
Abstract
Years of research have highlighted the importance of the immune system in Alzheimer’s disease (AD), a system that, if manipulated during strategic time windows, could potentially be tackled to treat this disorder. However, to minimize adverse effects, it is essential to first grasp which exact aspect of it may be targeted. Several clues have been collected over the years regarding specific immune players strongly modulated during different stages of AD progression. However, the inherent complexity of the immune system as well as conflicting data make it quite challenging to pinpoint a specific immune target in AD. In this review, we discuss immune-related abnormalities observed in the periphery as well as in the brain of AD patients, in relation to known risk factors of AD such as genetics, type-2 diabetes or obesity, aging, physical inactivity and hypertension. Although not investigated yet in clinical trials, C5 complement system component, CD40/CD40L interactions and the CXCR2 pathway are altered in AD patients and may represent potential therapeutic targets. Immunotherapies tested in a clinical context, those aiming to attenuate the innate immune response and those used to facilitate the removal of pathological proteins, are further discussed to try and understand the causes of the limited success reached. The prevailing eagerness to move basic research data to clinic should not overshadow the fact that a careful preclinical characterization of a drug is still required to ultimately improve the chance of clinical success. Finally, specific elements to consider prior to initiate large-scale trials are highlighted and include the replication of preclinical data, the use of small-scale human studies, the sub-typing of AD patients and the determination of pharmacokinetic and pharmacodynamics parameters such as brain bioavailability and target engagement.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
General introduction
To this day, the mechanisms driving the pathophysiology of Alzheimer’s disease (AD) remain largely elusive but will inevitably necessitate a considerable understanding if treatment development and efficacy are to be reached. Genetic and epidemiological studies have identified a number of important risk factors associated with the sporadic forms of AD, including the expression of the Apolipoprotein E (APOE) 4 allele, aging, diabetes, hypertension, physical and intellectual inactivity, among others [4]. However, none of these risk factors, taken singly or in combination, are fully accountable for the cerebral pathology that characterizes AD, especially when it relates to the events that take place years before diagnosis [142, 152]. The data collected since the first description of the disease [3], more than 100 years ago, has depicted a portrait of AD of the highest complexity; a heterogeneous condition not only emerging from a plethora of causes but manifesting in several unique clinical features [152].
While it is widely accepted that inflammation and the immune system as a whole are intimately linked to AD pathology, their direct contribution to disease onset and progression is still much debated [58, 173]. A number of strategies aiming to modulate the immune response have nonetheless already been tested on patients but have been met with limited success [15, 17, 20, 36, 76, 97, 119, 131]. The objective here is to provide an overview of immune-related abnormalities reported in humans with sporadic AD, discuss the preclinical and clinical-based evidence for an association between immunity and the risk of AD, and reevaluate the different therapeutic strategies related to the immune response that have been tested thus far.
Evidence of immune-related abnormalities in human AD
Virtually all pathways of activation, control and signaling of the immune response show some degree of defect in AD. In the periphery, alterations in the response to activation of lymphocytes, monocytes and granulocytes, and in the cytokine and chemokine expression and secretion, complement system factor levels and toll-like receptor (TLR) expression have all been described in AD individuals (Suppl. Table 1 for details and references). Similarly, postmortem studies in brain samples of individuals presenting with AD-related neuropathologies have unveiled modifications in receptors and proteins of the complement system, cytokine and chemokine levels, infiltration of lymphocytes, and modulations of TLR expression (Suppl. Table 2 for details and references).
The complement system
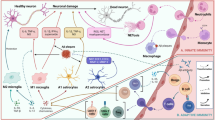
The complement system encompasses over 30 soluble proteins, cell receptors and control proteins. This central element of innate immunity promotes inflammation, annihilates microorganisms, removes apoptotic cells and immune complexes, and is altogether pivotal in the regulation of adaptive responses [129]. Three activation pathways have been unveiled: classical (initiated by antigen–antibody complex), alternative (initiated by activating surfaces such as microbial fragments, tumor cells, and intracellular organelles) and lectin pathways (initiated by the fusion of mannose binding lectin (MBL) to carbohydrates from the surface of bacteria), all converging to the cleavage of C3 fragment, ultimately leading to the formation of the membrane attack complex C5b-9 and cytolysis [129, 175]. In the brain of AD patients, modulations of RNA and/or protein levels from elements of all three activation pathways as well as colocalisation of complement factors with amyloid plaques or neurofibrillary tangles have been observed (Fig. 1 and Suppl. Table 2). Their contribution to AD neuropathogenesis could be on two opposite levels. On the one hand, the observed increased levels of the C3 convertase inhibitors—Factor I and H—and the inactive form of C3b–iC3b—[88, 89] may suggest diminished activation of pathways involved in the clearance of pathologic proteins such as tau and Aβ. On the other hand, the higher levels of C5 to C9 mRNAs [138], along with the detection of the membrane attack complex C5b-9 in AD brains, could potentiate neuronal death [178]. To decrease the membrane attack complex formation without compromising the pro-phagocytic proprieties of the complement system, anti-C5 monoclonal antibody, such as the FDA-approved eculizumab, could be tested in preclinical and clinical trials [124].

Implications of the complement factors in AD. The complement system, comprising more than 30 different factors, is a focal element of innate immunity. Activation of the complement by immune complexes (classical pathway), mannan (lectin pathway) or via spontaneous hydrolysis of C3 and foreign surfaces with a low sialic acid content (alternative pathway) all result in the opsonization and phagocytosis of target, leukocyte recruitment and cell lysis. All activation pathways converge into the formation of unstable protease complexes, the C3-convertases (C4bC2a in case of classical and lectin pathways and C3bBb for the alternative pathway), which cleaves C3 in C3b, C3a (a chemokine) and other cleavage products. C3b plays 2 major roles in complement activation. First, it can serve as an opsonin, and second, the binding of C3b to C3-convertase will generate the C5-convertase (C4bC2aC3b for the classical and lectin pathways; and C3bBbC3b for the alternative pathway). The cleavage of C5 by C5-convertase will produce C3b and C5a, a powerful chemokine that binds to C5a receptors: C5aR1 and C5L2. The recruitment of C6, C7, C8 and C9 by C5b ultimately forms the membrane attack complex within the target cell, inducing cell lysis. The activation of the complement cascade is tightly regulated to control autoimmunity and to minimize damage to host cells. Under normal conditions, factor H binds host-associated C3b and accelerates the decay of the alternative pathway C3-convertase. C3b can also be degraded in its inactive form, iC3b, in a reaction that requires factor I, and a co-factor such as factor H. Therefore, the complement system is involved in the opsonization and phagocytosis of antigens, which may participate to the clearance of Aβ oligomers. Additionally, it also enhances the inflammatory response and contributes to cell death by lysing targeted cells. In the brains of AD patients, several complement factor proteins (blue) and mRNAs (green) are increased. Components of the complement system have also been identified in pathological structures such as neurofibrillary tangles (red) and amyloid plaques (yellow). Molecules of the complement further colocalize with pyramidal neurons (brown) in AD. Given that the activation of C5 and downstream complement components may lead to cell death in the brain, the inhibition of chronic C5 activation could potentially lead to beneficial effects in AD (For references, please see Suppl. Tables 2). Abbreviations AD Alzheimer’s disease, MASPs mannose binding lectin-associated serine proteases, MBL mannose binding lectin
The adaptive immune system
Blood leukocytes originate from the specific differentiation of hematopoietic stem cells into the myeloid or the lymphoid cell lines. The myeloid cell line gives rise to monocytes and granulocytes, whereas cells of the lymphoid pathway are destined to become lymphocytes [43]. Since circulating leukocytes are easily accessible and probed using inexpensive methods, these cells have been investigated in numerous studies attempting to identify AD biomarkers [121]. Differences in the number of cells and in their response to activation observed in AD patients have been associated with the pathogenesis and disease progression (Suppl. Table 1). Predominantly because of methodological issues, many studies have focused on peripheral blood mononuclear cells (PBMC), which are easily purified and stored allowing extensive characterization.
Among the PBMCs, B and T lymphocytes are the main components of the adaptive immune system. Experimental studies and clinical observations provide ample evidence of a deficient adaptive immune response in AD [77, 113, 141, 146]. This response relies on the activation of T lymphocytes following antigen presentation of pathological/foreign molecules by dendritic cells, macrophages and B lymphocytes, and subsequent costimulation [25]. The adaptive immune response not only is a prerequisite for long-lasting protection against pathogens, cancer cells and toxic molecules, but also plays a key role in the development of an adequate immune response to misfolded proteins such as tau and Aβ [6].
Different T lymphocyte subtypes have been associated with a variety of cell functions. Helper T lymphocytes assist the proliferation, differentiation or antibody production by B lymphocytes, or the phagocytic mononuclear cells to eliminate pathogens [25]. Cytotoxic T lymphocytes, on the other hand, are responsible for the destruction of cells infected by intracellular pathogens. Naïve T lymphocytes are mature cells that differentiate in the bone marrow and subsequently undergo positive and negative selection processes in the thymus, having never met their antigen [68]. This cell population is essential to the defense against new pathogens or neoplastic cells. In contrast, memory T lymphocytes have previously met their antigens. Lower exposure to costimulating molecules is needed for their activation, which is therefore more rapid upon a secondary infection [68].
A lower number of naïve T lymphocytes have been observed in AD [77, 113, 132], along with increased activation of circulating lymphocytes [113, 141]. However, findings obtained in the context of ex vivo stimulation contradict this. Indeed, studies using mitogen activation of AD lymphocytes reveal decreased [123, 146] or increased [90, 118] mitogen activation levels. Lower numbers of B lymphocytes [115, 141, 174], in parallel with decreased apoptosis [13] and response to mitogenic activation [146] within this antibody-secreting cell population, have also been reported in AD.
Along with differences in lymphocyte numbers, the cluster of differentiation (CD)40/CD40 ligand (CD40L) costimulation pathway plays a role in homeostasis and immune control. CD40 is a cell surface molecule that regulates activation and differentiation of B lymphocytes, binding to its ligand CD40L located onto T lymphocytes [25]. Increased levels of soluble CD40 are notable in the blood of AD patients [2, 99] as well as in individuals suffering from mild cognitive impairments (MCI), up to 5 years before their evolution into clinical AD [19]. Elevated soluble levels of CD40L (sCD40L) in the plasma of AD patients have been reported as well [2, 35]. In the brain, this pathway is critical for the activation of microglial cells [44]. Incidentally, increased expression of CD40L and CD40, by astrocytes and microglia respectively—suggesting increased activation of both cell types—are detected in AD brains using immunostaining [23, 155]. Taken together, these findings argue for a role of the CD40/CD40L pathway in AD. From a therapeutic point of view, pilot studies with the CD40-antagonist monoclonal antibody FFP104 in primary biliary cirrhosis and Crohn’s disease were recently initiated (clinicaltrials.gov, number NCT02193360 and NCT02465944, respectively). Positive outcomes on pharmacological activity and safety could support the initiation of clinical trials in AD as well.
Results from animal studies have provided strong evidence in favor of a role of the immune system in the manifestation of cognitive deficits [34, 70, 159, 171]. For example, young mice (3- to 4-month-old) injected with the plasma of 18- to 20-month-old animals suffer from learning and memory impairments; an observation that is reproducible by the injection of eotaxin [159]. In this study, eotaxin, a chemokine involved in allergic responses, was shown to inhibit adult neurogenesis and impair learning and memory, suggesting that systemic immune-related factors contribute to the susceptibility of the aging brain to cognitive impairments [159]. A recent study further showed that eotaxin promotes microglial migration and induces neuronal death by triggering the production of reactive oxygen species by microglia [109]. Moreover, while T lymphocyte depletion decreases neurogenesis via a CD4+-T-lymphocyte-dependent mechanism [171], adoptive transfer of splenocytes from wild-type mice ameliorates cognitive performances in transgenic mice deficient in T lymphocytes [70]. Following training in the Morris water maze, meningeal accumulation of T lymphocytes is associated with cognitive improvement [34]. In the PSAPP mouse model of AD, injections of PBMC from human umbilical cord blood reduce amyloid neuropathology and neuroinflammation by a CD40/CD40L-dependent mechanism [105]. Taken together, animal work supports observations collected in AD patients, demonstrating a protective effect of peripheral adaptive immunity on cognition and suggesting that peripheral immune impairments could be linked to disease exacerbation. Although several preclinical findings suggest that adaptive immunity could be a valid target for therapeutic interventions, very few clinical trials have specifically addressed this.
