
Abstract
Here we review the genetic causes and risks for Alzheimer’s disease (AD). Early work identified mutations in three genes that cause AD: APP, PSEN1 and PSEN2. Although mutations in these genes are rare causes of AD, their discovery had a major impact on our understanding of molecular mechanisms of AD. Early work also revealed the ε4 allele of the APOE as a strong risk factor for AD. Subsequently, SORL1 also was identified as an AD risk gene. More recently, advances in our knowledge of the human genome, made possible by technological advances and methods to analyze genomic data, permit systematic identification of genes that contribute to AD risk. This work, so far accomplished through single nucleotide polymorphism arrays, has revealed nine new genes implicated in AD risk (ABCA7, BIN1, CD33, CD2AP, CLU, CR1, EPHA1, MS4A4E/MS4A6A, and PICALM). We review the relationship between these mutations and genetic variants and the neuropathologic features of AD and related disorders. Together, these discoveries point toward a new era in neurodegenerative disease research that impacts not only AD but also related illnesses that produce cognitive and behavioral deficits.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
What disease will a person get and when? How severe will it be and how rapidly will it progress? Who will or will not respond to specific treatments? What is the initial trigger that starts the process of Alzheimer’s disease (AD) and neurodegeneration and what are underlying mechanisms? Where can we intervene to stop or prevent AD? These are medically and scientifically relevant questions that we could answer better with a more complete understanding of the genetics of both rare and common forms of AD and other neurodegenerative diseases.
For AD, early work between 1988 and 1995 identified three genes that harbor mutations that cause the disease. The term “cause” is used because individuals with a mutation in one of these genes inevitably develop AD, often at a remarkable young age. While mutations in these genes are rare causes of AD, these initial discoveries had a major impact on our understanding of molecular mechanisms and led to multiple therapeutic approaches that are currently being tested. In the early 1990 s, the technology and knowledge were available to identify causative genes, but not to identify systematically genes that just contribute to an individual’s risk of developing AD. In the past 5 years, advances in our knowledge of the human genome, the technology to interrogate the genome, and methods to analyze genetic data have made it possible to identify genes that contribute to risk. In this manuscript, we review early genetic findings and what these initial gene discovery efforts taught us, and then move to more recent findings on genetic risk factors for AD. The implications of recent findings are unknown and the focus of intense interest.
In addition to an overview of the genetic causes and risks for AD, we also review the relationship between these mutations and genetic variants and the neuropathologic features of AD and related disorders. AD has two core neuropathologic features: the formation of various forms of amyloid deposits often referred to as senile plaques and neurofibrillary degeneration that is typified by neurofibrillary tangles, neuropil threads, and dystrophic neurites in neuritic plaques (Fig. 1). Other changes that are characteristic of AD include cerebral amyloid angiopathy (CAA), brain regional atrophy and neuron loss, and glial activation. Still other neuropathologic changes may be observed in some but not all cases of AD; examples include Lewy body disease (LBD), vascular brain injury, hippocampal sclerosis, and TDP-43 inclusion formation. Since these last neuropathologic changes also are observed in other clinical contexts, they often are considered co-morbid, especially in the elderly; however, their co-occurrence in younger individuals with autosomal dominant forms of AD at least suggests that some may share common pathogenic mechanisms with AD.

Photomicrograph of neuritic plaque and neurofibrillary tangle. Histologic section of neocortex from a patient who died of dementia that has been stained with a modified Bielschowsky method to reveal the hallmark lesions of AD: a large extracellular plaque with amyloid core and abnormal neuritic processes (neuritic plaque) and a skein of darkly staining threads that encircle a neuron’s nucleus and extend down a process (neurofibrillary tangle)
Autosomal dominant Alzheimer’s disease
Families with large numbers of early-onset AD (onset <60 years) were reported as early as the 1930s. Because AD with this early-onset age is rare, the cause of AD in these families was clearly inherited in a simple Mendelian autosomal dominant pattern, meaning children of an affected parent were at 50 % risk for developing early-onset AD. It was not until the mid-1980s that methods became available to identify the responsible genes. In relatively rapid succession, three autosomal dominant genes were identified: amyloid precursor protein (APP), presenilin 1 (PSEN1), and presenilin 2 (PSEN2). While not all early-onset AD is caused by inheritance of mutations in one of these three genes, it is unlikely that another autosomal dominant causative gene for AD is yet to be discovered.
APP
Identification of the APP gene was made possible by the seminal work of Glenner and Wong [36] who, in 1984, determined a partial amino acid sequence of a peptide isolated from AD brain cerebrovascular amyloid. This partial sequence and additional sequence determined by others [107] were subsequently used by Kang et al. [68] to clone APP (chromosome 21). The predicted protein, designated APP, is a ubiquitously expressed, single-pass transmembrane protein that contains an internal 39–43 amino acid sequence corresponding to the Aβ peptide isolated by Glenner and Wong. Aβ peptides are generated from APP (Fig. 2) by two endoproteolytic cleavages catalyzed by β and γ secretases; α secretase catalyzes endoproteolysis of APP within the Aβ sequence [120]. Aβ peptides are the primary component of amyloid deposits that form in brain parenchyma (called “plaques”) and in the walls of cerebral vessels (called “cerebral amyloid angiopathy”, CAA); both are key features of AD neuropathologic changes.

APP protein structure and processing. The APP protein is a single-pass transmembrane protein with an extracellular globular domain (top of the figure). The extracellular segment is illustrated here in blue. The transmembrane segment is composed in part of sequence that makes up the Aβ peptide (red). The amino acid sequence of Aβ peptides and flanking regions are shown at the bottom along with the position and amino acid changes of different APP mutations. Locations of the α-, β-, and γ-secretase sites are shown. The γ-secretase site is shown with a red triangle because cleavage at this site can leave different C-terminal amino acids
APP mutations
Progress in AD genetics began with the identification of autosomal dominant mutations in APP [37]. To date, 24 APP single nucleotide mutations are known that cause AD (http://www.molgen.ua.ac.be/ADmutations). While APP can encode multiple isoforms with the longest being 750 amino acids, all AD mutations are clustered within a 54 amino acid segment, either adjacent to or within the sequence that encodes Aβ peptides. It is instructive to review how different mutations inform us about the molecular pathogenesis of AD.
The Swedish mutation is a double substitution changing the two amino acids immediately before the beginning of the Aβ peptide sequence. The normal sequence, lysine-methionine is replaced by asparagine-leucine. The functional consequence is that the amount of Aβ peptide produced from the mutant APP is two to threefold higher than that produced from non-mutated APP, presumably by affecting the efficiency of β-secretase cleavage. Since the Swedish mutation is outside the sequence encoding Aβ peptides, the amino acid sequence of the Aβ peptide produced from this double substitution is normal. The implication is that excess Aβ production is sufficient to cause AD. This conclusion is supported by the long-standing observation that individuals with trisomy 21 (Down syndrome; DS) develop extensive AD neuropathologic changes [173] that can occur very early in life [90]. By age 40, virtually all individuals with DS have full expression of AD neuropathologic features. Dementia is also common in individuals with DS with a prevalence of 15 % after age 45 that increases to 75 % after age 65 [20, 163]. Finally, consistent with the implication that an extra copy of APP is sufficient to cause AD neuropathologic changes, duplications of small segments of chromosome 21 that do not cause DS but that contain an additional copy of APP are sufficient to cause AD [69, 136, 146].
