ALS and sporadic ALS, rather than being amyloid diseases, are conformational diseases that involve amorphous aggregation of misfolded protein. This knowledge will provide new insights into subcellular events that cause misfolding, aggregation and toxicity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The presence of inclusion bodies containing aggregated proteins in motor neurons is a universal pathological hallmark of amyotrophic lateral sclerosis (ALS), a devastating adult-onset neurodegenerative disease that results in paralysis and death [14]. ALS is characterized by degeneration of both upper and lower motor neurons, and death due to respiratory failure usually occurs 3–5 years after onset [24]. While the causes of ~90% of ALS cases are unknown (i.e., “sporadic” ALS/sALS), the remaining 10% are familial, with an autosomal dominant pattern of inheritance [41]. Mutations in Cu/Zn superoxide dismutase (SOD1) are responsible for ~20% of familial cases (SOD1-ALS) [41]. SOD1 is a 32 kDa homodimeric enzyme composed of two monomeric eight-stranded beta barrels, as well as binding sites for one copper ion and one zinc ion per monomer [28] (Fig. 1). SOD1 normally plays a role in cellular antioxidant defense, converting superoxide into hydrogen peroxide and water (see Ref. [28]). Over 100 SOD1 mutations that cause ALS have been identified, and many of these do not result in significant loss of enzymatic activity [41]. Furthermore, SOD1 knock-out mice do not develop motor neuron degeneration [32]. The finding that overexpression of mutant SOD1 in mice causes an ALS-like disease even in the presence of endogenous mouse SOD1 [3, 11, 39] has led to the conclusion that the disease results from a toxic gain of function (reviewed in Ref. [4]). The finding that intraneuronal inclusion bodies in the motor neurons of SOD1-ALS patients contain SOD1 protein [19, 22] has further suggested that the toxic property of mutant SOD1 is an increased propensity to misfold and/or aggregate. Recently, the TAR DNA binding protein-43 (TDP-43) was indentified as a core component of ubiquitinated inclusions in sALS [1, 26], and mutations in the TDP-43 gene (TARDP) were subsequently found in a subset of familial cases [16, 36]. The mechanism leading to TDP-43 aggregation and whether or not this involves misfolding of the TDP-43 polypeptide remains unknown. However, the presence of inclusion bodies containing SOD1 or TDP-43 indicates that protein misfolding and aggregation are common features of both sporadic and familial ALS.

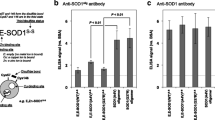
Design and validation of USOD antibody. a–d Location of epitopes of USOD and SEDI antibodies in the 3D structure of SOD1. SEDI epitope (yellow) and USOD epitope (red) are highlighted in each structure. a Surface representation of native dimeric SOD1 (PDB code 1spd). b The two monomers from the structure in a are separated, revealing the SEDI epitope. d A hypothetical structure for unfolded monomeric SOD1, revealing both the SEDI and USOD epitopes. c Ribbon diagram of native dimeric SOD1 with bound copper (blue) and zinc (gray). e First panel Indirect ELISA showing specificity of USOD and SEDI for misfolded SOD1. e Second–third panels Competition ELISAs for SEDI and USOD, with misfolded SOD1 as the competitor
There are wide variety of other diseases, including several neurodegenerative diseases (such as Alzheimer’s, Parkinson’s, and Huntington’s) that also have protein misfolding and/or aggregation as central pathological features. These diseases are referred to as conformational diseases [5], and can be categorized according to the nature of the conformational change that precedes the aggregation of the disease-causing protein, as well as the nature of the aggregates. One category comprises those diseases in which the relevant protein undergoes a significant loss of native structure, followed by aggregation into ordered amyloid fibrils [20, 40]. For a protein aggregate to be identified as amyloid, it must satisfy several structural/tinctorial criteria including: (1) the presence of long, unbranched 5–10 nm fibrils detected by negative-stain electron microscopy or atomic force microscopy, (2) the presence of β-sheet secondary structure as determined by circular dichroism or infrared spectroscopy, (3) a “cross-β” X-ray fiber diffraction pattern, indicating that the β-strands run perpendicular to the long axis of the fibrils, and (4) Congo Red (CR) staining with apple-green birefringence when viewed through crossed polarizers [35]. Binding of the dyes Thioflavin S (ThS) and/or Thioflavin T (ThT), resulting in significant enhancement of their fluorescence, is also an indicator of amyloid [8]; however, it has been recommended that in addition to positive ThS/T staining, one or more of the other criteria listed above (such as CR staining/birefringence) must also be satisfied to confidently identify a structure as amyloid [35]. Many disease-causing proteins are known to form amyloid fibrils, including Aβ (Alzheimer’s disease), α-synuclein (Parkinson’s disease [12]), Huntingtin (Huntington’s disease [23]), IgG light chains (light chain amyloidosis), transthyretin (senile systemic amyloidosis), and prion protein (spongiform encephalopathies) [35]. In another category of conformational diseases, significant loss of native structure is also presumed to occur, but the resulting aggregates are largely disordered and amorphous. Examples of such diseases are the hemoglobinopathies, in which mutations in hemoglobin destabilize protein structure, leading to misfolding and aggregation into Heinz bodies under stressed conditions [46]. In a third category of conformational disease, only a subtle structural change results in polymerization of the nearly-native protein into an insoluble (non-amyloid) form. These are exemplified by the serpinopathies, in which an intermolecular exchange of β-strands leads to polymerization and intracellular aggregation of serpins [2].