Microglia and astrocytes
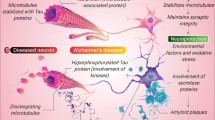
Evidence from animal studies suggests that in early stages of the disease, microglial activation may contribute to Aβ removal and prevent plaque formation [116]. However, in later stages, pro-inflammatory and dysfunctional microglia would rather promote tau pathology and neuropathological progression [116]. Except for an increase in the anti-inflammatory cytokine IL-10, other modulations of cytokines or receptors documented in the brain of AD patients suggest glial activation and upregulation of pro-inflammatory pathways (Fig. 2; Suppl. Table 2). The CD200-CD200R interaction that maintains microglia in a quiescent state is compromised in AD by a decreased expression of both of these molecules [150, 162]. Likewise, the increased expression of CD40/CD40L costimulation molecules further supports an increased activation of immune cells in the brain of AD patients [23, 155]. In vitro activation of microglial cells and astrocytes with Aβ peptide leads to secretion of a number of cytokines such as interleukin (IL)-1β, IL-6, monocyte chemoattractant protein-1 (MCP-1, also called CCL2) and IL-8 [37], the levels of which are increased in AD [140, 161]. A common receptor for IL-8 and the growth-regulated oncogenes (GRO)-α, GRO-β and GRO-γ—other molecules for which the mRNA level are increased in AD—is CXC chemokine receptor 2 (CXCR2) [150]. This receptor is expressed by neurons, astrocytes and microglial cells and is involved in leukocyte recruitment and transmigration. Therefore, CXCR2 binding with IL-8 and/or GRO could explain the presence of T lymphocytes in AD brains [172, 179]. Furthermore, CXCR2 activation has been reported to be involved in the γ–secretase expression and activity, increasing Aβ production and associated glial activation [154]. SCH52123, a CXCR2 antagonist that has already been tested in clinical trials for chronic obstructive pulmonary disease and asthma, may perhaps represent a new treatment target for AD [102, 120]. From a therapeutic point of view however, caution should be exercised before targeting immune pathways for AD therapies. The possibility remains that these modifications can have both beneficial and detrimental consequences on AD pathogenesis. Indeed, despite MCP-1 increases in the brain of AD patients, animal studies have demonstrated acceleration of plaque formation, and exacerbation of Aβ oligomerization in MCP-1-deficient APP/PS1 mice [72].

Immune involvement in AD neuropathogenesis: evidence from postmortem human brain analyses. 1 Cleavage of APP by γ- and β-secretase produces Aβ peptides that are prone to oligomerization and assemble as Aβ oligomers. 2 These oligomers can in turn trigger the activation of microglia and astrocytes. Consequently, these cells produce increased levels of IL-1β, IL-6, IL-8, IL-10, MCP-1 proteins as well as GRO mRNA. In cerebral tissues from the brain of AD patients, rising amounts of CD40L on astrocytes and CD40 on microglia, along with increased levels of CD74, and reduced quantities of CD200 and CD200R all support pro-activation pathways. 3 Increased expression of IL-8 and GRO protein can bind to CXCR2. In the brain, this receptor is expressed by microglia, astrocytes and neurons and its activation modulates the expression and activity of γ-secretase. 4 Aβ oligomers also affect synaptic integrity as well as cell-to-cell communication in neurons. 5 The end product of Aβ aggregation is the formation of amyloid plaques, one of the hallmarks of AD. CD74+ microglia are associated with amyloid plaques in AD brains. 6 Hyperphosphorylated tau concentration is increased in AD neurons. As a result, neurofibrillary tangles—another hallmark of AD—also accumulate in the brains of AD patients and are colocalized with CD74+ microglia. 7 Endothelial cells of the BBB express molecules such as CD40 and IL-8 that could be implicated in immune cell transmigration. Decreased levels of tight junction proteins and increased levels of extravascular IgG have been reported and support BBB impairments in AD. 8 Some of the Ig+ neurons express the active form of caspase-3, a marker of cell death, whereas caspase-3 immunoreactivity is absent from Ig- neurons. 9 In the hippocampus of AD patients, increased numbers of auxiliary (CD4+) and cytotoxic (CD8+) T lymphocytes, as well as higher levels of cytotoxic over auxiliary cells have been reported. Overall, the data collected from human brains demonstrate that the immune response in AD favors immune-related cell activation and support CXCR2 (i.e., SCH527123, a CXCR2 antagonist) or CD40/CD40L (i.e., FFP104, a CD40-antagonist antibody) pathways as potential new pharmacological targets (For references, please see Suppl. Tables 1 and 2). Abbreviations AD Alzheimer’s disease, APP amyloid β precursor protein, BBB blood–brain barrier, CD cluster of differentiation, CXCR2 CXC chemokine receptor 2, GRO growth-regulated oncogene, Ig immunoglobulin, IL interleukin, MCP-1 monocyte chemoattractant protein-1, MHC II major histocompatibility complex class II, mRNA messenger ribonucleic acid
The immune system and the blood–brain barrier (BBB)
Although for decades the brain was believed to be sheltered from the peripheral immune system, we now know that there is continuous communication between the brain and immune blood cells [101]. At the interface between the brain and the blood stands the BBB, a vital active element in the regulation of the brain immune response [101]. In AD, disruptions of the tight junctions, morphological anomalies of the microvasculature, decreased cerebral blood flow, presence of blood-borne compounds in the cerebrospinal fluid (CSF), increased transcytosis and/or enzymatic degradation of basal lamina proteins have been proposed as possible indicators of a dysfunction of the BBB [66, 180], some of which have been replicated in animal models [16]. Although evidence of enhanced BBB permeability in AD has been published, the extent by which this affect drug distribution or AD pathogenesis per se remains controversial [38]. Cerebrovascular deposits of Aβ peptides in small arteries, arterioles as well as capillaries, known as cerebral amyloid angiopathy, may result in cognitive deficits and are frequently observed in elderly with and without AD [9, 71]. In a subset of the cerebral amyloid angiopathy cases, evidence of inflammation and edema on magnetic resonance imaging are accompanied with rapid cognitive decline [71]. Interestingly, this encephalopathy is reminiscent of the autoimmune inflammation following anti-Aβ immunization therapy and is reversible with immunosuppressive or corticosteroid treatments, further emphasizing the need for adequate diagnosis of cognitive impairments in elderly [71].
It has been suggested that these BBB anomalies are associated with modifications of the neurovascular unit, which can impact cerebral immunity by favoring immune cell transmigration into the paravascular space, particularly in inflammatory conditions [101]. Evidence from the experimental autoimmune encephalopathy (EAE) model of multiple sclerosis suggests that the migration of blood mononuclear cells to the brain is possible even in the presence of relatively intact tight junctions [170]. Moreover, in the EAE model or in cultured endothelial cells, transcellular migration is dependent upon the expression, by endothelial cells or leukocytes, of a plethora of factors including ninjurin-1, α4-integrin, activated leukocyte cell adhesion molecule (ALCAM) and intercellular adhesion molecule (ICAM) [28, 59, 60, 98].
Such interactions are hard to confirm in human brains, but data obtained from both in vitro models of BBB and in vivo models of brain amyloidopathy (via intracranial injection of Aβ peptides) suggest that increased chemokine and cytokine secretion can promote leukocyte migration to the brain in the context of AD neuropathology [40, 86, 91, 176]. Observations of increased expression of CD40 on cerebrovascular cells [155] in AD further support a role in immune activation. Therefore, the anomalies of the BBB found in neurodegenerative diseases such as AD may thus facilitate the transmigration of peripheral leukocytes to the brain as well as the activation of the immune cells within the brain [180].
Risk factors of AD and inflammation: confounding variables
The sporadic forms of AD are likely to originate from a convoluted interplay between genetic and environmental risk factors, in which immune dysfunctions play a role. Indeed, the majority of AD risk factors are themselves accompanied by important deficits of the immune system. For example, immune impairments have been reported in aging, APOE4 allele carriage, obesity and diabetes, hypertension as well as physical inactivity. These confounding variables must therefore be kept in mind when attempting to dissect the role of inflammation in AD pathogenesis through the known immune-related changes associated with the main risk factors of sporadic AD.
Genetic risk factors
Genetic vulnerabilities associated with sporadic AD appear to be driven by different allelic forms of a variety of genes [151]. APOE [133, 147] was the first discovered and remains, to this day, the most prominent genetic factor in sporadic AD [151]. In humans, the apoE isoform expressed by immune cells has been shown to modify cell response to immune stimuli. For example, in response to ex vivo stimulation using TLR2, TLR4 or TLR5 ligands, blood cells from APOE3/APOE4 carriers produce increased cytokine levels [42]. Furthermore, susceptibility to apoptosis upon stimulation is increased in macrophages from mice expressing the human apoE4 protein compared to human apoE3 [26]. Genome wide association studies (GWAS) and other complementary approaches focusing on the recognition of genetic risk factors for late onset sporadic AD have led to the identification of nine additional genetic loci [134, 151]. Among these genes, six code for proteins that can be assigned to the immune response, namely ABCA7, CD33, CLU, CR1, EphA1, and MS4A [134]. The role of a rare variant of the gene TREM2, rs75932628, was more recently reported [50, 64]. A study performed in Icelandic individuals has shown that this variant, affecting 0.63 % of the population, encodes an arginine-to-histidine substitution at position 47, which seemingly accelerates the disease onset by 3.18 years. This variant further confers a relative risk of 2.92, similar to that of heterozygosity for the APOE4 allele [64]. TREM2 codes for a membrane protein, which is up-regulated in myeloid cells accumulating in human AD brains and mouse models, along with decreased levels in the CSF of patients with AD and frontotemporal dementia [62, 73]. Of note, lentiviral-mediated overexpression of TREM2 decreases brain amyloid burden and rescues spatial cognitive impairments in APPswe/PS1dE9 mice [63]. Surprisingly, TREM2-deficient mice led to contrasting findings, either exacerbating or reducing amyloid pathologies [62, 163]. These studies used different AD mouse models; nevertheless, such different outcomes emphasize the need to repeat preclinical investigations in multiple models and assess cognitive decline when developing new therapeutic targets for AD.
Aging
Old age is a common predictive factor to both familial and sporadic forms of AD. The elderly often display qualitative and quantitative modifications of the immune response—also referred to as immunosenescence—which are associated to a higher susceptibility to infections, neoplasia and autoimmune events [164]. Decreasing proportion of regulatory and naïve T cells, increasing concentrations of circulating IL-6, tumor necrosis factor (TNF)α and C-reactive protein (CRP), functional deficits of antigenic presentation, reduction of antibody production and reduction of cytotoxic function of natural killer (NK) cells have all been detected with aging [113, 130, 164].
Physical activity
Exercise may impact multiple aspects of the immune response such as T cell phenotype and proliferation, immune response to vaccination and cytokine production upon activation [137]. It can decrease levels of C-reactive protein and IL-6 in patients at risk for heart disease and reduce infection rate in elderly [126]. In this population, moderate exercise has also been proposed to counteract age-related immunosenescence such as reduced response to vaccination and low-grade inflammation [126]. Individuals at risk of AD could therefore benefit from regular physical activity in terms of improved immunological health, as was corroborated by a recent study demonstrating a reduced concentration of TNFα and IL-6 and improved cognition after a 16-week exercise program in elderly with MCI [103].