Other APP mutations that cause AD cluster at or after the C-terminal amino acids of Aβ peptides, including the most common mutation, p.Val717Ile (Fig. 2). These mutations alter the activity of γ-secretase cleavage. When proteolysis of normal APP is catalyzed by γ-secretase, the predominant species formed is 40 amino acids long (Aβ40) with smaller amounts of Aβ that is 42 amino acids long (Aβ42). APP mutations at the C-terminal end of Aβ shift proteolysis to produce more Aβ42 at the expense of Aβ40, resulting in an increased Aβ42/Aβ40 ratio but not necessarily a change in the total amount of Aβ peptides formed [8, 138]. Aβ42 is more amyloidogenic and more prone to aggregate than shorter forms of Aβ [62]. These were among the first data to suggest that Aβ aggregation is key to AD pathogenesis.
Another mutation of note is p.Glu693Gly [66] also known as the Arctic mutation [118]. This missense mutation does not alter the amount of Aβ formed nor does it alter the γ-secretase cleavage site or the Aβ40/Aβ42 ratio. However, this amino acid change within Aβ does increase the aggregation rate of the mutant peptide, adding to the evidence that APP mutations can result in Aβ peptides with altered aggregation properties and that this process is critical to AD pathogenesis.
APP mutations and the neuropathology of individuals with three copies of this gene provide conclusive evidence that an alteration in either the amount of Aβ produced or the amino acid sequence of Aβ is sufficient to cause AD and strongly support the proposal that a multimeric form of Aβ is critical for AD pathogenesis.
Neuropathologic changes observed with APP mutations
Several studies have demonstrated variation in the core neuropathologic features of AD among patients with mutations in APP. At the time of autopsy, patients with APP mutations tend to have greater amounts of neocortical senile plaques than patients who had “sporadic” AD, although these data come from relatively few cases and there is broad overlap [57, 61, 104]; there is no clear trend for a difference in neurofibrillary degeneration [96, 153, 162]. The ratio of accumulated Aβ42/Aβ40 may be higher in cases of autosomal dominant AD from APP mutations, but again there are a limited number of examples and wide variation in results [116, 156–158].
In addition to variation in the amount of core neuropathologic features of AD, APP mutations also are associated with morphologic variants. Several APP mutations have been associated with variation in the structure of Aβ deposits. These include the p.Ala692Gly (Flemish) APP mutation that is associated with large, dense plaques [23, 77] and the p.Glu693Gly (Arctic) APP mutation with ring-like plaques [5]. CAA is a common feature of AD caused by mutations in APP. The p.Asp694Asn (Iowa) and p.Ala713Thr mutations are associated with exceptionally severe CAA [39, 135] in the context of the core neuropathologic features of AD. The p.Glu693Gln (Dutch) APP mutation produces CAA without significant accumulation of the core features of AD; in these individuals, progressive cognitive impairment is linked to vascular brain injury [11, 117].
Several cases of AD caused by mutations in APP also have LBD, which may be observed by histochemical stains of pigmented brainstem neurons or demonstrated in more widespread brain regions, including limbic structures and neocortex, by α-synuclein immunoreactive Lewy bodies and Lewy neurites. Some investigators have demonstrated only brainstem LBD using histochemical methods [43] while others have demonstrated limbic LBD [41], and still others neocortical LBD [54, 82] in cases of AD caused by mutations in APP.
PSEN1 and PSEN2
Mutations in either of these two genes cause early-onset autosomal dominant AD. Over 180 mutations in PSEN1 are known to cause autosomal dominant AD with onset as early as the late 1920s (http://www.molgen.ua.ac.be/ADmutations). Penetrance of PSEN1 mutations is complete by age 60–65 years, meaning all mutation carriers develop early-onset AD. Fewer than 15 known mutations in PSEN2 also can cause early-onset autosomal dominant AD, but penetrance is more variable than PSEN1. In the Volga German kindreds, the families that facilitated identification of PSEN2 mutations, onset ranges from the early 1940s to the 7th and 8th decade of life [91].
Mechanism
PSEN1 and PSEN2 encode closely related proteins that are part of the γ-secretase complex (Fig. 3). Either presenilin 1 or 2, together with nicastrin, APH1, and PEN2, form a complex that catalyze the cleave a number of different membrane proteins [169] at sites embedded in the lipid bilayer. In the case of APP, the presenilin-containing γ-secretase catalyzes endoproteolysis at the C-terminal end of the Aβ peptide sequence. This proteolytic event, along with a second cleavage at the N-terminal end of this sequence, is required for production of Aβ peptide from APP.

The γ-secretase complex. The complex is composed of four membrane proteins: nicastrin, aph-1, Pen-2, and presenilin (1 or 2). The actual catalytic site is in presenilin. The gap in presenilin represents cleavage of the protein that is part of normal processing. Cleavage of APP and other substrates occurs in the membrane. As illustrated here, when there is an APP mutation that causes AD or related disease, the cleavage site is altered by approximately two amino acids (short yellow segment at the end of the red Aβ peptide). While only a single PSEN mutation is shown in the figure, as noted in the text, there are numerous PSEN1 and PSEN2 mutations that cause early-onset AD
How do PSEN1 or 2 mutations cause AD? γ-Secretase containing mutation-altered presenilin still catalyzes cleavage of APP, but the proteolytic site is altered. Normal γ-secretase yields predominantly Aβ40 with smaller amounts of Aβ42. Mutant γ-secretase produces more Aβ42, a result consistently obtained across multiple studies. As noted above, Aβ42 is more amyloidogenic and more prone to aggregate than Aβ40 [62].
Neuropathologic changes observed with PSEN1 or PSEN2 mutations
As with APP mutations, individuals who died from AD caused by mutations in PSEN1 or PSEN2 tend to have a greater amount of neocortical senile plaques [57, 103, 105, 106] and a shift toward a higher Aβ42/Aβ40 ratio [53, 110, 156] than patients who had “sporadic” AD, although again these data derive from relatively few examples with wide variation even within the same family. The relationship between PSEN1 or PSEN2 mutations and neurofibrillary degeneration is more complex. Overall, there is no difference between the amount of neurofibrillary tangles present in AD resulting from PSEN1 or PSEN2 mutations and in “sporadic” AD [38, 96, 162]. One PSEN1 mutation, a substitution in exon 8 (leucine271valine) that results in transcripts lacking exon 8, has been associated with lack of neuritic change in senile plaques [78]. In contrast, increased accumulation of tau species other than neurofibrillary change has been reported in some cases caused by a PSEN1 mutation [144].
PSEN1 mutations also are associated with a morphologic variant in Aβ plaques: the so-called cotton wool plaque (Fig. 4). Cotton wool plaques occur in the same regions of brain as senile plaques and have been associated with multiple PSEN1 mutations. Cotton wool plaques typically have limited fluorescence following staining with thioflavin S and tend to be more immunoreactive for Aβ42 than for Aβ40 [151, 155, 165]. It is important to stress that although highly characteristic of PSEN1 mutation, cotton wool plaques also have been reported in apparently sporadic AD [84, 177].

Photomicrograph of cotton wool plaques. Histologic section of frontal cortex from a patient with a strong family history of AD, who inherited PSEN1 mutation, and died of dementia. Hematoxylin and eosin stain (×200) of cerebral cortex shows marked neuron loss, vacuolization, gliosis, and large amorphous eosinophilic structures that are called cotton wool plaques. Modified Bielschowsky stain (inset) further highlights cotton wool plaques
Phenotypic heterogeneity also occurs between PSEN1 and PSEN2 mutation carriers [8]. The clinical and neuropathologic variability could be related to the mechanism of action of different PSEN1 and 2 mutations, or comorbidity with other neurodegenerative disease conditions.
LBD also has been observed in individuals who inherited a mutation in PSEN1 or PSEN2 [67, 95, 131]. A recent detailed survey of LBD reported that virtually all of the 24 cases of AD caused by nine different mutations in PSEN1 harbored LBD in limbic structures; 11 also had diffuse LBD that also involved regions of neocortex [89]. The same study examined 14 cases of AD from a PSEN2 mutation (asparagine141isoleucine). These had less frequent limbic or neocortical LBD, but a similar frequency of brainstem LBD as the PSEN1 mutation cases. In some cases, diffuse LBD occurs in the context of the expected core neuropathologic features of AD [148], while in others there is some restriction of usual AD changes. For example, a three base pair deletion in exon 12 of PSEN1 was associated with diffuse LBD and increased Aβ accumulation in the form of cotton wool plaques, but not other forms of senile plaques [58].