The category of conformational disease in which sALS, SOD1-ALS, and familial ALS caused by mutations in proteins other than SOD1 belong is currently unknown. It is crucial to determine what conformation(s) inclusion-forming proteins (e.g., SOD1, TDP-43) adopt in vivo, in order to identify the possible cellular/molecular events that precede misfolding/aggregation, as well as to understand how altered protein conformational states lead to cellular toxicity. Many studies have shed light on possible aggregation mechanisms of SOD1 in vitro, demonstrating, e.g., that monomerization is an early event in SOD1 misfolding [25] and generally precedes aggregation [21, 29]. Until recently, however, it has not been possible to evaluate the pathological relevance of such mechanisms, largely due to the inherent difficulty in studying the conformation of a protein in vivo, and determining the mechanism by which it unfolds and aggregates. Our approach to this problem involves histochemistry using conformation-specific dyes and immunohistochemistry using antibodies that specifically recognize certain misfolded forms of SOD1.
We previously reported the design of the SEDI (SOD1 Exposed Dimer Interface) antibody, which only recognizes SOD1 conformations in which the dimer interface is exposed [31]. Using this antibody, we were able to detect dimer interface-exposed SOD1 in disease-affected tissues from SOD1-ALS patients, as well as from mouse models of SOD1-ALS [22, 31]. Those studies were the first in vivo conformational characterizations of SOD1 at the residue-specific level. While they provided information on the status of the dimer interface, it is unknown whether misfolded SOD1 in ALS retains a native-like beta barrel structure, or if a greater degree of unfolding occurs. There is accumulating evidence that disruption of the dimer interface of wild-type SOD1 does not occur in either sALS or non-SOD1-ALS, and the involvement of SOD1 misfolding in sALS is under debate [22]. Whether SOD1-ALS can be grouped in with other amyloid diseases like Alzheimer’s and Parkinson’s disease has not been established. Furthermore, the conformations that are adopted by proteins in ubiquitin- and/or TDP-43-positive inclusions are unknown.
Here, we report investigations of the conformational make-up of inclusion bodies in SOD1-ALS and sALS. In order to characterize the structures of SOD1 in both SOD1-ALS and sALS, we have used conformation-specific antibodies that recognize varying degrees of SOD1 misfolding. We have employed the amyloid-specific dyes CR and ThS to evaluate the conformational composition of ubiquitin- and TDP-43-positive inclusion bodies. We find that in SOD1-ALS, the SOD1 molecules present in inclusions adopt misfolded conformations that expose the most buried core of the molecule, while in sALS, misfolded SOD1 is not detected. We also find that SOD1-ALS and sALS, rather than being amyloid diseases, belong to that class of conformational disease that involve protein misfolding and formation of disordered amorphous aggregates.
Materials and methods
Antibody generation
We synthesized a multiple antigenic peptide (MAP) on a [Fmoc-Lys(Fmoc)]4-Lys2-Lys-Cys(Acm)-β-Ala-Wang resin (Advanced ChemTech, SM5104) using Fmoc-protected amino acids (Advanced ChemTech, Novabiochem and Applied Biosystems). The sequence was acetyl-GGLHGFHVHGG (residues in bold are residues 42–48 of SOD1); we verified composition and sequence with amino acid analysis. We sent the MAP to Sigma-Genosys for rabbit antiserum production, which followed standard protocols (Sigma-Genosys) and was in accordance with the Animal Welfare Act (USA). The USOD antibody was affinity-purified as described in the online resource (supporting information).
Preparation of SOD1
Folded (native holo) SOD1 was prepared as described previously by Mulligan et al. [25]. Unfolded SOD1 was prepared by dissolving folded SOD1 (final concentration 15 μM) in 6 M GuHCl containing 2 mM DTT and 1 mM EDTA and incubating at room temperature for at least 2 h.
Enzyme-linked immunosorbent assay (ELISA)
For indirect ELISAs, 96-well plates were coated with either folded SOD1 or unfolded SOD1 (50 ng/well) overnight. After blocking with PBS + 1% BSA w/v, plates were incubated with 100 μl/well of affinity-purified USOD (1 μg/ml) or SEDI (1:200 dilution of a 50% ammonium sulfate cut) for 2 h followed by secondary antibody (Sheep anti-rabbit HRP conjugate, Calbiochem: 100 μl/well of a 1:5,000 dilution) for 2 h. TMB (Sigma) was used for detection. For competition ELISAs, wells were coated with unfolded SOD1. Prior to addition to the plate, the primary antibody (i.e., either SEDI or USOD at the concentrations stated above) was mixed with unfolded SOD1 over a concentration range of 0–1 μg/ml and incubated for 1 h before being added to the plate. The residual concentration of GuHCl in these samples was less than 40 mM, and control experiments confirmed that this amount of GuHCl had no effect on the ELISA signal.
Immunohistochemistry
Six-micrometer sections of formalin-fixed, paraffin-embedded spinal cords were incubated with affinity-purified USOD (0.3 μg/ml) or SEDI (1:1,000), or with the SOD100 antibody (Stressgen: 1:10,000) overnight at 4°C. After incubation with secondary antibodies, immunoreactivity was developed using 3,3′-diaminobenzidine and counterstained with hematoxylin. For peptide competition, USOD antibody was pre-incubated with a linear peptide containing the USOD epitope (1,000× molar excess) for 1 h at room temperature prior to incubating with tissue. Some sections from sALS cases were labeled with polyclonal rabbit anti-ubiquitin antibody (1:800, Dako) or mouse anti-phospho TDP-43 (pS409/410) (1:2,000, Cosmo Bio Co.) to reveal inclusions.