Obesity and type 2 diabetes
Epidemiological studies have established a link between obesity, insulin resistance, type 2 diabetes and pro-inflammatory factors which include increased number of circulating leukocytes and higher plasmatic concentrations of IL-6, plasminogen inhibitor-1 and CRP [74, 80]. Along these lines, increases in IL-6 and IL-8 in the plasma of obese individuals correlate with insulin resistance [18]. Immune cells infiltrating the adipose tissue and the liver, such as macrophages and T cells, are also important mediators of inflammation [74]. Whereas impairments in the adaptive immune system, possibly contributing to cognitive deficits, have been reported in AD [77, 113, 141, 146], these data support an increased activation of the innate immune response and tissue infiltration by leukocytes in obesity and type 2 diabetes [74, 80]. It is tempting to speculate that inflammatory components of these metabolic diseases may help trigger AD neuropathology within the CNS, as data collected in animal studies suggests [65, 157, 158]. While this hypothesis remains to be validated, mid-life obesity and type 2 diabetes are now well-known risk factors of AD and therefore must be factored in when studying immunity in this disease context.
Hypertension
Hypertension affects approximately one-third of the western population, and its prevalence increases with age, reaching up to 70 % of individuals by the age of 70 [96]. Evidence on a causal role of immunity in the development of hypertension in humans is limited; however, it has been associated with accumulation of T cells and monocytes/macrophages in vessels and kidney [5, 96]. A significant linear relationship between blood pressure and levels of soluble ICAM-1 or IL-6 have been observed in cohorts of healthy men [5]. In the blood of newly diagnosed, treatment-naïve patients with hypertension, increased levels of immunosenescent cytotoxic T cells secreting higher amounts of perforin, granzyme and interferon (IFN)γ have been reported [177]. A T cell-dependent pro-inflammatory response is further supported by increased levels of plasmatic IL-17A [5, 96]. Interestingly, as in AD cases, sCD40L levels are also increased in hypertensive individuals [2, 5, 35].
Immunotherapies in AD: One step forward, two steps back?
Immunotherapeutic strategies to treat AD can be classified under two main headings: (1) strategies aiming to attenuate the innate, pro-inflammatory immune response or (2) strategies designed to modulate adaptive immunity to facilitate CNS Aβ clearance. Many of the therapeutic agents under preclinical or clinical investigation in AD have the properties to interfere with inflammation or other immune-related processes. These include nonsteroidal anti-inflammatory drugs (NSAID), passive and active immunization, statins, TNFα antagonists, omega-3 fatty acids as well as inhibitors of acetylcholinesterase [21, 78]. Here, we opted to focus our discussion on NSAID and immunization, which have been the favored strategy in the majority of immune-related clinical trials.
NSAID
The first evidence for the potential benefits following NSAID treatment comes from epidemiological data showing that prolonged intake of NSAID decreases the risk of developing AD [95] with a stronger association in ibuprofen users [160]. In contrast, epidemiological studies on cohorts of older individuals (median age: 74–75 year old at recruitment) indicate that the use of NSAID does not correlate with such positive outcome [17, 61].
Clinically used NSAID fall into two categories: those inhibiting equally cyclooxygenase (COX)-1 and COX-2 (indomethacin, naproxen, ibuprofen, diclofenac) or those that selectively inhibit COX-2 (celecoxib, meloxicam, rofecoxib) [128]. COX are enzymes catalyzing the conversion of arachidonic acid into prostaglandin (PG) G2 and H2 [128]. The PGE2, which is produced from PGH2, is one of the most abundant PG in the body and is implicated in a broad-spectrum of functions, which include regulation of the immune response, blood pressure, gastro-intestinal integrity and fertility [122]. In AD, COX-2 mRNA and protein levels are increased in the frontal cortex [111] and hippocampus [55]. In vitro assays as well as studies conducted in the Tg2576 and APPsw mouse models of AD have corroborated that NSAID (diclofenac, fenoprofen, sulindac, indomethacin, ibuprofen, flurbiprofen and meclofen) could reduce Aβ42 production [29], which has further lead to the initiation of clinical trials. Unfortunately, the treatment of AD using acetylsalicylic acid, NSAID and steroidal anti-inflammatory drugs all failed to improve primary outcome measures which included decline in cognitive function, as well as depression, activity of daily living and neuropsychiatric symptoms [61]. In MCI individuals, however, triflusal, an analog of acetylsalicylic acid, attenuated the rate of conversion to dementia, although these results must be interpreted with caution given that the study was terminated prematurely due to recruitment issues [45]. Moreover, studies in animal models with ibuprofen or flurbiprofen indicate that the Aβ lowering effects of NSAID may be independent from their COX-related anti-inflammatory action and emphasize the fact that a careful preclinical selection of a drug improves the chance of clinical success [30, 97].
It is now widely accepted that AD pathogenesis begins years before the manifestation of initial symptoms. ADAPT (Alzheimer’s Disease Anti-inflammatory Prevention Trial), a double-blind study conducted to evaluate the efficacy of NSAID in the prevention of AD, tackled this concept [97]. This study not only failed to show benefits of NSAID, but revealed cardiovascular toxicity associated with COX-2 inhibitors (celecoxib) [97, 128] and suggested a slight detrimental effect of naproxen on cognition [93]. Of note, naproxen—used instead of ibuprofen due to placebo concerns—was one of the NSAID devoid of Aβ lowering effects [30]. An additional 2 years of monitoring revealed that NSAID have an adverse effect on AD pathogenesis in advanced AD but treatment during the presymptomatic stage for more than 2–3 years reduces the incidence of the pathology [17]. These results highlight the necessity to begin clinical trials at the earliest stage of the disease and to extend duration of treatment as well as the follow-up [1, 24, 52] (Fig. 3).

Potential windows of treatment in immunotherapeutic clinical trials. Immune-related therapies have been proposed as disease-modifying treatments that will slow or halt the progression of AD. The normal progression of AD is represented as a black curve. It is hypothesized that the clinical benefits of disease-modifying trials (red and blue curves) will take time to be detected, but will increase with treatment duration. A disease-modifying effect of immunization is also expected to persist beyond the end of the treatment. The cumulative therapeutic effect of disease-modifying therapies will likely decrease with disease progression. The corollary is that an early therapeutic intervention in the pre-symptomatic or early symptomatic stage of the disease (blue curve) will be particularly more impactful than late intervention (red curve) in a disease-modifying paradigm. Therefore, both early interventions and increased trial duration would be critical to ultimate clinical efficacy. Abbreviations AD Alzheimer’s disease, APOE apolipoprotein E, PK pharmacokinetic, PD pharmacodynamics
Active immunization
While NSAID attenuate the pro-inflammatory response of the immune system, the objective of active and passive immunization strategies is to take advantage of the immune system to decrease the amyloid or tau burden and thereby halt or reverse cognitive decline/disease progression. In transgenic models of brain amyloid pathology, active immunization (i.e., vaccination) have resulted in a significant reduction in the number of amyloid plaques, increased synaptic density and improvement of cognitive performance in most published studies [22, 106, 135]. These results were quickly translated into clinical initiatives that have been completed or are currently underway (Table 1), with the very first trial using active immunization initiated in 2000. The AN1792 vaccine used a synthetic Aβ42 peptide and the immunogenic adjuvant QS-21. Although immunogenicity was obtained in 50 % of treated individuals, the development of aseptic meningoencephalitis in 6 % of the participants led to the termination of the development of this vaccine [107]. Infiltration of T lymphocytes has in fact been identified as the main cause of the adverse effects of AN1792 immunization [104]. Despite a significant reduction of the amyloid burden identified by postmortem histological analyses, the assessment of the cognitive performance of remaining participants to the AN1792 study failed to highlight an impact on disease progression [56, 104].
The development of second-generation vaccines soon emerged from these initial investigations with the goal to prompt a strong antibody production in absence of inflammatory and cytotoxic Aβ-specific T lymphocytes. These new strategies are based on the use of modified antigens, such as truncated Aβ containing the immunodominant B lymphocyte epitope [11, 32, 49, 85, 92], at times coupled to virus-like particles [11, 92], GPGPG spacer [49] or a foreign epitope [32, 85]. A number of these second-generation vaccines are currently being tested in clinical trials (Table 1).
Passive immunization
An alternative to bypass inflammatory and autoimmune adverse effects of active immunization is to directly administer monoclonal antibodies targeting Aβ peptides or other AD-related targets. Although this approach requires repeated injections of the antibody preparation, it allows immediate treatment cessation in the event of adverse effects. Preclinical studies testing passive immunization, also currently tested in the clinical setting, have shown the capacity to decrease amyloid pathology in AD mouse models, some groups further reporting improvement of cognitive functions [27, 79, 81, 168] (Table 1). Similarly to other approaches, passive immunization has been associated with adverse effects. For example, cerebral microhemorrhages have been reported in mice [83, 114, 166, 167]. Administration of the anti-Aβ monoclonal antibody bapineuzumab induced vasogenic edema in AD patients (Table 1). The absence of benefits in a phase III clinical trial involving bapineuzumab also prompted abortion of the study [131]. Nonetheless, some positive results have been reported, such as a reduction of cognitive and functional decline in mild AD treated with solanezumab or with specific doses of BIIB037 (3 and 10 mg/kg but not 1 and 6 mg/kg treatment regimens) [112, 139] (Table 1). Based on preliminary data released from the BIIB037 phase II trial, 26- and 54-week treatments led to significant decreases of 18F-florbetapir binding in the brain, as measured with positron emission tomography (PET) [112]. Such a clear effect on an in vivo amyloid-related biomarker would argue for a higher target engagement for BIIB027 compared to previous immunotherapy trials. For example, analyses from the two phase 3 studies of bapineuzumab revealed differences in amyloid deposits evaluated with Pittsburg B—PET analyses between placebo and treatment groups in APOE4 carriers only, where the difference was due to an increased amyloid level in the placebo group rather than a decrease in the treatment group [131]. Interestingly, the more recent passive immunotherapies seek to target specific Aβ species such as oligomers (crenezumab), protofibrils (BAN2401 and SAR228810) and insoluble fibrils (BII037), rather than Aβ monomers.
As an alternative to monoclonal antibodies, polyclonal intravenous immunoglobulin (IVIg) prepared from the plasma of healthy human donors and used for the treatment of immunodeficiency and autoimmune diseases [51] has also been tested in patients (Table 1). The clinical trials for AD included low-dose IVIg treatment regimen (0.1 to 0.8 g/kg for AD vs. up to 4 g/kg for autoimmune diseases) [36, 119]. Despite promising results in the initial phases I and II trials, the largest clinical study reported thus far, covering an 18-month period and including over 350 participants, did not support the use of IVIg in the treatment of AD, with the possible exception of APOE4 carriers and moderately impaired AD patients [119]. Extended monitoring of these subsets of individuals would be of great value and could further help decipher the benefits of IVIg in this population.
In the wake of these clinical trials, a number of preclinical studies were initiated to investigate the potential mechanisms of action of IVIg (Table 2). Pharmacokinetic analyses suggest that IVIg reach limited, but therapeutically relevant concentrations in cerebral tissue [144]. In line with the immunomodulatory effects of IVIg in immune disorders, these animal studies underscore a large range of immune-related action of IVIg in mouse models of AD. Indeed, decreased CX3CR1 expression in bone marrow cells, modification of blood CD4+/CD8+ T cell ratio, increased microglial activation and elevated brain levels of C5a have been observed in IVIg-treated AD animal models [47, 117, 145, 148]. Despite in vitro work proposing IVIg as an alternative to Aβ-lowering antibodies [149], the results obtained from preclinical studies are inconsistent when it comes to Aβ peptide levels and plaque counts [47, 117, 145, 148]. Nevertheless, beneficial effects of IVIg on synaptic plasticity, Aβ oligomer concentrations and neurogenesis were reported in mouse models of AD [47, 117, 145]. IVIg also generated improvements on recognition memory and percentage of freezing episodes in the fear-conditioning test in old IVIg-treated mice (16–26 month old) [47, 145]. Although the results from clinical trials were not as favorable as expected, preclinical studies did unveil a number of immune- and non-immune-related mechanisms of action for IVIg in AD, which emerge as promising drug targets.