A series of 11 cases of autosomal dominant AD from mutations in APP or PSEN1 had one example of TDP-43 inclusion formation; this case also showed hippocampal sclerosis [27]. TDP-43 neuronal inclusions are uncommon in cases of AD from the PSEN2 asparagine141isoleucine (Volga German) mutation and were limited to amygdala [63].
Other phenotypes associated with mutations in PSEN1, or other genes encoding components of γ-secretase
NCSTN encodes nicastrin, and PSENEN encodes PEN2; both also are components of the γ-secretase complex (Fig. 3). Mutations in PSEN1, NCSTN, or PSENEN cause a rare autosomal dominant chronic skin disease called acne inversa [170], also called hidradenitis suppurativa, which is characterized by follicular inclusion with secondary inflammation [132]. Three different types of mutations cause this disorder: single base deletions that result in frame shifts and premature stop codons, a mutation that alters a splice site and results in exon skipping and an altered protein, and a nonsense mutation. Each of these mutations is predicted to cause loss of function due to haploinsufficiency and reduced γ-secretase activity. Some work suggests that at least some PSEN1 AD-causing mutations also could be loss of function mutations [45, 142]; however, none of the elderly acne inversa mutation carriers developed early-onset AD, even though there were 15 subjects over age 50. Likewise, subjects with AD-causing PSEN1 mutations do not have severe acne.
Late-onset AD susceptibility genes
While the mutated genes that cause early-onset AD inform about molecular mechanisms, they account for only a fraction of AD cases. The majority of AD patients are late-onset with risk approximately doubling every 5 years after age 65. Thus understanding late-onset AD genetics is imperative if we are to illuminate the mechanisms of neurodegeneration in older individuals. Except for a handful of PSEN2 mutation carriers, late-onset AD very rarely results from highly penetrant dominantly inherited mutations [70]. Rather, evolving data indicate that multiple genes contribute to susceptibility or risk for late-onset AD.
Susceptibility genes are identified by genetic association studies in which allele frequencies for polymorphisms at or near a gene are compared between cases and controls. Susceptibility genes are revealed when case and control frequencies differ significantly. Early attempts to identify these types of susceptibility genes were driven by selecting candidate genes based on the existing knowledge of disease pathogenesis. Until about 2005, due to technical limitations, only small numbers of genes and modest populations could be evaluated. Over 1,000 candidate genes were tested for AD susceptibility. With few exceptions, most studies of this type could not be replicated likely because of inadequately evaluated population substructure, sample sizes that were too small to detect meaningful associations, and P value thresholds that were insufficiently stringent.
Recent advances now make it possible to evaluate essentially all genes and all regions between genes in a single experiment, a method called genome-wide association studies (GWAS). This method has led to an explosion in susceptibility gene discovery for a large number of diseases, disease-related traits, and associated phenotypes [123]. For brain diseases, successful GWAS results have been reported for AD [114], Parkinson’s disease, amyotrophic lateral sclerosis, progressive supranuclear palsy [48], and multiple sclerosis, to name a few.
Advances in a number of key technological and analytical areas have driven rapid progress in complex disorder genetics using GWAS. First, advances in genotyping platforms permit interrogation of most of the genome for disease-associated variation in a single experiment. Current genotyping arrays contain assays between 600,000 and 2.5 million single nucleotide polymorphisms (SNPs) and cost less than $500/subject. To accompany this cost-effective genotyping methodology, the HapMap Project and the more recent 1,000 Genomes Project identified over 20 million SNPs and provide key information on linkage disequilibrium (LD), or correlation, between neighboring SNPs. Understanding LD not only allows the construction of haplotypes, but also provides the ability to impute the genotypes of nearby unobserved (not genotyped) SNPs using directly observed genotypes. Imputing not only provides additional information, but also facilitates merging data from different genotyping platforms with incomplete overlap, an advance that makes initial selection of genotyping platform less critical.
Analytical advances also have been made in methods for evaluating population substructure and for matching cases and controls. Even when using approaches to avoid a mismatch between cases and controls, such as collecting ethnicity information, it is still possible to have cryptic population substructure in case–control samples. Now, principal component methods are routinely used in GWAS to match cases and controls, avoiding false-positive results. This is particularly important as we move toward methods that detect rare variants (see below). Other advances in quality-control methods for managing large data sets are now firmly established. These include standards for identifying problematic SNPs and poor quality samples that can yield unreliable results.
Two susceptibility genes for AD had been identified prior to large GWAS: APOE and SORL1. We will discuss these genes separately from those identified by GWAS methods.
APOE
This gene encodes apolipoprotein E (ApoE), a protein originally extensively studied for its role in lipid metabolism but now recognized to be a pleiotropic molecule with important (perhaps overlapping) actions in lipid transport, Aβ trafficking, synaptic function, immune regulation, and intracellular signaling [101, 102]. ApoE, like other apolipoproteins, is a protein component of lipoprotein particles that binds to the cell surface receptors. Humans are unlike other mammals in that we have three common alleles of APOE called ε2, ε3, and ε4 alleles that encode corresponding 299 amino acid-containing protein isoforms. Early work identified APOE genotypes as contributing to risk for heart disease [55, 100]. In 1991, Pericak-Vance and colleagues [124], using family-based methods, identified a genetic linkage between AD and a region of chromosome 19 that harbors APOE. Subsequent work showed that polymorphisms within the APOE gene were strongly associated with AD [152]. The polymorphisms showing association are the ε2, ε3, and ε4 alleles. These alleles represent haplotypes of two SNPs in the coding region of APOE. The ε2 allele encodes a cysteine (Cys) both at amino acid position 112 and at position 158. Allele ε3 encodes a Cys at 112 and an arginine (Arg) at 158, while the ε4 allele encodes an Arg both at 112 and at 158. The ε4 allele is the ancestral and high-risk form [100], the ε3 allele is the most common in humans and the neutral allele, and the ε2 allele is associated with decreased risk of AD. On a population level, people with an ε4 allele have lower average onset age and those with an ε2 allele have a higher average onset age for AD [21]. The AD risk associated with these alleles is additive. A person with a genotype of ε4/ε4 is at higher risk than someone with an ε3/ε4 or ε2/ε4 genotype. Likewise, ε2/ε2 genotype is more protective than genotypes where only one ε2 allele is inherited. This risk pattern has been replicated in many publications and in all ethnic groups studied [34]. While earlier work observed that amyloid plaques had ApoE immunoreactivity [115] and that APOE expression is up-regulated in activated astrocytes in AD brain [30], it was the genetic discovery that clearly linked ApoE isoforms to AD pathogenesis.
APOE genotypes also influence onset ages in carriers of PSEN1 [122] or PSEN2 [171] mutations. Less compelling evidence suggests that APOE genotypes also affect age of onset in subjects carrying APP mutations [150]. Several publications reported that SNPs other than the APOE ε2/ε3/ε4 alleles might influence risk by modifying the effects of these three alleles. Early work suggested that polymorphisms within the promoter of APOE influence risk, though these findings were not replicated in large data sets. A recent publication, using a modest sample size, suggests that a poly-T track in TOMM40, the gene adjacent to APOE (Fig. 5) affects AD onset age [134]. The poly-T track in TOMM40 is variable in length; long poly-T length alleles associated with the ε3 APOE allele were reported to have an earlier onset age than short poly-T track alleles. In subsequent work using substantially larger samples, this relationship between poly-T track length and onset age was not replicated, and when APOE genotypes were accounted for, there was no significant association to TOMM40 [18, 26].