CR staining was performed under aqueous conditions for 15–20 min, followed by differentiation in alkaline alcohol solution. ThS staining was performed using a 0.05% solution in 50% alcohol. Sections were stained for 8 min in the dark, followed by washes with 80% alcohol and distilled water.
Immunoprecipitations
Frozen lumbar spinal cord (~100 mg) from ALS cases or from a symptomatic G93A transgenic mouse was homogenized in 10 volumes of RIPA buffer (50 mM Tris–HCl pH 7.5, 150 mM NaCl, 1% NP-40, 0.25% Na deoxycholate, 1 mM EDTA, 0.1% SDS) with protease inhibitor cocktail (Roche), and centrifuged at 15,000g for 15 min at 4°C. Two milligrams of protein from the soluble fraction was incubated with 2 μg affinity-purified USOD antibody and 20 μl of 50% (w/v) Protein A-Agarose (Sigma) overnight at 4°C. Immunoprecipitates were eluted with SDS sample buffer and analyzed by SDS-PAGE and Western blotting as described in the online resource (supporting information).
Additional details are given in the online resource (supporting information).
Results
Design and validation of the USOD antibody
We previously described the design of the SEDI antibody, which recognizes any SOD1 conformation in which the dimer interface of SOD1 has been disrupted [31]; these conformations included folded monomeric SOD1 (Fig. 1b) and more extensively misfolded SOD1 (Fig. 1d). We have now designed and characterized a second antibody that should bind extensively misfolded SOD1 conformations that have an exposed hydrophobic core (Fig. 1d), but should not bind to native dimeric SOD1 or folded monomeric SOD1. We raised a polyclonal antibody against the sequence SOD1 42–48, which comprises residues in the fourth beta strand of the beta barrel, including two histidine residues that are involved in chelation of the copper ion (Fig. 1c). This sequence of SOD1 is buried in the hydrophobic core and is inaccessible in both native dimeric SOD1 (Fig. 1a) and folded monomeric SOD1 (Fig. 1b). Antibodies against this epitope would only be able to recognize SOD1 molecules in which the copper ion has been lost, the beta barrel structure has been disrupted, and the hydrophobic core is exposed (Fig. 1d). We refer to the polyclonal antibody raised against this epitope as USOD (for “unfolded” SOD1). The SEDI antibody and the USOD antibody can be used in conjunction to gauge the extent of SOD1 misfolding in ALS.
We used ELISAs to verify that the USOD antibody was specific for unfolded SOD1. As expected, USOD did not recognize natively folded holoSOD1, but showed strong recognition of SOD1 that had been unfolded, de-metallated and reduced using 6 M GuHCl, 1 mM EDTA and 2 mM DTT (Fig. 1e). In competition ELISAs, unfolded SOD1 showed concentration-dependent competition for both SEDI and USOD (Fig. 1e, middle and right panels). Fitting of the data in these ELISAs gave nearly identical IC50 values for the two antibodies (~15 nM for SEDI and ~17 nM for USOD), suggesting that USOD and SEDI have very similar affinities for their respective epitopes.
To verify that USOD can be used to specifically label misfolded SOD1 in tissue sections, we carried out immunohistochemistry on spinal cord sections from symptomatic G93A mice. We previously used the SEDI antibody to demonstrate that misfolded SOD1 accumulates in the ventral horn and ventral roots of these mice [31]. Although SOD1 is present throughout the spinal cord (Fig. S1, right panels), USOD specifically labeled vacuolated structures in the anterior horn, as well as some axons in the ventral root (Fig. S1, left panels), closely resembling the labeling previously obtained with SEDI [31]. This indicates that USOD can be used in immunohistochemistry to distinguish between folded and misfolded SOD1. This also suggests that misfolding of SOD1 in the G93A mouse involves disruption of the SOD1 beta barrel structure and exposure of the hydrophobic core, in addition to disruption of the dimer interface.
Misfolded SOD1 is detected in intraneuronal inclusions in SOD1-ALS, but not in sALS
Various antibodies against SOD1 have been used to show that intraneuronal inclusions in SOD1-ALS contain SOD1 protein [19], but these antibodies have generally not reported on the conformation of the aggregated protein. Using the SEDI antibody, we have previously shown that SOD1 deposited in inclusions is conformationally altered—the normally buried dimer interface is exposed and available for antibody interactions [22, 31]. In order to further characterize the conformation of SOD1 in inclusions, we performed USOD immunohistochemistry on spinal cord sections from two SOD1-ALS cases, one resulting from the A4V mutation, and the other resulting from a deletion of the sequence G27/P28 [48].