Alternative immunotherapeutic targets
Tauopathy is a key constituent of AD neuropathology as it correlates particularly well with clinical symptoms [48, 156]. Hence, passive or active immunization strategies aiming at reducing the levels of neurofibrillary tangles (NFT) or tau oligomers are currently under investigation (clinicaltrials.gov). In the transgenic animal model P301L, immunization with a fragment of phosphorylated tau (Tau 379–408 [P-Ser396,404]) induced an increase in soluble tau and a decrease in insoluble tau, suggesting a possible mobilization of insoluble tau for subsequent elimination [8]. In this particular study, tau immunization improved motor functions although it failed to delay the progression of the pathology. Consistent data were generated in other preclinical studies, confirming the potential benefits of tau-based immunotherapy in AD [54], and clinical trials for anti-tau active immunization have been initiated (AADvac1, Table 1). Similarly to Aβ-driven immunization, adverse events have been reported for tau immunotherapy. Using full-length human tau (highly homologous to murin tau) to immunize C57Bl/6 mice, Rosenmann and colleagues reported increased gliosis, brain infiltration of monocytes, axonal damage, NFT-like pathology and neurological symptoms similar to those associated to EAE [127], further highlighting the challenges in setting in motion an immune response against an endogenous cerebral protein.
Other players of the amyloid cascade have been targeted for the development of alternative passive immunization. For example, the administration of a BACE1-specific monoclonal antibody reduces CNS concentrations of Aβ peptides in rodents and primates [10]. As a major genetic risk factor, apoE has also been proposed as a suitable target for immunotherapy [69, 84].
Food for thought for future immunotherapy of AD
Despite the impressive amount of clinical and preclinical data available, we still struggle to explain the failure or limited success of immunotherapies in AD. In spite of the enormous amount of data that has been derived from animal work and human studies, the exact role of the inflammatory/immune responses in AD remains unclear [52, 53]. To what extent are these responses beneficial or harmful? What is the relationship between disease progression and immune-related abnormalities observed in AD patients? Are these responses a cause or a consequence of the pathology? Without answers to these critical questions, the development of immune-related therapies may indeed be destined to fail.
Albeit some limitations, postmortem investigations in the brain as well as analysis of blood and CSF markers provide the bulk of evidence for immune dysfunctions in AD [46] (Suppl. Tables 1 and 2). Although the results of these studies may not directly reflect the CNS immunological state, the consistencies on some of the changes observed both in the periphery and CNS—such as CD40 and CD40L increases in plasma and AD brain cells [2, 19, 23, 35, 99, 155]—indicates that indeed, these changes likely relate to the pathology per se. To specifically tackle the pathways involved in AD-specific immune dysfunctions, future studies should (1) take into account the coinciding health issues including risk factors, medication and other comorbidities, (2) establish the AD diagnosis based on multiple scoring tests and neuroimaging data (as well as neuropathology, when available) and (3) include validation of the primary findings on large-scale populations. The identification of immune mechanisms specifically linked to the pathogenesis of AD, at least in subgroups of patients, is the basis for the development of successful immunity-based therapeutic strategies.
To this day, both suppressors (e.g., anti-inflammatory drugs) and activators (e.g., immunization) of the immune response have been tested in the clinic, and both have led to limited benefits. Clinical failure may therefore also be due to the choice of intervention, which has been mostly empirical [57]. The multifactorial and heterogeneous nature of AD suggests that a “one therapy fits all” paradigm may not be the solution, particularly when targeting the immune system. The absence of subclassification of the AD population involved in clinical trials may also explain the overall negative outcomes reached from these studies. Therefore, an in-depth characterization of the different subtypes of AD patients at the levels of biomarkers, genetic risk factors, disease progression, immune phenotype, comorbidities and diversity of clinical symptoms must be taken into account in the design of future immune-related interventions [24, 41, 153].
It is generally recognized that AD neuropathology starts to develop years or decades before the onset of the disease [142, 152]. One of the challenges of AD therapy is to accurately identify preclinical stages in the hope of initiating treatment to stop or slow neuronal damages before the onset of symptoms [142]. However, differential diagnosis of AD is still complicated, the criteria for diagnosis of definite AD requiring histopathologic evidence from biopsy or autopsy [152]. The need of reliable biomarkers for AD thus remains urgent to improve the design and setup of clinical trials aimed at detecting disease modification [142]. Significant progress has been made toward the identification of such biomarkers. The ones currently available are separated in two categories according to whether they relate to cerebral measures (detection of amyloid deposits with 18F-florbetapir or Pittsburgh B compounds, decreased metabolism in parietal and temporal cortex evaluated by 18flurodeoxyglucose using PET imaging, or cortical atrophy using magnetic resonance imaging) or CSF assessments (reduction of Aβ42 as well as increased hyperphosphorylated or total tau). Biomarkers measured in the CSF allow for an AD diagnosis with >85 % specificity [67], and their levels are modified more than 15 years before the onset of symptoms in carriers of autosomal dominant mutations for familial AD [14]. The use of these new tools will considerably improve the diagnosis of preclinical/early-stage AD for the setup of clinical trials with new compounds, for enabling sub-typing of AD patients, determining target engagement and monitoring therapeutic response.
Using biomarker-based advanced characterization of patients in neuropathologically relevant subclasses, immunotherapies will presumably be more effective in well-selected patients and during the preclinical phase of the disease, when the neurodegenerative process may still be reversible (Fig. 3). With the help of new biomarkers, it may be tempting to launch preventive treatment in populations at risk of developing AD, as was tested for NSAID in the ADAPT trial or for the ongoing the Dominantly Inherited Alzheimer Network Trial in individuals with familial AD (Table 1, solanezumab and gantenerumab). For now, the frequency of adverse effects would argue against a broad preventive vaccination trial (Table 1), although this conclusion would need to be revisited with the future development of safe and well-tolerated anti-AD vaccines. Interestingly, although the first immunotherapy trials focused mainly on mild-to-moderate AD, more recent trials included groups with prodromal AD, MCI and cognitively normal individuals carrying familial AD-causing mutation (Table 1). Hopefully, these study designs will yield more positive outcomes.
Reiterating a point made above, it will be imperative to separate abnormalities relating to the risk factors of the disease to those relating to AD per se in order to pinpoint the immunological pathways contributing to AD. The complexity of the interplay between AD, comorbidities and the immune response makes it nearly impossible to fully control all these different parameters in clinical intervention studies. It is at this point that animal models and preclinical studies come into play and are required to formulate hypotheses, on one hand, and provide mechanistic insights to human data, on the other. The use of animal models can help understand key aspects of immune-related mechanisms, including causal relationships. However, the translational potential of these studies remains limited, given the intrinsic differences between human AD and animal models [12, 75]. Nevertheless, one of the lessons learned is that performing extensive preclinical pharmacokinetic and pharmacodynamics characterization of drug before moving into expensive clinical trials is probably a cost-effective idea. In addition to animal models and preclinical studies, pilot or feasibility studies can provide invaluable data to prompt large-scale clinical trials. These small-scale investigations can test important parameters such as mechanisms of recruitment, randomization, treatment and follow-up assessments, as well as providing staff training [165]. However, it is important to note that, due to their small sizes, these trials are not designed to compare groups or to evaluate safety, efficacy of treatment, but rather to enhance the likelihood of success of the main studies [165].
In active and passive immunization, immunoglobulin (Ig) is either produced or injected to AD patients but although considerable financial investments has been made to devise Ig-based treatments for CNS disease, little is known regarding the concentrations that truly gets into the brain. Indeed, Ig are large molecules that cannot diffuse much through the BBB. Although quantitative experiments to determine their brain bioavailability in vivo remain scarce, available data suggest limited access with lower than 0.01 % of administered Ig reaching the brain in mice [144]. Thus, Ig-related clinical trials may also have fallen short of providing cognitive benefits due to poor BBB passage of these therapeutic molecules rather than pharmacodynamic issues [24, 110].
Finally, results from previous AD clinical trials also argue that early initiation of treatment during presymptomatic phases and extended follow-up periods are critical as well for successful disease-modifying treatments (Fig. 3). A thorough retrospective analyses of the results obtained from failed trials would provide invaluable information as to how to design future preclinical and clinical studies [24, 33, 119, 136].
Conclusion
It is still early in the history of immunotherapy for neurodegenerative diseases to determine if it is worth the effort and money invested, and if it truly represents a viable alternative to current pharmacologic strategies for the effective treatment of AD. Immunotherapies tested so far generally fall in two categories: attenuation of the immune response or potentiation of Aβ and tau clearance from the brain (Table 1). However, few have attempted to actually correct any observed changes or anomalies of specific immune pathways, although specific therapeutic compounds have already been tested in clinical trials for other pathologies (i.e., anti-C5 or anti-CD40 antibodies, and CXCR2 antagonist) (Figs. 1, 2). Despite the negative results and adverse events observed with the first immunotherapeutic interventions, studies focused on the immune-mediated removal of pathological proteins still receive most of the attention from the scientific community and pharmaceutical companies. Regardless of the limited success of these trials, in-depth knowledge of immune-related anomalies in AD combined with thorough analysis of the results from preclinical and clinical investigations will definitely provide invaluable data for a better understanding of this devastating disease.