APOE gene structure and the ε2/ε3/ε4 allele system. The top of the figure shows the relationship between APOE and adjacent genes TOMM40 and APOC1 (not drawn to scale). In the middle of the figure, APOE exon structure and location of the polymorphic nucleotides that are responsible for the APOE ε2/ε3/ε4 allele system are shown. At the bottom, nucleotide and the resulting amino acid changes corresponding to these alleles are shown
APOE is by far the strongest risk locus in GWAS of AD. It is observed as a significant signal even in GWAS with limited power [1, 6, 9, 40, 92]. Unfortunately, the SNPs that define the APOE ε2/ε3/ε4 alleles were not on the early genotyping arrays. These SNPs are present on more recent arrays but the genotypes are not reliable. Often, the results reported are for surrogate SNPs that do not capture the complete genetic information for the APOE tri-allelic system. Still, odds ratios (OR’s) for APOE from surrogate SNPs, ranging from 2.3 to 4.01 [15, 19, 141], are much larger than other AD risk loci that have been identified by GWAS (see below). Because the sample size used in these studies is very large (>10,000 cases and 10,000 controls), and because APOE has such a large effect, there are many SNPs with genome-wide significance (P < 5 × 10−8) in the APOE region and these can span over 400 kb [114]. Additional SNPs spanning a larger region show nominal significance. The signal from many, if not most, of these adjacent SNPs disappears when the analysis is conditioned on APOE ε2/ε3/ε4 genotypes. However, in one study of SNP rs597668, which is 296 kb from APOE (between BLOC1S3 and EXOC3L), adjusting for APOE genotypes still left a signal with an OR = 1.18, 95 % confidence interval (CI) of 1.08–1.24, and P = 3.9 × 10−4. This finding was not replicated in other GWAS [65, 172]. Additional work is needed to determine whether it is an independent AD risk factor, or it is due to extended LD between APOE and the BLOC1S3/EXOC3L region.
SORL1
The gene product of the SORL1 gene is a receptor that participates in trafficking vesicles from the cell surface to the Golgi-endoplasmic reticulum, a pathway thought to be important for APP processing and the generation of Aβ peptide. For this reason, Rogaeva and co-workers selected SORL1 and related genes as candidates for AD susceptibility loci [133]. In the original report, there was a complex association between AD and SNPs clustered at two locations within SORL1. As with many candidate gene associations, initial attempts to replicate these findings were mixed with both positive [10, 75, 76, 83, 86, 87, 109, 159] and negative [93, 98, 111, 139] findings. Not all of these studies had adequate sample size or sufficient power for replication.
A recent unbiased meta-analysis of all previous SORL1 association studies reported significant association between both clusters of SNPs and AD in Caucasians [129]. Only results from the 3’ end of the gene were significant from an Asian cohort. This meta-analysis used a large sample consisting of 11,592 cases and 17,048 controls for the Caucasian analysis and 872 cases and 881 controls for the Asian analysis. Additional support for an association between SORL1 variants and AD also comes from several GWAS [6, 114, 141], although the sample set partially overlaps with the SORL1 meta-analysis. Thus, there is strong evidence for SORL1 as an AD risk gene. Further evidence awaits meta-analysis of all GWAS from a combined meta-analysis of the four major AD genetics consortia. The Alzheimer’s Disease Genetics Consortium (ADGC), the Genetic and Environmental Risk in Alzheimer’s disease (GERAD) consortium, the European Alzheimer Disease Initiative (EADI) group, and the Cohort for Heart and Aging Research in Genomic Epidemiology (CHARGE) are undertaking this analysis as part of the International Alzheimer Genomic Project (IGAP) [52].
Unbiased GWAS of AD
The candidate gene approach was successful for identifying APOE and SORL1 as risk loci. However, over 1,000 other candidate genes for late-onset AD were reported, and most, if not all, were false positives. Initial GWAS of AD had limited power [1, 6, 9, 15, 19, 40, 92, 128], but two findings were clear. First, as a proof of principle, APOE was easily detected with P values ranging from 5.3 × 10−34 to 3.7 × 10−120. Second, there was no other locus of similar effect size to APOE. In two of these studies, loci reaching genome-wide significance were reported. Carrasquillo et al. [15] highlighted the X-linked gene PCDH11X with an OR = 1.30 (CI 1.18–1.43). Evidence was stronger in females than males using a sample of 2,356 cases and 2,384 controls. Reiman et al. [128], in a combined cohort of 861 cases and 550 controls, observed that SNPs in GAB2 reached genome-wide significance (9.66 × 10−11, OR = 4.06, CI = 2.81–14.68), but only in APOE ε4 carriers. In subsequent much larger GWAS, the signal at PCDH11X did not replicate and the signal for GAB2 was only nominally significant (P = 0.017) [7, 114].
In the past 3–4 years, well-powered GWAS were performed and new genes significantly associated with late-onset AD were discovered and replicated in independent samples. The advantages of this genome-wide approach are several fold: (1) gene discovery by this method is not predicated on previous knowledge about AD pathogenesis, and thus has the capacity to identify new genes and molecular pathways relevant to AD; (2) GWAS methods can be used with case–control populations that were collected in earlier studies, leveraging pre-existing resources; (3) rigorous quality control and statistical methods coupled with sufficient sample size can lead to high reproducibility; (4) methods are available so that investigators can recognize and account for cryptic population substructures, contributing to reproducibility. Disadvantages of this approach are: (1) signals are often ambiguous and may span multiple genes (see MS4A gene cluster described below and Fig. 5); (2) signals can be in intergenic regions making assessment of the functional relevance difficult; (3) genetic methods often cannot identify which single nucleotide variant is pathogenic; (4) most signals are from small effect loci.
It is worthwhile to pause on this last point of small effect size for risk associated with many GWAS-identified loci. The small effect size of most GWAS signals determines that each will add only incrementally risk assessment; nevertheless, small effect loci can be tremendously insightful by revealing relevant players in molecular pathogenesis and thereby highlighting potential therapeutic targets. An excellent example is the quantitative lipid trait on chromosome 1p13 where the signal detected initially spanned multiple genes and, when further refined, was in an intergenic regulatory element [161]. Additional studies identified SORT1 as the relevant gene, revealing a new pathway for VLDL secretion [112]. This work shows that despite the intergenic nature of the signal, the relevant gene can be identified, and despite the small contribution of genetic variation to the trait, the molecular mechanisms may be illuminated.
The following sections review recent AD GWAS work.
Clusterin (CLU), phosphatidylinositol binding clathrin Assembly protein (PICALM), and complement receptor type 1 (CR1)
The first GWAS to report loci that could be replicated were published in 2009 [44, 97]. The GERAD consortium [44] employed a two-stage design. Stage 1 consisted of 3,941 AD cases and 7,848 controls, of which 2,078 were screened, healthy elderly and the remaining controls were unscreened subjects whose mean age was younger than the age when AD risk begins. Genotypes for 529,205 SNPs, obtained using a commercial genotyping array, were analyzed for association with AD. In this stage, the most significant SNPs were close to APOE with the most significant result coming from a SNP in TOMM40 (P = 1.8 × 10−157, OR = 2.53, CI = 2.37–2.71). Note that the SNPs responsible for the APOE ε2/ε3/ε4 genotypes were not on the SNP array used, and these APOE SNPs are incompletely tagged by other SNPs in the region. Thus the fact that SNPs in TOMM40 are the SNPs yielding the lowest P value, most likely reflects the strong LD between TOMM40 SNPs and APOE genotypes, and not that TOMM40 is a better candidate for the signal in this region. Two other signals reached the threshold for genome-wide significance (P < 5 × 10−8), one in CLU (P = 1.4 × 10−9, OR = 0.84, CI = 0.79–0.89) and one near PICALM (P = 1.9 × 10−8, OR = 0.85, CI = 0.80–0.90). In stage 2, an additional 2,023 AD cases and 2,340 age-matched cognitively normal controls were genotyped and evidence supporting CLU and PICALM as AD risk genes was obtained.