Like SEDI, USOD strongly labeled many intraneuronal inclusions in spinal cord motor neurons of the A4V (Figs. 2a, b, e, g, 3a; Fig. S2a) and ΔG27/P28 cases (Fig. 2c, d). Although the two SOD1-ALS cases had few motor neurons remaining (an average of ~10 motor neurons per transverse section; Table S1), we found that approximately 40% of all neurons we observed had large hyaline conglomerate inclusions that stained strongly with USOD. Specificity of labeling was confirmed by competition of the antibody with antigenic peptide, which completely abrogated labeling (Fig. 2e, f). We also observed vascular labeling (Fig. 2b, arrow) which was not disease-specific, as it was present in every ALS case tested as well as the Alzheimer’s disease (AD) and corticobasal degeneration (CBD) controls. To determine if USOD and SEDI label the same material, we labeled pairs of serial spinal cord sections from the A4V case with USOD and SEDI. We found that inclusions that were labeled with one antibody were also labeled with the other (Fig. 2g, h; Fig. S2a, b). These results suggest that the conformation of SOD1 that is deposited in inclusions is significantly different from the native conformation, showing disruption of the dimer interface (SEDI), as well as the beta barrel, the hydrophobic core, and the copper binding site (USOD).

USOD immunohistochemistry of SOD1-ALS cases. First row USOD labeling of inclusions in motor neurons of the A4V case (a, b) and the ΔG27/P28 case (c, d). The arrow in b indicates vascular labeling that was present in all human cases and controls. e, f Two serial sections from the A4V case showing the elimination of labeling by competition with antigenic peptide. g, h Two serial sections from the A4V case showing labeling of the same motor neuron with USOD and SEDI. Scale bar All panels except e, f 35 μm, e, f 70 μm

USOD immunohistochemistry of sALS cases and pathological controls. a An example of USOD labeling of a motor neuron from the A4V case. b–f Show the absence of USOD labeling of motor neurons in five sALS cases. g, h Show the absence of USOD labeling of motor neurons in the AD and CBD cases, respectively. Some motor neurons are highlighted by arrows. Scale bar a 35 μm, b–h 70 μm, i Top gel Immunoprecipitation of misfolded SOD1 from spinal cord extracts of the A4V case, the ΔG27/P28 case and a G93A mouse, but not from sALS cases. Bottom gel IP supernatants (0.1%) were analyzed by immunoblotting with anti-SOD1 antibody to show equivalencies of samples used for the immunoprecipitations
It has been hypothesized that misfolding of wild-type SOD1 could be involved in the pathogenesis of sALS [9, 29]. However, we have previously demonstrated the absence of misfolded SOD1 in inclusions of sALS using the SEDI antibody [22]. To determine if that result was specific for the SEDI epitope, we performed USOD immunohistochemistry on spinal cord sections from the same sALS cases that were investigated in our previous work. While there were many intact motor neurons remaining in the sALS cases (an average of ~30 motor neurons per transverse section; Table S1), none of them stained with USOD compared to the two SOD1-ALS cases (Fig. 3b–f; Fig. S2c–g). Taken together, these results suggest that there is no detectable accumulation of misfolded SOD1 in motor neurons in sALS. We also did not detect misfolded SOD1 using USOD in a case of AD and a case of CBD (Fig. 3g, h), further showing that SOD1 misfolding is unique to SOD1-ALS.
We assessed the presence or absence of misfolded SOD1 in the detergent-soluble fractions of frozen spinal cord tissue from the same SOD1-ALS and sALS cases that were investigated by immunohistochemistry. Full-length misfolded SOD1 was immunoprecipitated by USOD from tissue extracts of the two SOD1-ALS cases, but not the sALS cases (Fig. 3i). This is consistent with results previously obtained with SEDI [22] and further supports the conclusion that misfolded SOD1 is present in motor neurons in SOD1-ALS, but not in sALS.
Inclusions in SOD1-ALS and sALS do not contain amyloid
It has been suggested, based on the results of in vitro experiments on SOD1, that mutant SOD1 forms amyloid in ALS [6, 10, 27]. Amyloid can be detected in situ using the dye CR. When this dye is bound to amyloid, which contains long-range regular structure, it exhibits apple-green birefringence, which can be detected by observing the stained tissue through crossed polarizers [13]. We, therefore, used CR staining to determine if motor neurons/inclusions in ALS contain amyloid. In the A4V case, we identified misfolded SOD1-containing inclusions by staining consecutive serial sections with USOD and SEDI (Fig. 4e, f). A third serial section was then stained with CR. Inclusions that were labeled by USOD and SEDI did not stain with CR (Fig. 4g), nor did we observe any apple-green birefringence when the sections were viewed through crossed polarizers (Fig. 4h). In contrast, senile plaques in AD brain tissue gave strong CR staining and apple-green birefringence (Fig. 4a, b), indicating that our staining technique is indeed capable of detecting amyloid where it is present.

Absence of amyloid in SOD1- and sporadic ALS. Top row A plaque from AD brain stained with CR (a), viewed through crossed polarizers (b) and stained with ThS (c), as well as neurofibrillary tangles from AD brain stained with ThS (d). Second row Four consecutive serial sections of a motor neuron from the A4V case. The first section was labeled with USOD (e), the second with SEDI (f), the third stained with CR (g the neuron shows no staining), and viewed through crossed polarizers (h), and the fourth stained with ThS (i). Third and fourth rows Each row shows the results of staining of a set of three consecutive serial spinal cord sections from a sALS case. The first section was labeled either with anti-TDP-43 antibody (j) or anti-ubiquitin antibody (n); the second was stained with CR (k, o) and viewed through crossed polarizers (l, p); the third was stained with ThS (m, q). In rows 3–4, arrows indicate the locations of immunopositive inclusions (j–l and n–p) or lipofuscin (m and q)
We also used CR staining to determine if TDP-43-positive and ubiquitin-positive inclusions in sALS contain amyloid. In sets of serial sections from several sALS cases, the first section from each set was labeled with an anti-TDP 43 antibody (Fig. 4j; Fig. S3a, e) or an anti-ubiquitin antibody (Fig. 4n; Fig. S3i) to reveal the locations of TDP-43/ubiquitin-positive inclusions. The second section from each set was stained with CR. As was the finding with the SOD1-ALS case, there was no labeling of either TDP-43-positive or ubiquitin-positive inclusion with CR (Fig. 4k, o; Fig S3b, f, j) nor did we observe apple-green birefringence when the sections were viewed through crossed polarizers (Fig. 4l, p; Fig. S3c, g, k).