Abbreviations
- Aβ:
-
Amyloid-β peptide
- AD:
-
Alzheimer’s disease
- ALCAM:
-
Activated leukocyte cell adhesion molecule
- APOE:
-
Apolipoprotein E
- APP:
-
Amyloid precursor protein
- BACE:
-
β-Site APP cleaving enzyme
- BBB:
-
Blood–brain barrier
- CD:
-
Cluster of differentiation
- CNS:
-
Central nervous system
- COX:
-
Cyclooxygenase
- CRP:
-
C reactive protein
- CSF:
-
Cerebrospinal fluid
- CX3CR1:
-
Chemokine (C-X3-C motif) receptor 1
- CXCR2:
-
CXC chemokine receptor 2
- EAE:
-
Experimental autoimmune encephalomyelitis
- ICAM:
-
Intercellular adhesion molecule
- IFN:
-
Interferon
- IL:
-
Interleukin
- IVIg:
-
Intravenous immunoglobulin
- MASPs:
-
Mannose binding lectin-associated serine proteases
- MBL:
-
Mannose binding lectin
- MCP-1:
-
Monocyte chemoattractant protein-1
- MCI:
-
Mild cognitive impairment
- MIP:
-
Macrophage inflammatory protein
- NFT:
-
Neurofibrillary tangle
- NK:
-
Natural killer
- NSAID:
-
Nonsteroidal anti-inflammatory drug
- PBMC:
-
Peripheral blood mononuclear cells
- PECAM:
-
Platelet endothelial cell adhesion molecule
- PET:
-
Positron emission tomography
- PS:
-
Presenilin
- RNA:
-
Ribonucleic acid
- s:
-
Soluble
- TLR:
-
Toll-like receptor
- TNF:
-
Tumor necrosis factor
- VCAM:
-
Vascular cell adhesion molecule
References
ADAPT-FS research group (2015) Follow-up evaluation of cognitive function in the randomized Alzheimer’s disease anti-inflammatory prevention trial and its follow-up study. Alzheimers Dement 11(216–25):e1
Ait-ghezala G, Abdullah L, Volmar C-H, Paris D, Luis CA, Quadros A, Mouzon B, Mullan MA, Keegan AP, Parrish J, Crawford FC, Mathura VS, Mullan MJ (2008) Diagnostic utility of APOE, soluble CD40, CD40L, and Abeta1-40 levels in plasma in Alzheimer’s disease. Cytokine 44:283–287
Alzheimer A, Stelzmann RA, Schnitzlein HN, Murtagh FR (1995) An English translation of Alzheimer’s 1907 paper, “Uber eine eigenartige Erkankung der Hirnrinde”. Clin Anat 8:429–431
Association A (2012) Alzheimer’s disease facts and figures. Alzheimers Dement 2012(2):1–72
Anders HJ, Baumann M, Tripepi G, Mallamaci F (2015) Immunity in arterial hypertension: Associations or causalities? Nephrol Dial Transplant 11 Mar 2015. pii: gfv057 [Epub ahead of print]
Anderson KM, Olson KE, Estes KA, Flanagan K, Gendelman HE, Mosley RL (2014) Dual destructive and protective roles of adaptive immunity in neurodegenerative disorders. Transl Neurodegener 3:25
Arai H, Umemura K, Ichimiya Y, Iseki E, Eto K, Miyakawa K, Kirino E, Shibata N, Baba H, Tsuchiwata S (2015) Safety and pharmacokinetics of bapineuzumab in a single ascending-dose study in Japanese patients with mild to moderate Alzheimer’s disease. Geriatr Gerontol Int. doi:10.1111/ggi.12516
Asuni AA, Boutajangout A, Quartermain D, Sigurdsson EM (2007) Immunotherapy targeting pathological tau conformers in a tangle mouse model reduces brain pathology with associated functional improvements. J Neurosci 27:9115–9129
Attems J, Jellinger K, Thal DR, Van Nostrand W (2011) Review: sporadic cerebral amyloid angiopathy. Neuropathol Appl Neurobiol 37:75–93
Atwal JK, Chen Y, Chiu C, Mortensen DL, Meilandt WJ, Liu Y, Heise CE, Hoyte K, Luk W, Lu Y, Peng K, Wu P, Rouge L, Zhang Y, Lazarus RA, Scearce-Levie K, Wang W, Wu Y, Tessier-Lavigne M, Watts RJ (2011) A therapeutic antibody targeting BACE1 inhibits amyloid-{beta} production in vivo. Sci Transl Med 3:84ra43
Bach P, Tschäpe J-A, Kopietz F, Braun G, Baade JK, Wiederhold K-H, Staufenbiel M, Prinz M, Deller T, Kalinke U, Buchholz CJ, Müller UC (2009) Vaccination with Abeta-displaying virus-like particles reduces soluble and insoluble cerebral Abeta and lowers plaque burden in APP transgenic mice. J Immunol 182:7613–7624
Bales KR (2012) The value and limitations of transgenic mouse models used in drug discovery for Alzheimer’s disease: an update. Expert Opin Drug Discov 7:281–297
Bartolome F, de Las Cuevas N, Munoz U, Bermejo F, Martin-Requero A (2007) Impaired apoptosis in lymphoblasts from Alzheimer’s disease patients: cross-talk of Ca2+/calmodulin and ERK1/2 signaling pathways. Cell Mol Life Sci 64:1437–1448
Bateman RJ, Xiong C, Benzinger TL, Fagan AM, Goate A, Fox NC, Marcus DS, Cairns NJ, Xie X, Blazey TM, Holtzman DM, Santacruz A, Buckles V, Oliver A, Moulder K, Aisen PS, Ghetti B, Klunk WE, McDade E, Martins RN, Masters CL, Mayeux R, Ringman JM, Rossor MN, Schofield PR, Sperling RA, Salloway S, Morris JC (2012) Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med 367:795–804
Bayer AJ, Bullock R, Jones RW, Wilkinson D, Paterson KR, Jenkins L, Millais SB, Donoghue S (2005) Evaluation of the safety and immunogenicity of synthetic Abeta42 (AN1792) in patients with AD. Neurology 64:94–101
Bourasset F, Ouellet M, Tremblay C, Julien C, Do TM, Oddo S, Laferla F, Calon F (2009) Reduction of the cerebrovascular volume in a transgenic mouse model of Alzheimer’s disease. Neuropharmacology 56:808–813
Breitner JC, Baker LD, Montine TJ, Meinert CL, Lyketsos CG, Ashe KH, Brandt J, Craft S, Evans DE, Green RC, Ismail MS, Martin BK, Mullan MJ, Sabbagh M, Tariot PN, R. G. ADAPT (2011) Extended results of the Alzheimer’s disease anti-inflammatory prevention trial. Alzheimers Dement 7:402–411
Bruun JM, Verdich C, Toubro S, Astrup A, Richelsen B (2003) Association between measures of insulin sensitivity and circulating levels of interleukin-8, interleukin-6 and tumor necrosis factor-alpha. Effect of weight loss in obese men. Eur J Endocrinol 148:535–542
Buchhave P, Janciauskiene S, Zetterberg H, Blennow K, Minthon L, Hansson O (2009) Elevated plasma levels of soluble CD40 in incipient Alzheimer’s disease. Neurosci Lett 450:56–59
Burstein AH, Zhao Q, Ross J, Styren S, Landen JW, Ma WW, McCush F, Alvey C, Kupiec JW, Bednar MM (2013) Safety and pharmacology of ponezumab (PF-04360365) after a single 10-minute intravenous infusion in subjects with mild to moderate Alzheimer disease. Clin Neuropharmacol 36:8–13
Butchart J, Holmes C (2012) Systemic and central immunity in Alzheimer’s disease: therapeutic implications. CNS Neurosci Ther 18:64–76
Buttini M, Masliah E, Barbour R, Grajeda H, Motter R, Johnson-Wood K, Khan K, Seubert P, Freedman S, Schenk D, Games D (2005) Beta-amyloid immunotherapy prevents synaptic degeneration in a mouse model of Alzheimer’s disease. J Neurosci 25:9096–9101
Calingasan NY, Erdely HA, Altar CA (2002) Identification of CD40 ligand in Alzheimer’s disease and in animal models of Alzheimer’s disease and brain injury. Neurobiol Aging 23:31–39
Calon F (2011) Omega-3 polyunsaturated fatty acids in Alzheimer’s disease: key questions and partial answers. Curr Alzheimer Res 8:470–478
Cantrell D (2015) Signaling in lymphocyte activation. Cold Spring Harb Perspect Biol 7:a018788
Cash JG, Kuhel DG, Basford JE, Jaeschke A, Chatterjee TK, Weintraub NL, Hui DY (2012) Apolipoprotein E4 impairs macrophage efferocytosis and potentiates apoptosis by accelerating endoplasmic reticulum stress. J Biol Chem 287:27876–27884
Cattepoel S, Hanenberg M, Kulic L, Nitsch RM (2011) Chronic intranasal treatment with an anti-Abeta(30–42) scFv antibody ameliorates amyloid pathology in a transgenic mouse model of Alzheimer’s disease. PLoS ONE 6:e18296
Cayrol R, Wosik K, Berard JL, Dodelet-Devillers A, Ifergan I, Kebir H, Haqqani AS, Kreymborg K, Krug S, Moumdjian R, Bouthillier A, Becher B, Arbour N, David S, Stanimirovic D, Prat A (2008) Activated leukocyte cell adhesion molecule promotes leukocyte trafficking into the central nervous system. Nat Immunol 9:137–145
Cole GM, Frautschy SA (2010) Mechanisms of action of non-steroidal anti-inflammatory drugs for the prevention of Alzheimer’s disease. CNS Neurol Disord: Drug Targets 9:140–148
Cole GM, Morihara T, Lim GP, Yang F, Begum A, Frautschy SA (2004) NSAID and antioxidant prevention of Alzheimer’s disease: lessons from in vitro and animal models. Ann N Y Acad Sci 1035:68–84
Counts SE, Perez SE, He B, Mufson EJ (2014) Intravenous immunoglobulin reduces tau pathology and preserves neuroplastic gene expression in the 3xTg mouse model of Alzheimer’s disease. Curr Alzheimer Res 11:655–663
Davtyan H, Ghochikyan A, Petrushina I, Hovakimyan A, Davtyan A, Poghosyan A, Marleau AM, Movsesyan N, Kiyatkin A, Rasool S, Larsen AK, Madsen PJ, Wegener KM, Ditlevsen DK, Cribbs DH, Pedersen LO, Agadjanyan MG (2013) Immunogenicity, efficacy, safety, and mechanism of action of epitope vaccine (Lu AF20513) for Alzheimer’s disease: prelude to a clinical trial. J Neurosci 33:4923–4934
De Strooper B, Chavez Gutierrez L (2015) Learning by failing: ideas and concepts to tackle gamma-secretases in Alzheimer’s disease and beyond. Annu Rev Pharmacol Toxicol 55:419–437
Derecki NC, Cardani AN, Yang CH, Quinnies KM, Crihfield A, Lynch KR, Kipnis J (2010) Regulation of learning and memory by meningeal immunity: a key role for IL-4. J Exp Med 207:1067–1080
Desideri G, Cipollone F, Necozione S, Marini C, Lechiara MC, Taglieri G, Zuliani G, Fellin R, Mezzetti A, di Orio F, Ferri C (2008) Enhanced soluble CD40 ligand and Alzheimer’s disease: evidence of a possible pathogenetic role. Neurobiol Aging 29:348–356
Dodel R, Rominger A, Bartenstein P, Barkhof F, Blennow K, Forster S, Winter Y, Bach JP, Popp J, Alferink J, Wiltfang J, Buerger K, Otto M, Antuono P, Jacoby M, Richter R, Stevens J, Melamed I, Goldstein J, Haag S, Wietek S, Farlow M, Jessen F (2013) Intravenous immunoglobulin for treatment of mild-to-moderate Alzheimer’s disease: a phase 2, randomised, double-blind, placebo-controlled, dose-finding trial. Lancet Neurol 12:233–243
Doens D, Fernandez PL (2014) Microglia receptors and their implications in the response to amyloid beta for Alzheimer’s disease pathogenesis. J Neuroinflammation 11:48
Farrall AJ, Wardlaw JM (2009) Blood–brain barrier: ageing and microvascular disease–systematic review and meta-analysis. Neurobiol Aging 30:337–352
Ferrer I, Rovira MB, Guerra MLS, Rey MJ, Costa-Jussa F (2004) Neuropathology and pathogenesis of encephalitis following amyloid beta immunization in Alzheimer’s disease. Brain Pathol 14:11–20
Fiala M, Zhang L, Gan X, Sherry B, Taub D, Graves MC, Hama S, Way D, Weinand M, Witte M, Lorton D, Kuo YM, Roher AE (1998) Amyloid-beta induces chemokine secretion and monocyte migration across a human blood–brain barrier model. Mol Med 4:480–489