Another AD GWAS by the EADI [80], published at the same time as GERAD consortium [112], used an independent set of samples also with a 2-stage approach for 2,025 cases and 5,328 controls in stage 1 and 3,803 cases and 3,097 controls in stage 2. Stage 1 yielded an association between late-onset AD and a SNP in CLU that approached genome-wide significance and marginal results for several other loci. Select SNPs were genotyped for the stage 2 cohort and supporting evidence obtained for CLU and a second site at CR1. The combined stage 1 + 2 analysis yielded significant results for CLU (P = 7.5 × 10−9, OR = 0.86, CI = 0.81–0.90), consistent with the findings of Harold et al. [112] for this locus. The signal at CR1 also reached genome-wide significance (P = 3.5 × 10−9, OR = 1.21, CI = 1.14–1.29) in the stage 1 + 2 analysis. Evidence supporting PICALM as an AD risk locus also was reported.
Results from these two GWAS demonstrated that reproducible genetic associations could be obtained with AD as long as sample sizes were sufficiently large and rigorous criteria for genome-wide significance were employed. The association with CR1, CLU, and PICALM has been confirmed by a number of investigators [65, 172], including one GWAS reviewed below that used a very large independent sample [114]. Also, these reports clearly demonstrate that there are no common variants with effect sizes comparable to APOE, and that remaining AD risk loci will be small effect genes.
Bridging integrator protein-1 (BIN1)
A subsequent manuscript, which included population-based cohorts from the CHARGE consortium, reported strong evidence that BIN1 is an AD risk locus [141]. In stage 1, incident cases and normal cohort subjects were analyzed along with an autopsy case–control cohort previously described by Reiman et al. [19, 128] and a second case–control group previously described by Carrasquillo et al. [15]. Stage 1 had 973 incident AD cases and 2,033 prevalent cases from both the population-based and the case–control studies. These were compared to 14,649 dementia-free controls. In stage 1, the only SNPs yielding significant results were those in the APOE region with the most significant SNP being again in TOMM40 (P = 3.18 × 10−68, OR = 2.23, CI = 2.04–2.44). SNPs with P values <10−3 were carried on to stage 2, which used samples from EADI [80]. SNPs with a P value <10−5 were carried on to stage 3, which used samples from GERAD [44]. Meta-analysis of all three stages showed genome-wide significance for two genes: BIN1 (P = 1.59 × 10−11, OR = 1.15, CI = 1.11–1.20) and exocyst complex component 3-like 2 (EXOC3L2; P = 6.45 × 10−9, OR = 1.17, CI = 1.11–1.23). Because EXOC3L2 is about 300 kb from APOE, conditional analysis was performed, using either the APOE ε2/ε3/ε4 or a surrogate SNP in TOMM40. For both analyses, an attenuated marginal signal remained that was not genome-wide significant. Additional work is needed to determine if a gene in the EXOC3L2 region is a risk factor independent of APOE (see below).
ABC transporter member 7 (ABCA7), membrane-spanning 4-domains, subfamily A, member 4 (MS4A4A)/membrane-spanning 4-domains, subfamily A, member 4E (MS4A4E)/membrane-spanning 4-domains, subfamily A, member 6A (MS4A6A), ephrin receptor EphA1 (EPHA1), CD33 antigen (CD33), and CD2-associated protein (CD2AP)
In 2011, two simultaneously published manuscripts described strong evidence for five new AD risk loci. The manuscript of Naj et al. [114] from the National Institute on Aging (NIA) AD Genetics Consortium (ADGC) reported a three stage design. Stage 1 consisted of 8,309 AD cases and 7,366 elderly screened normal. Some subjects (1,604 cases and 675 controls) were from NIA-funded Alzheimer’s Disease Centers (ADC’s) across the US that contributed material and data to the ADGC. The National Alzheimer Coordinating Center (NACC) collects the phenotypes for these subjects and the tissue/DNA is centralized at the National Cell Repository for Alzheimer’s Disease (NCRAD). The remaining subjects were from other published [176] and unpublished case–control and population-based studies. This stage yielded genome-wide significance for the previously associated loci APOE, CR1, and BIN1, and two novel loci: EPHA1 and MS4A4A/MS4A4E/MS4A6A. Loci with at P < 10−6 were carried on to stage 2 that added samples from the NIA ADCs and from several other case–control and population-based studies (3,531 cases, 3,565 controls). Combined analysis of stages 1 + 2 yielded genome-wide significance for two additional loci, PICALM and CLU, confirming previous results. The five novel loci that had suggestive or genome-wide significance were carried on to stage 3 where data were supplied from CHARGE, EADI, and GERAD consortia. Four of the five loci tested in stage 3 yielded genome-wide significance when all three stages were combined in a meta-analysis. These were CD2AP (P < 8.6 × 10−9, OR = 1.11, CI = 1.07–1.15), EPHA1 (P < 6.0 × 10−10, OR = 0.90, CI = 0.86–0.93), MS4A4A (P < 8.2 × 10−12, OR = 0.91, CI = 0.88–0.93), and CD33 (1.6 × 10−9, OR = 0.91, CI = 0.88–0.93). A fifth gene, ARID5B, did not replicate. This work also reported suggestive results for ABCA7 (5.0 × 10−7, OR = 1.15, CI = 1.09–1.21) with the peak signal at a non-synonymous SNP that is potentially the pathogenic variant (p.Gly1527Ala). These data were supplied to the CHARGE, EADI, and GERAD consortia to support evidence for this locus as an AD risk gene. As a summary of this work, the five previous AD risk loci (APOE, CLU, CR1, PICALM, and BIN1) were confirmed with genome-wide significance in a completely independent sample. Note that in this manuscript, and the one described in the following paragraph, investigators from all groups communicated to avoid using identical or overlapping datasets for the final analyses, an important precaution because some samples and datasets are broadly shared.
A second manuscript by Hollingworth et al. [50] reports the findings of the CHARGE, GERAD, and EADI consortia. Again a multi-stage approach was used. In stage 1, there were 6,688 cases and 13,685 controls from the GERAD group, consisting of both screened and un-screened control subjects. Genome-wide significance was obtained in stage 1 for MS4A6A and suggestive results for ABCA7. Select SNPs for these loci were tested in stage 2 (4,896 cases and 4,903 controls) and support was obtained for both. Select SNPs were carried on to stage 3, which used data from the EADI and CHARGE consortia and another case–control series. The combined stage 1 + 2+3 analysis produced results with genome-wide significance for ABCA7 (P = 5.0 × 10−21, OR = 1.23, CI = 1.17–1.28), and MS4A6A/MS4A4E (P = 1.2 × 10−16, OR = 0.91, CI = 0.88–0.93). In addition, in collaboration with the ADGC, the EADI, GERAD, CHARGE groups contributed data to support that CD33, EPHA1, and CD2AP are AD risk loci.
Other GWAS reports
A number of additional GWAS reports have been published that evaluate AD case versus controls as the primary phenotype [1, 85, 92, 113, 172], as well as other endophenotypes [17, 28, 51, 81, 140, 143]. Of the AD case versus control studies, the most noteworthy is the report of Naj et al. [113] who reported a signal (P = 1.9 × 10−10, OR = 2.1, CI = 1.67–2.64) at MTHFD1L, which encodes a component of the tetrahydrofolate synthesis pathway. In another study, SNPs highlighting CUGBP2 produced genome-wide significance in APOE ε4 homozygotes [172]. The findings from both of these studies will require independent replication.