The fluorescent dye ThS is also often used for the histological detection of amyloid [8]. We were able to use ThS to detect amyloid in plaques and neurofibrillary tangles from AD brain tissue (Fig. 4c, d). However, ThS did not label misfolded SOD1-containing inclusions in motor neurons from the A4V case (Fig. 4i), and in the sALS cases, there was no ThS fluorescence associated with TDP-43-positive or ubiquitin-positive inclusions (Fig. 4m, q; Fig. S3c, f, i). We observed the diffuse, granular autofluorescence that is associated with lipofuscin deposits. The autofluorescence was confirmed by examining the emission at two different wavelengths (488 and 594 nm). ThS produces strong emission at 488 nm, but not at 594 nm [8]. However, we observed strong emission at both wavelengths (Fig. S3m, n), indicating that the source of the fluorescence was not ThS, and is likely to be lipofuscin [34]. We, therefore, conclude that the proteins present in inclusions in both sporadic and SOD1-ALS do not form amyloid.
Discussion
We have used a combination of conformation-specific antibodies and histological dyes in a systematic analysis of protein conformation in disease-affected tissues from ALS patients. Our results show that, in SOD1-ALS resulting from two different mutations (A4V and ΔG27/P28), SOD1 misfolding involves exposure of the hydrophobic core, disruption of the beta barrel and copper binding site, in addition to disruption of the dimer interface. This represents a significantly unfolded structure, in contrast to diseases such as the serpinopathies where the protein retains a near-native conformation in the aggregates. Our investigation of the in vivo conformation of two different regions of the SOD1 molecule allows us to go beyond a simple “folded/misfolded” description, and provides detailed structural information at the residue-specific level. Such information will be valuable in evaluating the relevance of in vitro misfolding/aggregation models to the disease process in vivo. In sALS, detectable misfolded SOD1 does not accumulate in inclusion bodies. The absence of CR staining/birefringence in both SOD1-ALS and sALS indicates that the inclusion-forming proteins in these diseases (such as SOD1 and TDP-43) do not form amyloid fibrils in vivo. This implies that ALS falls into the category of conformational diseases that involve significant loss of native structure, followed by formation of disordered amorphous aggregates. This differentiates ALS from other neurodegenerative diseases, such as Alzheimer’s, Parkinson’s and Huntington’s where the formation of ordered amyloid aggregates is a key feature.
In addition to detecting USOD- and SEDI-immunoreactive SOD1 in inclusion bodies, we also detect similar species of SOD1 in the detergent-soluble fraction of SOD1-ALS spinal cord tissue [22] (Fig. 3i). This indicates that some misfolded SOD1 in these ALS cases is present in soluble states, which may be precursors of the large insoluble inclusion bodies. This is consistent with earlier in vitro studies of SOD1 aggregation, in which the monomerization of SOD1 is an early event [21, 29]. Importantly, both USOD and SEDI only immunoprecipitate full-length SOD1 of the expected molecular weight from the detergent-soluble fraction, whereas Wang et al. [42] also detected low molecular weight cleavage products and ubiquitinated species of SOD1 in the detergent-insoluble fraction. This suggests that the soluble species that are immunoprecipitated by USOD and SEDI represent early intermediates in the process of inclusion body formation. An improved understanding of the process would, therefore, require more detailed characterization of these soluble species, including their size, the presence of proteins other than SOD1, and further analysis of SOD1 conformation.
Our immunohistochemistry procedure involves pre-treatment of tissue sections with heat and EDTA-containing buffer, and we utilize a detergent/EDTA-containing buffer in our immunoprecipitations. While the possibility exists that these treatments/buffers may have influenced the conformation of SOD1 prior to interaction with USOD, the totality of our experience with SEDI and USOD suggests that this is unlikely. Similar pre-treatment of tissue sections was used in our SEDI immunohistochemistry experiments on G93A mouse tissue [31]. Despite widespread expression of the mutant protein, SEDI staining is not observed at all in young, presymptomatic mice [31], indicating that mutant SOD1 is refractory to the pre-treatment. In symptomatic mice, USOD (Fig. S1) and SEDI [31] staining is only observed in very specific areas of the spinal cord, further indicating that the majority of the mutant protein, even in disease-affected tissue, is not structurally affected by the pre-treatment. The buffer used for immunoprecipitations is mild enough to maintain protein–protein interactions (e.g., antibody–antigen interactions), and although it contains EDTA, removal of tightly bound metals from SOD1 is unlikely, and would generally not unfold the protein (i.e., apo mutant SOD1s are folded proteins). We conclude that our immunohistochemistry and immunoprecipitation experiments faithfully report on the in vivo conformation of SOD1.