Franklin EE, Perrin RJ, Vincent B, Baxter M, Morris JC, Cairns NJ (2015) Brain collection, standardized neuropathologic assessment, and comorbidity in Alzheimer’s disease neuroimaging initiative 2 participants. Alzheimers Dement 11:815–822
Gale SC, Gao L, Mikacenic C, Coyle SM, Rafaels N, Murray Dudenkov T, Madenspacher JH, Draper DW, Ge W, Aloor JJ, Azzam KM, Lai L, Blackshear PJ, Calvano SE, Barnes KC, Lowry SF, Corbett S, Wurfel MM, Fessler MB (2014) APOepsilon4 is associated with enhanced in vivo innate immune responses in human subjects. J Allergy Clin Immunol 134:127–134
Geiger H, de Haan G, Florian MC (2013) The ageing haematopoietic stem cell compartment. Nat Rev Immunol 13:376–389
Giunta B, Rezai-Zadeh K, Tan J (2010) Impact of the CD40-CD40L dyad in Alzheimer’s disease. CNS Neurol Disord: Drug Targets 9:149–155
Gomez-Isla T, Blesa R, Boada M, Clarimon J, Del Ser T, Domenech G, Ferro JM, Gomez-Anson B, Manubens JM, Martinez-Lage JM, Munoz D, Pena-Casanova J, Torres F (2008) A randomized, double-blind, placebo controlled-trial of triflusal in mild cognitive impairment: the TRIMCI study. Alzheimer Dis Assoc Disord 22:21–29
Gomez-Nicola D, Boche D (2015) Post-mortem analysis of neuroinflammatory changes in human Alzheimer’s disease. Alzheimers Res Ther 7:42
Gong B, Pan Y, Zhao W, Knable L, Vempati P, Begum S, Ho L, Wang J, Yemul S, Barnum S, Bilski A, Gong BY, Pasinetti GM (2013) IVIG immunotherapy protects against synaptic dysfunction in Alzheimer’s disease through complement anaphylatoxin C5a-mediated AMPA-CREB-C/EBP signaling pathway. Mol Immunol 56:619–629
Guadagna S, Esiri MM, Williams RJ, Francis PT (2012) Tau phosphorylation in human brain: relationship to behavioral disturbance in dementia. Neurobiol Aging 33:2798–2806
Guan X, Zou J, Gu H, Yao Z (2012) Short amyloid-beta immunogens with spacer-enhanced immunogenicity without junctional epitopes for Alzheimer’s disease immunotherapy. NeuroReport 23:879–884
Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, Cruchaga C, Sassi C, Kauwe JS, Younkin S, Hazrati L, Collinge J, Pocock J, Lashley T, Williams J, Lambert JC, Amouyel P, Goate A, Rademakers R, Morgan K, Powell J, St George-Hyslop P, Singleton A, Hardy J (2013) TREM2 variants in Alzheimer’s disease. N Engl J Med 368:117–127
Hartung H-P, Mouthon L, Ahmed R, Jordan S, Laupland KB, Jolles S (2009) Clinical applications of intravenous immunoglobulins (IVIg)–beyond immunodeficiencies and neurology. Clin Exp Immunol 158(Suppl 1):23–33
Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, Jacobs AH, Wyss-Coray T, Vitorica J, Ransohoff RM, Herrup K, Frautschy SA, Finsen B, Brown GC, Verkhratsky A, Yamanaka K, Koistinaho J, Latz E, Halle A, Petzold GC, Town T, Morgan D, Shinohara ML, Perry VH, Holmes C, Bazan NG, Brooks DJ, Hunot S, Joseph B, Deigendesch N, Garaschuk O, Boddeke E, Dinarello CA, Breitner JC, Cole GM, Golenbock DT, Kummer MP (2015) Neuroinflammation in Alzheimer’s disease. Lancet Neurol 14:388–405
Heppner FL, Ransohoff RM, Becher B (2015) Immune attack: the role of inflammation in Alzheimer disease. Nat Rev Neurosci 16:358–372
Herrmann A, Spires-Jones T (2015) Clearing the way for tau immunotherapy in Alzheimer’s disease. J Neurochem 132:1–4
Ho L, Pieroni C, Winger D, Purohit DP, Aisen PS, Pasinetti GM (1999) Regional distribution of cyclooxygenase-2 in the hippocampal formation in Alzheimer’s disease. J Neurosci Res 57:295–303
Holmes C, Boche D, Wilkinson D, Yadegarfar G, Hopkins V, Bayer A, Jones RW, Bullock R, Love S, Neal JW, Zotova E, Nicoll JA (2008) Long-term effects of Abeta42 immunisation in Alzheimer’s disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet 372:216–223
Honig LS (2012) Translational research in neurology: dementia. Arch Neurol 69:969–977
Hoozemans JJ, Rozemuller AJ, van Haastert ES, Eikelenboom P, van Gool WA (2011) Neuroinflammation in Alzheimer’s disease wanes with age. J Neuroinflammation 8:171
Ifergan I, Kebir H, Alvarez JI, Marceau G, Bernard M, Bourbonniere L, Poirier J, Duquette P, Talbot PJ, Arbour N, Prat A (2011) Central nervous system recruitment of effector memory CD8+ T lymphocytes during neuroinflammation is dependent on alpha4 integrin. Brain 134:3560–3577
Ifergan I, Kebir H, Terouz S, Alvarez JI, Lecuyer MA, Gendron S, Bourbonniere L, Dunay IR, Bouthillier A, Moumdjian R, Fontana A, Haqqani A, Klopstein A, Prinz M, Lopez-Vales R, Birchler T, Prat A (2011) Role of Ninjurin-1 in the migration of myeloid cells to central nervous system inflammatory lesions. Ann Neurol 70:751–763
Jaturapatporn D, Isaac MG, McCleery J, Tabet N (2012) Aspirin, steroidal and non-steroidal anti-inflammatory drugs for the treatment of Alzheimer’s disease. Cochrane Database Syst Rev 2:CD006378
Jay TR, Miller CM, Cheng PJ, Graham LC, Bemiller S, Broihier ML, Xu G, Margevicius D, Karlo JC, Sousa GL, Cotleur AC, Butovsky O, Bekris L, Staugaitis SM, Leverenz JB, Pimplikar SW, Landreth GE, Howell GR, Ransohoff RM, Lamb BT (2015) TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer’s disease mouse models. J Exp Med 212:287–295
Jiang T, Tan L, Zhu XC, Zhang QQ, Cao L, Tan MS, Gu LZ, Wang HF, Ding ZZ, Zhang YD, Yu JT (2014) Upregulation of TREM2 ameliorates neuropathology and rescues spatial cognitive impairment in a transgenic mouse model of Alzheimer’s disease. Neuropsychopharmacology 39:2949–2962
Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, Bjornsson S, Huttenlocher J, Levey AI, Lah JJ, Rujescu D, Hampel H, Giegling I, Andreassen OA, Engedal K, Ulstein I, Djurovic S, Ibrahim-Verbaas C, Hofman A, Ikram MA, van Duijn CM, Thorsteinsdottir U, Kong A, Stefansson K (2013) Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med 368:107–116
Julien C, Tremblay C, Phivilay A, Berthiaume L, Emond V, Julien P, Calon F (2010) High-fat diet aggravates amyloid-beta and tau pathologies in the 3xTg-AD mouse model. Neurobiol Aging 31:1516–1531
Kalaria RN, Akinyemi R, Ihara M (2012) Does vascular pathology contribute to Alzheimer changes? J Neurol Sci 322:141–147
Kang JH, Korecka M, Toledo JB, Trojanowski JQ, Shaw LM (2013) Clinical utility and analytical challenges in measurement of cerebrospinal fluid amyloid-beta(1–42) and tau proteins as Alzheimer disease biomarkers. Clin Chem 59:903–916
Kedzierska K, Valkenburg SA, Doherty PC, Davenport MP, Venturi V (2012) Use it or lose it: establishment and persistence of T cell memory. Front Immunol 3:357
Kim J, Eltorai AE, Jiang H, Liao F, Verghese PB, Kim J, Stewart FR, Basak JM, Holtzman DM (2012) Anti-apoE immunotherapy inhibits amyloid accumulation in a transgenic mouse model of Abeta amyloidosis. J Exp Med 209:2149–2156
Kipnis J, Cohen H, Cardon M, Ziv Y, Schwartz M (2004) T cell deficiency leads to cognitive dysfunction: implications for therapeutic vaccination for schizophrenia and other psychiatric conditions. Proc Natl Acad Sci USA 101:8180–8185
Kirshner HS, Bradshaw M (2015) The inflammatory form of cerebral amyloid angiopathy or “cerebral amyloid angiopathy-related inflammation” (CAARI). Curr Neurol Neurosci Rep 15:54
Kiyota T, Gendelman HE, Weir RA, Higgins EE, Zhang G, Jain M (2013) CCL2 affects beta-amyloidosis and progressive neurocognitive dysfunction in a mouse model of Alzheimer’s disease. Neurobiol Aging 34:1060–1068
Kleinberger G, Yamanishi Y, Suarez-Calvet M, Czirr E, Lohmann E, Cuyvers E, Struyfs H, Pettkus N, Wenninger-Weinzierl A, Mazaheri F, Tahirovic S, Lleo A, Alcolea D, Fortea J, Willem M, Lammich S, Molinuevo JL, Sanchez-Valle R, Antonell A, Ramirez A, Heneka MT, Sleegers K, van der Zee J, Martin JJ, Engelborghs S, Demirtas-Tatlidede A, Zetterberg H, Van Broeckhoven C, Gurvit H, Wyss-Coray T, Hardy J, Colonna M, Haass C (2014) TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci Transl Med 6:243ra86
Kornete M, Mason ES, Piccirillo CA (2013) Immune regulation in T1D and T2D: prospective role of Foxp3+ Treg cells in disease pathogenesis and treatment. Front Endocrinol 4:76
LaFerla FM, Green KN (2012) Animal models of Alzheimer disease. Cold Spring Harb Perspect Med 2:a006320
Landen JW, Zhao Q, Cohen S, Borrie M, Woodward M, Billing CBJ, Bales K, Alvey C, McCush F, Yang J, Kupiec JW, Bednar MM (2013) Safety and pharmacology of a single intravenous dose of ponezumab in subjects with mild-to-moderate Alzheimer disease: a phase I, randomized, placebo-controlled, double-blind, dose-escalation study. Clin Neuropharmacol 36:14–23
Larbi A, Pawelec G, Witkowski JM, Schipper HM, Derhovanessian E, Goldeck D, Fulop T (2009) Dramatic shifts in circulating CD4 but not CD8 T cell subsets in mild Alzheimer’s disease. J Alzheimers Dis 17:91–103
Layé S, Madore C, St-Amour I, Delpech JC, Joffre C, Nadjar A, Calon F (2015) N-3 polyunsaturated fatty acid and neuroinflammation in aging and Alzheimer’s disease. Nutr Aging 3:33–47
Lee EB, Leng LZ, Zhang B, Kwong L, Trojanowski JQ, Abel T, Lee VM-Y (2006) Targeting amyloid-beta peptide (Abeta) oligomers by passive immunization with a conformation-selective monoclonal antibody improves learning and memory in Abeta precursor protein (APP) transgenic mice. J Biol Chem 281:4292–4299
Lee J (2013) Adipose tissue macrophages in the development of obesity-induced inflammation, insulin resistance and type 2 diabetes. Arch Pharm Res 36:208–222
Levites Y, Das P, Price RW, Rochette MJ, Kostura LA, McGowan EM, Murphy MP, Golde TE (2006) Anti-Abeta42- and anti-Abeta40-specific mAbs attenuate amyloid deposition in an Alzheimer disease mouse model. J Clin Invest 116:193–201
Leyhe T, Andreasen N, Simeoni M, Reich A, von Arnim CA, Tong X, Yeo A, Khan S, Loercher A, Chalker M, Hottenstein C, Zetterberg H, Hilpert J, Mistry P (2014) Modulation of beta-amyloid by a single dose of GSK933776 in patients with mild Alzheimer’s disease: a phase I study. Alzheimers Res Ther 6:19
Li Q, Lebson L, Lee DC, Nash K, Grimm J, Rosenthal A, Selenica ML, Morgan D, Gordon MN (2012) Chronological age impacts immunotherapy and monocyte uptake independent of amyloid load. J Neuroimmune Pharmacol 7:202–214