Other AD-related phenotypes that have been used in GWAS include MRI and cognitive measures [140, 143], age-related cognitive decline [17, 28], psychosis [50], and age-at-onset [83]. While none of these studies produced genome-wide significant loci, these phenotypes could be important in defining the risk gene architecture of AD when larger samples are assembled.
Functional implications of late-onset AD genes
The early genetic studies of APP, PSEN1, and PSEN2 led not only to the establishment of Aβ peptide as a critical molecule in AD pathogenesis, but also to therapeutic approaches that are being tested currently. This success came despite the normal role of APP remaining unknown. Also, when initially described, PSEN1 and PSEN2 were essentially orphan sequences with only limited homology with proteins with known functions. In contrast, for most of the late-onset AD genes described above, the normal functions are at least partially known. However, the role of each in AD pathogenesis will require more investigation. A few themes emerge that can focus future work on specific pathways.
Lipid metabolism and APOE, SORL1, CLU, and ABCA7
When APOE was first identified as a late-onset AD susceptibility gene, much was already known about the function of ApoE in cholesterol transport [101]. In brain, ApoE is the main cholesterol transport protein. Despite being an extensively studied protein, the role of ApoE isoforms in AD pathogenesis remains enigmatic [12, 72]. ApoE may influence AD through its role in cholesterol transport, either in the periphery and/or in the brain. In the brain, cholesterol is needed to support synaptogenesis and maintenance of synaptic connections. Alternatively, ApoE might bind Aβ and act as a chaperone to influence the rate of Aβ fibrillogenesis. Another hypothesis is that ApoE lipid particles bind Aβ and clear the extracellular peptide by endocytosis and subsequent degradation. In each of these proposed mechanisms, the functional difference between the ε2-, ε3-, and ε4-encoded isoforms is the basis of risk associated with APOE.
SORL1 was selected as a candidate gene because it encodes a protein, SorLA, that is important in intracellular APP trafficking [133]. When cell surface APP is re-cycled via the endocytic pathway, SorLA directs APP to processing by presenilins to produce Aβ. This hypothesis is supported by studies of SorLA and APP processing in cell-based systems, in vitro studies, and in mouse models [3, 4, 46, 149]. Another function of SorLA related to AD is as a lipoprotein receptor that can bind ApoE [154].
Clu, also known as apolipoprotein J (ApoJ), is another lipoprotein found in both the periphery and the brain [119]. Like ApoE, Clu is involved in lipid transport in both the periphery and the brain where it is highly expressed [174]. Clu also is hypothesized to act as an extracellular chaperone that influences Aβ-aggregation and receptor-mediated Aβ clearance by endocytosis [64, 119]. Unlike APOE, there are no known coding variants that account for the observed genetic association to CLU, suggesting that genetic variation in expression levels may be responsible for altered risk for AD.
ABCA7 encodes a protein belonging to ABC superfamily of transporters. The normal role of the ABCA7 protein is only partially understood. It appears to function in the efflux of phospholipids and possibly cholesterol [74], although this function is not supported by recent work with ABCA7 knockout mice. Another role for this transporter is in phagocytosis. ABCA7 levels are regulated by sterol-responsive/regulatory element binding protein in response to cholesterol levels, which, in turn, promotes ABCA7 regulated phagocytosis [60]. While ABCA7 is clearly involved in lipid metabolism, how this protein is connected to AD will require additional work.
Innate and adaptive immunity
A number of genes identified in GWAS highlight the innate and adaptive immune response. One is CR1, a cell surface receptor that is part of the complement system and plays a role in regulating the complement response. CR1 has binding sites for complement factors C3b and C4b and participates in clearing immune-complexes containing these two proteins. Since Aβ oligomers can bind C3b, CR1 may participate in the clearance of Aβ. CR1 may also play a role in neuroinflammation, which is a prominent feature in AD [24, 147]. Interestingly, Clu may play a role in this process as an inhibitor [108]. Finally, in addition to their other roles in lipid and Aβ trafficking, ApoE isoforms differentially regulate the innate immune response in brain [71].
Another immune system-related gene identified by an AD GWAS is CD33. This gene encodes a protein that is a member of a family of cell surface immune receptors that bind extracellular sialylated glycans and signal via a cytoplasmic domain called the immunoreceptor tyrosine inhibitory motif [14, 25, 168]. CD33 has primarily been studied in peripheral immune system where it is expressed on myeloid progenitors and monocytes and also in the brain. In the periphery, CD33 appears to inhibit proliferation of myeloid cells [167]. The role of CD33 in brain is unknown.
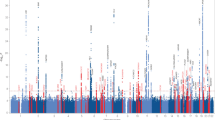
The MS4A4A/MS4A4E/MS4A6E locus is significantly associated with AD. These genes are part of a cluster of 15 MS4A genes on chromosome 11 [56, 94] and encode proteins with multiple membrane-spanning domains that were initially identified by their homology to CD20, a B-lymphocyte cell surface molecule. Little is known about the function of MS4A4A gene products; however, like CD33, MS4A4A is expressed on myeloid cells and monocytes. Based on this expression pattern and the homology to CD20, MS4A4A and possibly other members of this protein family may have an immune-related function. The GWAS signal identifying the MS4A4A/MS4A4E/MS4A6E locus cannot distinguish which of these three genes is responsible for the AD risk at this location (Fig. 6). Note that the top SNP is in an intergenic region, and that SNPs with linkage disequilibrium of r 2 > 0.8 with the top SNP also span several genes.

Manhattan plot for AD association for the MS4A region of chromosome 11. The association plot is for P values determined by a three-stage meta-analysis as reported in Naj et al. [114]. The X axis is the genomic location (NCBI Build 37.1) and the y axis is the −log10 of the P value. The black diamond is the P value for the SNP with the smallest P value in the discovery phase (stage 1). The purple diamond is the P values for the same SNP for the first replication phase (stage 1 + 2). The red diamond is the P value for the same SNP at the second replication phase (stage 1 + 2 + 3). The colors of the small diamonds (orange, yellow, gray and white) indicate the estimated linkage disequilibrium with the top SNP and are r 2 ≥ 0.8, 0.8 > r 2 ≥ 0.5, 0.5 > r 2 > 0.2, and r 2 < 0.2, respectively. Genes in the region are indicated by arrows at the bottom with the direction of the arrow indicating the direction of transcription and the length proportional to the size of the gene
Cell adhesion and EPHA1
This gene, implicated in AD risk, encodes a member of the ephrin family of receptor protein-tyrosine kinases [47, 121, 125]. This group of cell surface receptors interacts with ephrin ligands on adjacent cells to modulate cell adhesion. In brain development, these receptors are important in establishing appropriate connections between different brain regions and act by modulating cell adhesion, migration, and axon guidance. Ephrin receptors also are involved in synapse formation and in plasticity [79]. Another brain development function of ephrin receptors is the regulation of apoptosis of neural progenitor cells [29]. These receptors also function in other systems including epithelial branching morphogenesis, the blood and lymphatic vascular systems, and the peripheral inflammatory response [59].
While there is a substantial body of research on the function of ephrin receptors, less is known about the EPHA1 gene product. Like other ephrin receptors, it regulates cell morphology and motility [175]. Early work implicated this receptor in regulating vascular morphogenesis and angiogenesis [2]. Knockout of the mouse gene results in abnormal tail and reproductive tract development [32], but no defects were detected in other tissues including the brain. In mouse, expression is restricted to epithelial tissue; no expression was detected in the brain or spinal cord. In humans, EPHA1 is expressed by CD4-positive T lymphocytes [49], monocytes [137], intestinal epithelium, and colon [42]. There is no work on EPHA1 and neurodegenerative diseases. The lack of evidence for brain expression and the expression by lymphocytes and monocytes suggests that, like CD33, CR1, and MS4A4/MS4A6E (discussed above), the role of the EPHA1 gene product in AD is mediated though the immune system.