The absence of detectable misfolded SOD1 in sALS we observed is consistent with previous studies in which the accumulation of SOD1 in ALS inclusion bodies was not detected. In one study, an antibody raised against SOD1 residues 125–137 did not detect SOD1 in the inclusions of 17 sALS cases [45]. In our previous study, the SEDI antibody, raised against residues 143–151 (dimer interface), also did not detect SOD1 in sALS inclusions [22]. The USOD antibody used in the current study recognizes residues 42–48. These antibodies recognize significantly different regions of the SOD1 molecule (see Fig. 1), and they all readily detect SOD1 in inclusions of all SOD1-ALS cases examined. The existence of misfolded SOD1 species in which both the SEDI and USOD epitopes are sequestered is unlikely. The fact that none of the antibodies could detect misfolded or aggregated SOD1 in a total of 10 sALS cases indicates that these results are not simply due to the inaccessibility of a particular epitope—rather, they strongly support the conclusion that misfolding and aggregation of SOD1 do not occur in sALS. Furthermore, Wang et al. [42] could only detect insoluble SOD1 in spinal cord homogenates from SOD1-ALS patients, and not in sporadic or non-SOD1 familial ALS, further supporting this conclusion.
Several studies have shown that both wild-type SOD1 and mutant SOD1 can be induced to unfold and aggregate under similar conditions [21, 29, 37]. Some of the cytotoxic properties of mutant SOD1 can be conferred upon wild-type SOD1 by oxidation [9], and overexpression of wild-type SOD1 can exacerbate the disease phenotype in several transgenic mouse models of SOD1-ALS [7, 15, 44]. Such results have led to the hypothesis that misfolding and aggregation of wild-type SOD1 may be a key etiological feature of sALS [9, 29], implying that the relationship between SOD1-ALS and sALS is the same as that between familial and sporadic forms of Alzheimer’s and Parkinson’s disease, where the same protein (i.e., Aβ or alpha-synuclein) is seen to accumulate in insoluble form in both familial and sporadic cases. The results presented here, however, indicate that this is not the case with ALS—misfolding and aggregation of SOD1 are unique features of SOD1-ALS. Clinical and pathological similarities between SOD1-ALS and sALS cannot, therefore, be explained by the presence of misfolded SOD1 in both diseases. Rather, the similarities must result from other effects that are downstream of protein misfolding, such as oxidative stress, mitochondrial dysfunction and excitotoxicity [33]. The absence of detectable SOD1 misfolding in sALS also suggests that features of mouse models and cellular models of ALS that exhibit mutant SOD1 misfolding are likely not directly relevant to sALS.
The absence of amyloid in SOD1-ALS is consistent with previous ultrastructural studies of SOD1-positive inclusions. Electron microscopy of these inclusions shows the presence of 15–25-nm thick granule-coated filaments as the key ultrastructural feature [17–19]. Such filaments differ significantly in appearance from typical amyloid fibrils, which are comprised of long, unbranched 5–10 nm fibrils without granules [35]. It has been reported that some inclusions in mutant SOD1 mouse models are stained with ThS [43]. In transgenic mice, very high levels of protein overexpression could lead to misfolding and aggregation pathways that form some amyloid; however, in human cases, where the level of SOD1 expression is significantly lower, amyloidogenesis does not occur. It is also likely that the inclusions in the mutant SOD1 expressing mice that are stained by ThS contain protein(s) other than SOD1, as they do not label well with antibodies against SOD1 [43].
Several in vitro studies have shown that it is possible to create amyloid fibrils and amyloid-like aggregates of SOD1 under various conditions [6, 10, 27]. Such studies add to the growing body of literature showing that amyloid formation is an inherent property of a large number of proteins, including some that are not known to form amyloid in vivo [38]. It has been suggested that amyloid formation is an inherent tendency of the polypeptide backbone [38]. However, the absence of amyloid in ALS patient tissue suggests that the misfolding and aggregation processes that take place in vivo differ significantly from the in vitro processes that lead to SOD1 amyloid formation. Rather, aggregation mechanisms that result in non-amyloid aggregates may more closely resemble the in vivo behavior of mutant SOD1. We have previously shown that misfolding and aggregation of SOD1 induced by metal-catalyzed oxidation lead to the formation of non-amyloid (i.e., CR negative) aggregates that are ultrastructurally similar to the aggregates observed in vivo [29, 30]. Our current results, therefore, support our earlier hypothesis that metal-catalyzed oxidation (or oxidation in general) may be responsible for inducing SOD1 misfolding and aggregation in SOD1-ALS [30]. This hypothesis can be further tested by investigating the presence of oxidative modifications on misfolded SOD1 isolated from patient tissues.
Previous studies have shed light on the biochemical processes involved in the formation of TDP-43-positive ubiquitinated inclusion bodies. These include hyperphosphorylation of the C-terminal region, as well as the formation of C-terminal fragments [1, 26], possibly resulting from caspase cleavage [47]. Until now, however, the conformation(s) adopted by pathological TDP-43 in ALS has remained unknown. We have now shown that ubiquitin-positive/TDP-43-positive inclusions and their constituent proteins in ALS do not contain amyloid structure. Overall, our results indicate that the formation of amyloid per se is not a feature of either SOD1-ALS or sALS. An antibody-based approach, such as the one described in this work, should be useful for studying the in vivo conformation of TDP-43, thus allowing us to improve our understanding of the pathological mechanisms underlying this devastating disease. The approach used in the present study should also prove useful for studying the conformations of proteins present in other sALS-related inclusions, such as Bunina bodies and axonal spheroids.