Liao F, Hori Y, Hudry E, Bauer AQ, Jiang H, Mahan TE, Lefton KB, Zhang TJ, Dearborn JT, Kim J, Culver JP, Betensky R, Wozniak DF, Hyman BT, Holtzman DM (2014) Anti-ApoE antibody given after plaque onset decreases Abeta accumulation and improves brain function in a mouse model of Abeta amyloidosis. J Neurosci 34:7281–7292
Liu B, Frost JL, Sun J, Fu H, Grimes S, Blackburn P, Lemere CA (2013) MER5101, a novel Abeta1-15: DT conjugate vaccine, generates a robust anti-Abeta antibody response and attenuates Abeta pathology and cognitive deficits in APPswe/PS1DeltaE9 transgenic mice. J Neurosci 33:7027–7037
Liu YJ, Guo DW, Tian L, Shang DS, Zhao WD, Li B, Fang WG, Zhu L, Chen YH (2010) Peripheral T cells derived from Alzheimer’s disease patients overexpress CXCR2 contributing to its transendothelial migration, which is microglial TNF-alpha-dependent. Neurobiol Aging 31:175–188
Lobello K, Ryan JM, Liu E, Rippon G, Black R (2012) Targeting beta amyloid: a clinical review of immunotherapeutic approaches in Alzheimer’s disease. Int J Alzheimers Dis 2012:628070
Loeffler DA, Camp DM, Bennett DA (2008) Plaque complement activation and cognitive loss in Alzheimer’s disease. J Neuroinflammation 5:9
Loeffler DA, Camp DM, Schonberger MB, Singer DJ, LeWitt PA (2004) Early complement activation increases in the brain in some aged normal subjects. Neurobiol Aging 25:1001–1007
Lombardi VR, Garcia M, Rey L, Cacabelos R (1999) Characterization of cytokine production, screening of lymphocyte subset patterns and in vitro apoptosis in healthy and Alzheimer’s Disease (AD) individuals. J Neuroimmunol 97:163–171
Man S-M, Ma Y-R, Shang D-S, Zhao W-D, Li B, Guo D-W, Fang W-G, Zhu L, Chen Y-H (2007) Peripheral T cells overexpress MIP-1alpha to enhance its transendothelial migration in Alzheimer’s disease. Neurobiol Aging 28:485–496
Mantile F, Basile C, Cicatiello V, De Diana F, Caivano A, De Piergiuseppe B, Prisco A (2011) A multimeric immunogen for the induction of immune memory to beta-amyloid. Immunol Cell Biol 89:604–609
Martin BK, Szekely C, Brandt J, Piantadosi S, Breitner JC, Craft S, Evans D, Green R, Mullan M (2008) Cognitive function over time in the Alzheimer’s disease anti-inflammatory prevention trial (ADAPT): results of a randomized, controlled trial of naproxen and celecoxib. Arch Neurol 65:896–905
Masliah E, Hansen L, Adame A, Crews L, Bard F, Lee C, Seubert P, Games D, Kirby L, Schenk D (2005) Abeta vaccination effects on plaque pathology in the absence of encephalitis in Alzheimer disease. Neurology 64:129–131
McGeer PL, McGeer EG (2013) The amyloid cascade-inflammatory hypothesis of Alzheimer disease: implications for therapy. Acta Neuropathol 126:479–497
McMaster WG, Kirabo A, Madhur MS, Harrison DG (2015) Inflammation, immunity, and hypertensive end-organ damage. Circ Res 116:1022–1033
Meinert CL, McCaffrey LD, Breitner JC (2009) Alzheimer’s disease anti-inflammatory prevention trial: design, methods, and baseline results. Alzheimers Dement 5:93–104
Millan J, Hewlett L, Glyn M, Toomre D, Clark P, Ridley AJ (2006) Lymphocyte transcellular migration occurs through recruitment of endothelial ICAM-1 to caveola- and F-actin-rich domains. Nat Cell Biol 8:113–123
Mocali A, Cedrola S, Della Malva N, Bontempelli M, Mitidieri VA, Bavazzano A, Comolli R, Paoletti F, La Porta CA (2004) Increased plasma levels of soluble CD40, together with the decrease of TGF beta 1, as possible differential markers of Alzheimer disease. Exp Gerontol 39:1555–1561
Moreth J, Mavoungou C, Schindowski K (2013) Passive anti-amyloid immunotherapy in Alzheimer’s disease: What are the most promising targets? Immun Ageing 10:18
Muldoon LL, Alvarez JI, Begley DJ, Boado RJ, Del Zoppo GJ, Doolittle ND, Engelhardt B, Hallenbeck JM, Lonser RR, Ohlfest JR, Prat A, Scarpa M, Smeyne RJ, Drewes LR, Neuwelt EA (2013) Immunologic privilege in the central nervous system and the blood–brain barrier. J Cereb Blood Flow Metab 33:13–21
Nair P, Gaga M, Zervas E, Alagha K, Hargreave FE, O’Byrne PM, Stryszak P, Gann L, Sadeh J, Chanez P (2012) Safety and efficacy of a CXCR2 antagonist in patients with severe asthma and sputum neutrophils: a randomized, placebo-controlled clinical trial. Clin Exp Allergy 42:1097–1103
Nascimento CM, Pereira JR, de Andrade LP, Garuffi M, Talib LL, Forlenza OV, Cancela JM, Cominetti MR, Stella F (2014) Physical exercise in MCI elderly promotes reduction of pro-inflammatory cytokines and improvements on cognition and BDNF peripheral levels. Curr Alzheimer Res 11:799–805
Nicoll JA, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO (2003) Neuropathology of human Alzheimer disease after immunization with amyloid-beta peptide: a case report. Nat Med 9:448–452
Nikolic WV, Hou H, Town T, Zhu Y, Giunta B, Sanberg CD, Zeng J, Luo D, Ehrhart J, Mori T, Sanberg PR, Tan J (2008) Peripherally administered human umbilical cord blood cells reduce parenchymal and vascular beta-amyloid deposits in Alzheimer mice. Stem Cells Dev 17:423–439
Oddo S, Vasilevko V, Caccamo A, Kitazawa M, Cribbs DH, LaFerla FM (2006) Reduction of soluble Abeta and tau, but not soluble Abeta alone, ameliorates cognitive decline in transgenic mice with plaques and tangles. J Biol Chem 281:39413–39423
Orgogozo JM, Gilman S, Dartigues JF, Laurent B, Puel M, Kirby LC, Jouanny P, Dubois B, Eisner L, Flitman S, Michel BF, Boada M, Frank A, Hock C (2003) Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology 61:46–54
Ostrowitzki S, Deptula D, Thurfjell L, Barkhof F, Bohrmann B, Brooks DJ, Klunk WE, Ashford E, Yoo K, Xu ZX, Loetscher H, Santarelli L (2012) Mechanism of amyloid removal in patients with Alzheimer disease treated with gantenerumab. Arch Neurol 69:198–207
Parajuli B, Horiuchi H, Mizuno T, Takeuchi H, Suzumura A (2015) CCL11 enhances excitotoxic neuronal death by producing reactive oxygen species in microglia. Glia 63:2274–2284
Pardridge WM (2009) Alzheimer’s disease drug development and the problem of the blood–brain barrier. Alzheimers Dement 5:427–432
Pasinetti GM, Aisen PS (1998) Cyclooxygenase-2 expression is increased in frontal cortex of Alzheimer’s disease brain. Neuroscience 87:319–324
Patel KR (2015) Biogen’s aducanumab raises hope that Alzheimer’s can be treated at its source. Manag Care 24:19
Pellicano M, Larbi A, Goldeck D, Colonna-Romano G, Buffa S, Bulati M, Rubino G, Iemolo F, Candore G, Caruso C, Derhovanessian E, Pawelec G (2012) Immune profiling of Alzheimer patients. J Neuroimmunol 242:52–59
Petrushina I, Ghochikyan A, Mkrtichyan M, Mamikonyan G, Movsesyan N, Ajdari R, Vasilevko V, Karapetyan A, Lees A, Agadjanyan MG, Cribbs DH (2008) Mannan-Abeta28 conjugate prevents Abeta-plaque deposition, but increases microhemorrhages in the brains of vaccinated Tg2576 (APPsw) mice. J Neuroinflammation 5:42
Pirttila T, Mattinen S, Frey H (1992) The decrease of CD8-positive lymphocytes in Alzheimer’s disease. J Neurol Sci 107:160–165
Prokop S, Miller KR, Heppner FL (2013) Microglia actions in Alzheimer’s disease. Acta Neuropathol 126:461–477
Puli L, Pomeshchik Y, Olas K, Malm T, Koistinaho J, Tanila H (2012) Effects of human intravenous immunoglobulin on amyloid pathology and neuroinflammation in a mouse model of Alzheimer’s disease. J Neuroinflammation 9:105
Reale M, Iarlori C, Gambi F, Feliciani C, Salone A, Toma L, DeLuca G, Salvatore M, Conti P, Gambi D (2004) Treatment with an acetylcholinesterase inhibitor in Alzheimer patients modulates the expression and production of the pro-inflammatory and anti-inflammatory cytokines. J Neuroimmunol 148:162–171
Relkin N (2014) Clinical trials of intravenous immunoglobulin for Alzheimer’s disease. J Clin Immunol 34(Suppl 1):S74–S79
Rennard SI, Dale DC, Donohue JF, Kanniess F, Magnussen H, Sutherland ER, Watz H, Lu S, Stryszak P, Rosenberg E, Staudinger H (2015) CXCR2 antagonist MK-7123. A phase 2 proof-of-concept trial for chronic obstructive pulmonary disease. Am J Respir Crit Care Med 191:1001–1011
Rezai-Zadeh K, Gate D, Szekely CA, Town T (2009) Can peripheral leukocytes be used as Alzheimer’s disease biomarkers? Expert Rev Neurother 9:1623–1633
Ricciotti E, FitzGerald GA (2011) Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol 31:986–1000
Richartz E, Stransky E, Batra A, Simon P, Lewczuk P, Buchkremer G, Bartels M, Schott K (2005) Decline of immune responsiveness: A pathogenetic factor in Alzheimer’s disease? J Psychiatr Res 39:535–543
Ricklin D, Lambris JD (2013) Complement in immune and inflammatory disorders: therapeutic interventions. J Immunol 190:3839–3847
Robinson SR, Bishop GM, Lee H-G, Munch G (2004) Lessons from the AN 1792 Alzheimer vaccine: lest we forget. Neurobiol Aging 25:609–615
Romeo J, Warnberg J, Pozo T, Marcos A (2010) Physical activity, immunity and infection. Proc Nutr Soc 69:390–399
Rosenmann H, Grigoriadis N, Karussis D, Boimel M, Touloumi O, Ovadia H, Abramsky O (2006) Tauopathy-like abnormalities and neurologic deficits in mice immunized with neuronal tau protein. Arch Neurol 63:1459–1467
Rouzer CA, Marnett LJ (2009) Cyclooxygenases: structural and functional insights. J Lipid Res 50(Suppl):S29–S34
Rus H, Cudrici C, Niculescu F (2005) The role of the complement system in innate immunity. Immunol Res 33:103–112
Rymkiewicz PD, Heng YX, Vasudev A, Larbi A (2012) The immune system in the aging human. Immunol Res 53:235–250
Salloway S, Sperling R, Fox NC, Blennow K, Klunk W, Raskind M, Sabbagh M, Honig LS, Porsteinsson AP, Ferris S, Reichert M, Ketter N, Nejadnik B, Guenzler V, Miloslavsky M, Wang D, Lu Y, Lull J, Tudor IC, Liu E, Grundman M, Yuen E, Black R, Brashear HR (2014) Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N Engl J Med 370:322–333
Saresella M, Calabrese E, Marventano I, Piancone F, Gatti A, Alberoni M, Nemni R, Clerici M (2011) Increased activity of Th-17 and Th-9 lymphocytes and a skewing of the post-thymic differentiation pathway are seen in Alzheimer’s disease. Brain Behav Immun 25:539–547
Saunders AM, Strittmatter WJ, Schmechel D, George-Hyslop PH, Pericak-Vance MA, Joo SH, Rosi BL, Gusella JF, Crapper-MacLachlan DR, Alberts MJ et al (1993) Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology 43:1467–1472
Schellenberg GD, Montine TJ (2012) The genetics and neuropathology of Alzheimer’s disease. Acta Neuropathol 124:305–323
Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandevert C, Walker S, Wogulis M, Yednock T, Games D, Seubert P (1999) Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 400:173–177
Selkoe DJ (2013) The therapeutics of Alzheimer’s disease: where we stand and where we are heading. Ann Neurol 74:328–336
Senchina DS, Kohut ML (2007) Immunological outcomes of exercise in older adults. Clin Interv Aging 2:3–16
Shen Y, Li R, McGeer EG, McGeer PL (1997) Neuronal expression of mRNAs for complement proteins of the classical pathway in Alzheimer brain. Brain Res 769:391–395
Siemers ER, Sundell KL, Carlson C, Case M, Sethuraman G, Liu-Seifert H, Dowsett SA, Pontecorvo MJ, Dean RA, Demattos R (2015) Phase 3 solanezumab trials: secondary outcomes in mild Alzheimer’s disease patients. Alzheimers Dement. doi:10.1016/j.jalz.2015.06.1893
Sokolova A, Hill MD, Rahimi F, Warden LA, Halliday GM, Shepherd CE (2009) Monocyte chemoattractant protein-1 plays a dominant role in the chronic inflammation observed in Alzheimer’s disease. Brain Pathol 19:392–398
Speciale L, Calabrese E, Saresella M, Tinelli C, Mariani C, Sanvito L, Longhi R, Ferrante P (2007) Lymphocyte subset patterns and cytokine production in Alzheimer’s disease patients. Neurobiol Aging 28:1163–1169
Sperling R, Mormino E, Johnson K (2014) The evolution of preclinical Alzheimer’s disease: implications for prevention trials. Neuron 84:608–622
Sperling R, Salloway S, Brooks DJ, Tampieri D, Barakos J, Fox NC, Raskind M, Sabbagh M, Honig LS, Porsteinsson AP, Lieberburg I, Arrighi HM, Morris KA, Lu Y, Liu E, Gregg KM, Brashear HR, Kinney GG, Black R, Grundman M (2012) Amyloid-related imaging abnormalities in patients with Alzheimer’s disease treated with bapineuzumab: a retrospective analysis. Lancet Neurol 11:241–249
St-Amour I, Pare I, Alata W, Coulombe K, Ringuette-Goulet C, Drouin-Ouellet J, Vandal M, Soulet D, Bazin R, Calon F (2013) Brain bioavailability of human intravenous immunoglobulin and its transport through the murine blood–brain barrier. J Cereb Blood Flow Metab 33:1983–1992
St-Amour I, Pare I, Tremblay C, Coulombe K, Bazin R, Calon F (2014) IVIg protects the 3xTg-AD mouse model of Alzheimer’s disease from memory deficit and Abeta pathology. J Neuroinflammation 11:54
Stieler JT, Lederer C, Bruckner MK, Wolf H, Holzer M, Gertz HJ, Arendt T (2001) Impairment of mitogenic activation of peripheral blood lymphocytes in Alzheimer’s disease. NeuroReport 12:3969–3972
Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, Roses AD (1993) Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci USA 90:1977–1981
Sudduth TL, Greenstein A, Wilcock DM (2013) Intracranial injection of Gammagard, a human IVIg, modulates the inflammatory response of the brain and lowers Abeta in APP/PS1 mice along a different time course than anti-Abeta antibodies. J Neurosci 33:9684–9692
Szabo P, Relkin N, Weksler ME (2008) Natural human antibodies to amyloid beta peptide. Autoimmun Rev 7:415–420
Tambuyzer BR, Ponsaerts P, Nouwen EJ (2009) Microglia: gatekeepers of central nervous system immunology. J Leukoc Biol 85:352–370
Tanzi RE (2012) The genetics of Alzheimer disease. Cold Spring Harb Perspect Med 2:a006296
Tarawneh R, Holtzman DM (2012) The clinical problem of symptomatic Alzheimer disease and mild cognitive impairment. Cold Spring Harb Perspect Med 2:a006148
Teng E, Yamasaki TR, Tran M, Hsiao JJ, Sultzer DL, Mendez MF (2014) Cerebrospinal fluid biomarkers in clinical subtypes of early-onset Alzheimer’s disease. Dement Geriatr Cogn Disord 37:307–314
Thathiah A, De Strooper B (2011) The role of G protein-coupled receptors in the pathology of Alzheimer’s disease. Nat Rev Neurosci 12:73–87
Togo T, Akiyama H, Kondo H, Ikeda K, Kato M, Iseki E, Kosaka K (2000) Expression of CD40 in the brain of Alzheimer’s disease and other neurological diseases. Brain Res 885:117–121
Tremblay C, Pilote M, Phivilay A, Emond V, Bennett DA, Calon F (2007) Biochemical characterization of Abeta and tau pathologies in mild cognitive impairment and Alzheimer’s disease. J Alzheimers Dis 12:377–390
Vandal M, White PJ, Chevrier G, Tremblay C, St-Amour I, Planel E, Marette A, Calon F (2015) Age-dependent impairment of glucose tolerance in the 3xTg-AD mouse model of Alzheimer’s disease. FASEB J 29:4273–4284
Vandal M, White PJ, Tremblay C, St-Amour I, Chevrier G, Emond V, Lefrancois D, Virgili J, Planel E, Giguere Y, Marette A, Calon F (2014) Insulin reverses the high-fat diet-induced increase in brain Abeta and improves memory in an animal model of Alzheimer disease. Diabetes 63:4291–4301
Villeda SA, Luo J, Mosher KI, Zou B, Britschgi M, Bieri G, Stan TM, Fainberg N, Ding Z, Eggel A, Lucin KM, Czirr E, Park JS, Couillard-Despres S, Aigner L, Li G, Peskind ER, Kaye JA, Quinn JF, Galasko DR, Xie XS, Rando TA, Wyss-Coray T (2011) The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature 477:90–94
Vlad SC, Miller DR, Kowall NW, Felson DT (2008) Protective effects of NSAIDs on the development of Alzheimer disease. Neurology 70:1672–1677
Vukic V, Callaghan D, Walker D, Lue LF, Liu QY, Couraud PO, Romero IA, Weksler B, Stanimirovic DB, Zhang W (2009) Expression of inflammatory genes induced by beta-amyloid peptides in human brain endothelial cells and in Alzheimer’s brain is mediated by the JNK-AP1 signaling pathway. Neurobiol Dis 34:95–106
Walker DG, Dalsing-Hernandez JE, Campbell NA, Lue LF (2009) Decreased expression of CD200 and CD200 receptor in Alzheimer’s disease: a potential mechanism leading to chronic inflammation. Exp Neurol 215:5–19
Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, Gilfillan S, Krishnan GM, Sudhakar S, Zinselmeyer BH, Holtzman DM, Cirrito JR, Colonna M (2015) TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 160:1061–1071
Weiskopf D, Weinberger B, Grubeck-Loebenstein B (2009) The aging of the immune system. Transpl Int 22:1041–1050
Whitehead AL, Sully BG, Campbell MJ (2014) Pilot and feasibility studies: Is there a difference from each other and from a randomised controlled trial? Contemp Clin Trials 38:130–133
Wilcock D, Rojiani A, Rosenthal A, Subbarao S, Freeman M, Gordon M, Morgan D (2004) Passive immunotherapy against Abeta in aged APP-transgenic mice reverses cognitive deficits and depletes parenchymal amyloid deposits in spite of increased vascular amyloid and microhemorrhage. J Neuroinflammation. doi:10.1186/1742-2094-1-24
Wilcock DM, Jantzen PT, Li Q, Morgan D, Gordon MN (2007) Amyloid-beta vaccination, but not nitro-nonsteroidal anti-inflammatory drug treatment, increases vascular amyloid and microhemorrhage while both reduce parenchymal amyloid. Neuroscience 144:950–960
Wilcock DM, Rojiani A, Rosenthal A, Levkowitz G, Subbarao S, Alamed J, Wilson D, Wilson N, Freeman MJ, Gordon MN, Morgan D (2004) Passive amyloid immunotherapy clears amyloid and transiently activates microglia in a transgenic mouse model of amyloid deposition. J Neurosci 24:6144–6151
Winblad B, Andreasen N, Minthon L, Floesser A, Imbert G, Dumortier T, Maguire RP, Blennow K, Lundmark J, Staufenbiel M, Orgogozo JM, Graf A (2012) Safety, tolerability, and antibody response of active Abeta immunotherapy with CAD106 in patients with Alzheimer’s disease: randomised, double-blind, placebo-controlled, first-in-human study. Lancet Neurol 11:597–604
Wolburg H, Wolburg-Buchholz K, Engelhardt B (2005) Diapedesis of mononuclear cells across cerebral venules during experimental autoimmune encephalomyelitis leaves tight junctions intact. Acta Neuropathol 109:181–190
Wolf SA, Steiner B, Akpinarli A, Kammertoens T, Nassenstein C, Braun A, Blankenstein T, Kempermann G (2009) CD4-positive T lymphocytes provide a neuroimmunological link in the control of adult hippocampal neurogenesis. J Immunol 182:3979–3984
Wu F, Zhao Y, Jiao T, Shi D, Zhu X, Zhang M, Shi M, Zhou H (2015) CXCR2 is essential for cerebral endothelial activation and leukocyte recruitment during neuroinflammation. J Neuroinflammation 12:98
Wyss-Coray T, Rogers J (2012) Inflammation in Alzheimer disease-a brief review of the basic science and clinical literature. Cold Spring Harb Perspect Med 2:a006346
Xue SR, Xu DH, Yang XX, Dong WL (2009) Alterations in lymphocyte subset patterns and co-stimulatory molecules in patients with Alzheimer disease. Chin Med J 122:1469–1472
Yanamadala V, Friedlander RM (2010) Complement in neuroprotection and neurodegeneration. Trends Mol Med 16:69–76
Yang YM, Shang DS, Zhao WD, Fang WG, Chen YH (2013) Microglial TNF-alpha-dependent elevation of MHC class I expression on brain endothelium induced by amyloid-beta promotes T cell transendothelial migration. Neurochem Res 38:2295–2304
Youn JC, Yu HT, Lim BJ, Koh MJ, Lee J, Chang DY, Choi YS, Lee SH, Kang SM, Jang Y, Yoo OJ, Shin EC, Park S (2013) Immunosenescent CD8+ T cells and C-X-C chemokine receptor type 3 chemokines are increased in human hypertension. Hypertension 62:126–133
Zanjani H, Finch CE, Kemper C, Atkinson J, McKeel D, Morris JC, Price JL (2005) Complement activation in very early Alzheimer disease. Alzheimer Dis Assoc Disord 19:55–66
Zhang K, Tian L, Liu L, Feng Y, Dong YB, Li B, Shang DS, Fang WG, Cao YP, Chen YH (2013) CXCL1 contributes to beta-amyloid-induced transendothelial migration of monocytes in Alzheimer’s disease. PLoS ONE 8:e72744
Zlokovic BV (2011) Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat Rev Neurosci 12:723–738
Acknowledgments
IS-A is supported by a CIHR-Huntington Society of Canada postdoctoral fellowship. Fond de Recherche du Québec en Santé provided salary support to FCi and FCa. The authors are grateful to Mr. Alain St-Amour from Si Design & Web for the artwork. FCa has received research grant from Grifols (Mississauga, ON, Canada). The funding source had no involvement in the study design, and in the collection, analysis or interpretation of the data.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no other conflict of interest to declare.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
St-Amour, I., Cicchetti, F. & Calon, F. Immunotherapies in Alzheimer’s disease: Too much, too little, too late or off-target?. Acta Neuropathol 131, 481–504 (2016). https://doi.org/10.1007/s00401-015-1518-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-015-1518-9