Endocytosis pathways: BIN1, PICALM, and CD2AP
Several genes revealed in GWAS encode proteins involved in intracellular vesicle trafficking. Bin1 belongs to a family of proteins with BAR (Bin1/amphiphysin/RVS167) domains. These proteins are membrane adaptors that form complexes that act in clathrin-mediated endocytosis [31, 126, 127, 130]. The functions of Bin1 may be complex judging by the numerous splice variants that can be produced by this gene. PICALM also is involved in clathrin-mediated endocytosis and recruits clathrin and adaptor protein complex 2 to sites of vesicle assembly [160, 164]. How these endocytic genes are involved in AD pathogenesis is unknown.
The CD2 associated protein gene (CD2AP) encodes a scaffolding protein that binds directly to actin [88], nephrin, a cell–cell adhesion protein [145], and other proteins involved in cytoskeletal organization. This protein is implicated in dynamic actin remodeling during cytokinesis and endocytosis and has been extensively studied in kidney podocytes where it is a component of the slit diaphragm intercellular junction complex [99]. Haploinsufficiency of this protein causes focal segmental glomerulosclerosis [73]. In the immune system, CD2AP is required for synapse formation [33] in a process that involves clathrin-dependent actin polymerization [13]. While expressed in brain, the normal function of CD2AP in the CNS and its role in AD have not been studied.
Clinical implications of AD genetics findings
Genetic findings, once unambiguously identified, have the potential to predict an individual’s disease risk, phenotype, and possibly progression. They also may contribute to clinical management by prompting heightened surveillance, providing indication for preventive intervention, and guiding treatment selection. While there is still a long way to go in our understanding of the genetics of AD, our current knowledge about APP and PSEN1 mutations clearly shows that carriers will develop early-onset AD usually before the age of 60 years. PSEN2 mutations have a broader range of onset ages, limiting but not abrogating risk assessment. Since there are presently no preventive therapies for AD, detection of these autosomal dominant mutations is mostly used medically for making reproductive decisions. In the hopefully near future, when disease suppressing interventions become available, identifying carriers with one of these autosomal dominant mutations might be followed by biomarker or neuroimaging surveillance to determine when disease prevention or disease modifying treatment should begin.
The AD susceptibility genes, APOE, SORL1, and the genes recently identified by GWAS, have mixed utility for disease risk assessment. APOE ε4 is by far the strongest risk factor with the contributions by the other loci being minor by comparison. Of particular interest in terms of predictive testing is the APOE ε4/ε4 genotype. The lifetime risk for AD in individuals with this genotype is high, estimated as 33 or 32 % for men or women by the age of 75 years; by age 85 years, the risk climbs to 52 % for men and 68 % for women [35]. As recently pointed out by Genin et al., this is comparable to the lifetime risk by age 70 years for carcinoma of the breast in women with a BRCA1 mutation [16]. Fortunately, only 2 % of the (Caucasian) population has the APOE ε4/ε4 genotype. Considering the delayed penetrance of “sporadic” AD, lack of preventive therapies, and the potential for psychological harm, genetic testing for APOE is not recommended. However, when AD prevention becomes possible, then this recommendation will need to be reconsidered.
Summary of AD genetics
Tables 1 and 2 summarize findings of AD genetics studies. Several important conclusions can be drawn from these studies. First, APP, PSEN1, and PSEN2 genes are unambiguously established as autosomal dominant causes of AD, with evidence coming from numerous mutations in multiple families with early-onset AD. Second, remarkably, the evidence for the late-onset AD risk loci is the replicable across independent data sets using cohorts ascertained by a variety of approaches and cases diagnosed in numerous research centers. Thus, not only are the results robust, but also the diagnostic and analytic procedures are used across studies. Third, APOE is by far the largest effect locus for late-onset AD. Note that the ORs in Table 1 are from surrogate SNPs that do not completely capture the APOE ε2/ε3/ε4 genotypes. A better estimate of effect size comes from studies where the SNPs responsible for the ε2/ε3/ε4 alleles are directly genotyped and different genotypes considered separately. For example, in Farrer et al. [34], ORs (for Caucasians) are 1.0 for ε3/ε3 (referent genotype), 0.6–0.9 for ε2/ε2, 0.6 for ε2/ε3, 1.2–2.6 for ε2/ε4, 2.7–3.2 for ε3/ε4, and 12.5–14.9 for ε4/ε4. Fourth, it is highly likely that additional small effect risk loci will be identified for late-onset AD. This will require larger sample sizes. To achieve this, an international effort called the International Genomics Alzheimer’s Project (IGAP) is underway with participation of the ADGC, CHARGE, GERAD, and EADI consortia [52].
The remarkable progress in AD genetics that started in 1992 has led to candidate therapeutics currently under evaluation in clinical trials. Second-generation Aβ immunotherapies are now being evaluated in clinical trials, as are γ-secretase inhibitors. While no ApoE-based therapies are in clinical trials, recent work [22] suggests that bexarotene, a compound that induces APOE expression, may have therapeutic potential. The time from gene discovery to candidate therapy ready for testing in clinical trials is discouragingly long, and it is, therefore, not surprising that no therapies yet have been developed from the 10 AD risk genes found over the past 3 years. Also, not every gene discovered will have a product that is approachable therapeutically. Nevertheless, there are examples where GWAS identified small effect risk loci are highly effective drug targets. One quantitative trait GWAS of lipid levels [161], e.g., identified two risk genes, HMGCoA reductase and NPC1L1, that are the target of simvastatin and Zetia (ezetimibe), both drugs used to lower cholesterol and prevent heart disease. The genetic variation in these genes had a small effect on population cholesterol levels, yet the drugs that target the products of these genes are arguably the most important preventive medications in use today. Other diseases for which GWAS hits revealed important drug targets include autoimmune disorders such as multiple sclerosis, ankylosing spondylitis and rheumatoid arthritis [166]. These examples illustrate not only that GWAS can identify potential therapeutic targets, but also that the effect size observed is GWAS is not an indicator of the biologic or medical significance of the gene’s product.
Despite the remarkable progress over the last 3 years, additional approaches need to be applied to completely resolve AD genetics. First, endophenotypes including cerebrospinal fluid biomarker data, quantitative neuropathology measures, and the phenotypes described above need to be used in studies where the sample size is sufficiently large to detect additional AD risk loci. Second, massively parallel DNA sequencing methods need to be used to detect rare large effect alleles in genes already identified by GWAS methods as well as variants that modulate susceptibility to AD in only a small fraction of late-onset cases. Third, genome-wide evaluation of insertion and deletion polymorphisms (indels) and copy-number variation (CNV) needs to be performed. Currently, SNP arrays can be used to detect CNVs that span greater than ~ 5 kb. However, shorter CNVs or indels cannot routinely be detected and genome-wide scans of this type of genomic variation await large-scale whole genome sequencing methods applied to AD.
The new wave of genetic discoveries in AD is just beginning.