Concluding remarks
Through the use of novel conformation-specific antibodies and histochemical stains, we have demonstrated that in human cases of SOD1-ALS, motor neurons contain aggregates of misfolded SOD1 that has a disrupted dimer interface, an unfolded beta barrel, and an exposed hydrophobic core. These SOD1 aggregates are disordered, amorphous, and are not amyloid. In sALS, misfolded SOD1 was undetectable, and the misfolded proteins present in the ubiquitin- or TDP-43-positive inclusions are also present as disordered, non-amyloid, amorphous aggregates. In addition, the data indicate that SOD1-ALS and sALS are distinct from amyloid diseases like Alzheimer’s and Parkinson’s, instead they belong to that class of conformational disease with disordered amorphous aggregates of misfolded proteins.
Abbreviations
- AD:
-
Alzheimer’s disease
- ALS:
-
Amyotrophic lateral sclerosis
- sALS:
-
Sporadic amyotrophic lateral sclerosis
- CR:
-
Congo Red
- CBD:
-
Corticobasal degeneration
- DTT:
-
Dithiothreitol
- ELISA:
-
Enzyme-linked immunosorbent assay
- GuHCl:
-
Guanidine hydrochloride
- MALDI:
-
Matrix-assisted laser desorption/ionization
- PBS:
-
Phosphate-buffered saline
- SOD1:
-
Superoxide dismutase 1
- TDP-43:
-
TAR DNA binding protein-43
- ThS:
-
Thioflavin S
References
Arai T, Hasegawa M, Akiyama H et al (2006) TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 351:602–611
Belorgey D, Hägglöf P, Karlsson-Li S, Lomas DA (2007) Protein misfolding and the serpinopathies. Prion 1:15–20
Bruijn LI, Becher MW, Lee MK et al (1997) ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron 18:327–338
Bruijn LI, Miller TM, Cleveland DW (2004) Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu Rev Neurosci 27:723–749
Carrell RW, Lomas DA (1997) Conformational disease. Lancet 350:134–138
Chattopadhyay M, Durazo A, Sohn SH et al (2008) Initiation and elongation in fibrillation of ALS-linked superoxide dismutase. Proc Natl Acad Sci USA 105:18663–18668
Deng HX, Shi Y, Furukawa Y et al (2006) Conversion to the amyotrophic lateral sclerosis phenotype is associated with intermolecular linked insoluble aggregates of SOD1 in mitochondria. Proc Natl Acad Sci USA 103:7142–7147
Elghetany MT, Saleem A (1988) Methods for staining amyloid in tissues: a review. Stain Technol 63:201–212
Ezzi SA, Urushitani M, Julien JP (2007) Wild-type superoxide dismutase acquires binding and toxic properties of ALS-linked mutant forms through oxidation. J Neurochem 102:170–178
Furukawa Y, Kaneko K, Yamanaka K, O’Halloran TV, Nukina N (2008) Complete loss of post-translational modifications triggers fibrillar aggregation of SOD1 in the familial form of amyotrophic lateral sclerosis. J Biol Chem 283:24167–24176
Gurney ME, Pu H, Chiu AY et al (1994) Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science 264:1772–1775
Hashimoto M, Hsu LJ, Sisk A et al (1998) Human recombinant NACP/alpha-synuclein is aggregated and fibrillated in vitro: relevance for Lewy body disease. Brain Res 799:301–306
Howie AJ, Brewer DB (2009) Optical properties of amyloid stained by Congo red: history and mechanisms. Micron 40:285–301
Ince PG, Lowe J, Shaw PJ (1998) Neuropathol Appl Neurobiol 24:104–117
Jaarsma D, Haasdijk ED, Grashorn JA et al (2000) Human Cu/Zn superoxide dismutase (SOD1) overexpression in mice causes mitochondrial vacuolization, axonal degeneration, and premature motoneuron death and accelerates motoneuron disease in mice expressing a familial amyotrophic lateral sclerosis mutant SOD1. Neurobiol Dis 7:623–643
Kabashi E, Valdmanis PN, Dion P et al (2008) TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet 40:572–574
Kato S, Nakashima K, Horiuchi S et al (2001) Formation of advanced glycation end-product-modified superoxide dismutase-1 (SOD1) is one of the mechanisms responsible for inclusions common to familial amyotrophic lateral sclerosis patients with SOD1 gene mutation, and transgenic mice expressing human SOD1 gene mutation. Neuropathology 21:67–81
Kato S, Shimoda M, Watanabe Y et al (1996) Familial amyotrophic lateral sclerosis with a two base pair deletion in superoxide dismutase 1: gene multisystem degeneration with intracytoplasmic hyaline inclusions in astrocytes. J Neuropathol Exp Neurol 55:1089–1101
Kato S, Takikawa M, Nakashima K et al (2000) New consensus research on neuropathological aspects of familial amyotrophic lateral sclerosis with superoxide dismutase 1 (SOD1) gene mutations: inclusions containing SOD1 in neurons and astrocytes. Amyotroph Lateral Scler Other Motor Neuron Disord 1:163–184
Kelly JW (1998) The alternative conformations of amyloidogenic proteins and their multi-step assembly pathways. Curr Opin Struct Biol 8:101–106
Khare SD, Caplow M, Dokholyan NV (2004) The rate and equilibrium constants for a multistep reaction sequence for the aggregation of superoxide dismutase in amyotrophic lateral sclerosis. Proc Natl Acad Sci USA 101:15094–15099
Liu HN, Sanelli T, Horne P et al (2009) Lack of evidence of monomer/misfolded superoxide dismutase-1 in sporadic amyotrophic lateral sclerosis. Ann Neurol 66:75–80
McGowan DP, van Roon-Mom W, Holloway H et al (2000) Amyloid-like inclusions in Huntington’s disease. Neuroscience 100:677–680
Mitchell JD, Borasio GD (2007) Amyotrophic lateral sclerosis. Lancet 369:2031–2041
Mulligan VK, Kerman A, Ho S, Chakrabartty A (2008) Denaturational stress induces formation of zinc-deficient monomers of Cu,Zn superoxide dismutase: implications for pathogenesis in amyotrophic lateral sclerosis. J Mol Biol 383:424–436
Neumann M, Sampathu DM, Kwong LK et al (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314:130–133
Oztug Durer ZA, Cohlberg JA, Dinh P et al (2009) Loss of metal ions, disulfide reduction and mutations related to familial ALS promote formation of amyloid-like aggregates from superoxide dismutase. PLoS One 4:e5004
Rakhit R, Chakrabartty A (2006) Structure, folding, and misfolding of Cu,Zn superoxide dismutase in amyotrophic lateral sclerosis. Biochim Biophys Acta 1762:1025–1037
Rakhit R, Crow JP, Lepock JR et al (2004) Monomeric Cu, Zn-superoxide dismutase is a common misfolding intermediate in the oxidation models of sporadic and familial amyotrophic lateral sclerosis. J Biol Chem 279:15499–15504
Rakhit R, Cunningham P, Furtos-Matei A et al (2002) Oxidation-induced misfolding and aggregation of superoxide dismutase and its implications for amyotrophic lateral sclerosis. J Biol Chem 277:47551–47556
Rakhit R, Robertson J, Vande Velde C et al (2007) An immunological epitope selective for pathological monomer-misfolded SOD1 in ALS. Nat Med 13:754–759
Reaume AG, Elliott JL, Hoffman EK et al (1996) Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat Genet 13:43–47
Rothstein JD (2009) Current hypotheses for the underlying biology of amyotrophic lateral sclerosis. Ann Neurol 65(Suppl 1):S3–S9
Schnell SA, Staines WA, Wessendorf MW (1999) Reduction of lipofuscin-like autofluorescence in fluorescently labeled tissue. J Histochem Cytochem 47:719–730
Sipe JD (1992) Amyloidosis. Annu Rev Biochem 61:947–975
Sreedharan J, Blair IP, Tripathi VB et al (2008) TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319:1668–1672
Stathopulos PB, Rumfeldt JA, Scholz GA et al (2003) Cu/Zn superoxide dismutase mutants associated with amyotrophic lateral sclerosis show enhanced formation of aggregates in vitro. Proc Natl Acad Sci USA 100:7021–7026
Stefani M, Dobson CM (2003) Protein aggregation and aggregate toxicity: new insights into protein folding, misfolding diseases and biological evolution. J Mol Med 81:678–699
Tu PH, Raju P, Robinson KA et al (1996) Transgenic mice carrying a human mutant superoxide dismutase transgene develop neuronal cytoskeletal pathology resembling human amyotrophic lateral sclerosis lesions. Proc Natl Acad Sci USA 93:3155–3160
Uversky VN, Fink AL (2004) Conformational constraints for amyloid fibrillation: the importance of being unfolded. Biochim Biophys Acta 1698:131–153
Valdmanis PN, Rouleau GA (2008) Genetics of familial amyotrophic lateral sclerosis. Neurology 70:144–152
Wang J, Slunt H, Gonzales V et al (2003) Copper-binding-site-null SOD1 causes ALS in transgenic mice: aggregates of non-native SOD1 delineate a common feature. Hum Mol Genet 12:2753–2764
Wang J, Xu G, Gonzales V et al (2002) Fibrillar inclusions and motor neuron degeneration in transgenic mice expressing superoxide dismutase 1 with a disrupted copper-binding site. Neurobiol Dis 10:128–138
Wang L, Deng HX, Grisotti G et al (2009) Wild-type SOD1 overexpression accelerates disease onset of a G85R SOD1 mouse. Hum Mol Genet 18:1642–1651
Watanabe M, Dykes-Hoberg M, Culotta VC et al (2001) Histological evidence of protein aggregation in mutant SOD1 transgenic mice and in amyotrophic lateral sclerosis neural tissues. Neurobiol Dis 8:933–941
Williamson D (1993) The unstable haemoglobins. Blood Rev 7:146–163
Zhang YJ, Xu YF, Dickey CA et al (2007) Progranulin mediates caspase-dependent cleavage of TAR DNA binding protein-43. J Neurosci 27:10530–10534
Zinman L, Liu HN, Sato C et al (2009) A mechanism for low penetrance in an ALS family with a novel SOD1 deletion. Neurology 72:1153–1159
Acknowledgments
This study was supported by grants from the Canadian Institutes of Health Research to A.C. and J.R. We thank Nancy F.L. Ng for critical comments, and Kelvin So and Milan Ganguly for performing immunohistochemistry experiments.
Author information
Authors and Affiliations
Corresponding authors
Additional information
A. Kerman and H.-N. Liu contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Kerman, A., Liu, HN., Croul, S. et al. Amyotrophic lateral sclerosis is a non-amyloid disease in which extensive misfolding of SOD1 is unique to the familial form. Acta Neuropathol 119, 335–344 (2010). https://doi.org/10.1007/s00401-010-0646-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-010-0646-5