References
Abraham R, Moskvina V, Sims R, Hollingworth P et al (2008) A genome-wide association study for late-onset Alzheimer’s disease using DNA pooling. BMC Med Genomics 1:44
Adams RH, Klein R (2000) Eph receptors and ephrin ligands. Essential mediators of vascular development. Trends Cardiovasc Med 10:183–188
Andersen OM, Reiche J, Schmidt V, Gotthardt M et al (2005) Neuronal sorting protein-related receptor sorLA/LR11 regulates processing of the amyloid precursor protein. Proc Natl Acad Sci USA 102:13461–13466
Andersen OM, Schmidt V, Spoelgen R, Gliemann J et al (2006) Molecular dissection of the interaction between amyloid precursor protein and its neuronal trafficking receptor SorLA/LR11. Biochemistry 45:2618–2628
Basun H, Bogdanovic N, Ingelsson M, Almkvist O et al (2008) Clinical and neuropathological features of the Arctic APP gene mutation causing early-onset Alzheimer disease. Arch Neurol 65:499–505
Beecham GW, Martin ER, Li YJ, Slifer MA et al (2009) Genome-wide association study implicates a chromosome 12 risk locus for late-onset Alzheimer disease. Am J Hum Genet 84:35–43
Beecham GW, Naj AC, Gilbert JR, Haines JL et al (2010) PCDH11X variation is not associated with late-onset Alzheimer disease susceptibility. Psychiatr Genet 20:321–324
Bergmans BA, De Strooper B (2010) gamma-secretases: from cell biology to therapeutic strategies. Lancet Neurol 9:215–226
Bertram L, Lange C, Mullin K, Parkinson M et al (2008) Genome-wide association analysis reveals putative Alzheimer’s disease susceptibility loci in addition to APOE. Am J Hum Genet 83:623–632
Bettens K, Brouwers N, Engelborghs S, De Deyn PP et al (2008) SORL1 is genetically associated with increased risk for late-onset Alzheimer disease in the Belgian population. Hum Mutat 29:769–770
Bornebroek M, Haan J, MaatSchieman MLC, VanDuinen SG et al (1996) Hereditary cerebral hemorrhage with amyloidosis-Dutch type (HCHWA-D).1. A review of clinical, radiologic and genetic aspects. Brain Pathol 6:111–114
Bu GJ (2009) Apolipoprotein E and its receptors in Alzheimer’s disease: pathways, pathogenesis and therapy. Nat Rev Neurosci 10:333–344
Calabia-Linares C, Robles-Valero J, de la Fuente H, Perez-Martinez M et al (2011) Endosomal clathrin drives actin accumulation at the immunological synapse. J Cell Sci 124:820–830
Cao H, Crocker PR (2011) Evolution of CD33-related siglecs: regulating host immune functions and escaping pathogen exploitation? Immunology 132:18–26
Carrasquillo MM, Zou F, Pankratz VS, Wilcox SL et al (2009) Genetic variation in PCDH11X is associated with susceptibility to late-onset Alzheimer’s disease. Nat Genet 41:192–198
Chen S, Parmigiani G (2007) Meta-analysis of BRCA1 and BRCA2 penetrance. J Clin Oncol 25:1329–1333
Chibnik LB, Shulman JM, Leurgans SE, Schneider JA et al (2011) CR1 is associated with amyloid plaque burden and age-related cognitive decline. Ann Neurol 69:560–569
Chu SH, Roeder K, Ferrell RE, Devlin B et al (2011) TOMM40 poly-T repeat lengths, age of onset and psychosis risk in Alzheimer disease. Neurobiol Aging 32:2328.e1–2328.e9
Coon KD, Myers AJ, Craig DW, Webster JA et al (2007) A high-density whole-genome association study reveals that APOE is the major susceptibility gene for sporadic late-onset Alzheimer’s disease. J Clin Psychiatry 68:613–618
Coppus A, Evenhuis H, Verberne GJ, Visser F et al (2006) Dementia and mortality in persons with Down’s syndrome. J Intellect Disabil Res 50:768–777
Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE et al (1993) Gene dose of apolipoprotein-E Type-4 allele and the risk of Alzheimer’s disease in late onset families. Science 261:921–923
Cramer PE, Cirrito JR, Wesson DW, Lee CY et al (2012) ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models. Science 335:1503–1506
Cras P, van Harskamp F, Hendriks L, Ceuterick C et al (1998) Presenile Alzheimer dementia characterized by amyloid angiopathy and large amyloid core type senile plaques in the APP 692Ala → Gly mutation. Acta Neuropathol 96:253–260
Crehan H, Holton P, Wray S, Pocock J et al (2011) Complement receptor 1 (CR1) and Alzheimer’s disease. Immunobiology 217:244–250
Crocker PR, Paulson JC, Varki A (2007) Siglecs and their roles in the immune system. Nat Rev Immunol 7:255–266
Cruchaga C, Nowotny P, Kauwe JS, Ridge PG et al (2011) Association and expression analyses with single-nucleotide polymorphisms in TOMM40 in Alzheimer disease. Arch Neurol 68:1013–1019
Davidson YS, Raby S, Foulds PG, Robinson A et al (2011) TDP-43 pathological changes in early onset familial and sporadic Alzheimer’s disease, late onset Alzheimer’s disease and Down’s syndrome: association with age, hippocampal sclerosis and clinical phenotype. Acta Neuropathol 122:703–713
De Jager PL, Shulman JM, Chibnik LB, Keenan BT et al (2012) A genome-wide scan for common variants affecting the rate of age-related cognitive decline. Neurobiol Aging 33(5):1017.e1–e15
Depaepe V, Suarez-Gonzalez N, Dufour A, Passante L et al (2005) Ephrin signalling controls brain size by regulating apoptosis of neural progenitors. Nature 435:1244–1250
Diedrich JF, Minnigan H, Carp RI, Whitaker JN et al (1991) Neuropathological changes in scrapie and Alzheimer’s disease are associated with increased expression of apolipoprotein E and cathepsin D in astrocytes. J Virol 65:4759–4768
Dowling JJ, Gibbs EM, Feldman EL (2008) Membrane traffic and muscle: lessons from human disease. Traffic 9:1035–1043
Duffy SL, Coulthard MG, Spanevello MD, Herath NI et al (2008) Generation and characterization of EphA1 receptor tyrosine kinase reporter knockout mice. Genesis 46:553–561
Dustin ML, Olszowy MW, Holdorf AD, Li J et al (1998) A novel adaptor protein orchestrates receptor patterning and cytoskeletal polarity in T-cell contacts. Cell 94:667–677
Farrer LA, Cupples LA, Haines JL, Hyman B et al (1997) Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease: a meta-analysis. JAMA 278:1349–1356
Genin E, Hannequin D, Wallon D, Sleegers K et al (2011) APOE and Alzheimer disease: a major gene with semi-dominant inheritance. Mol Psychiatry 16:903–907
Glenner GG, Wong CW (1984) Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun 120:885–890
Goate A, Chartier-Harlin M-C, Mullan M, Brown J et al (1991) Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 349:704–706
Gomez-Isla T, Growdon WB, McNamara MJ, Nochlin D et al (1999) The impact of different presenilin 1 and presenilin 2 mutations on amyloid deposition, neurofibrillary changes and neuronal loss in the familial Alzheimer’s disease brain—evidence for other phenotype-modifying factors. Brain 122:1709–1719
Grabowski TJ, Cho HS, Vonsattel JPG, Rebeck GW et al (2001) Novel amyloid precursor protein mutation in an Iowa family with dementia and severe cerebral amyloid angiopathy. Ann Neurology 49:697–705
Grupe A, Abraham R, Li Y, Rowland C et al (2007) Evidence for novel susceptibility genes for late-onset Alzheimer’s disease from a genome-wide association study of putative functional variants. Hum Mol Genet 16:865–873
Guardia-Laguarta C, Pera M, Clarimon J, Molinuevo JL et al (2010) Clinical, neuropathologic, and biochemical profile of the amyloid precursor protein I716F mutation. J Neuropathol Exp Neurol 69:53–59
Hafner C, Meyer S, Langmann T, Schmitz G et al (2005) Ephrin-B2 is differentially expressed in the intestinal epithelium in Crohn’s disease and contributes to accelerated epithelial wound healing in vitro. World J Gastroenterol 11:4024–4031
Halliday G, Brooks W, Arthur H, Creasey H et al (1997) Further evidence for an association between a mutation in the APP gene and Lewy body formation. Neurosci Lett 227:49–52
Harold D, Abraham R, Hollingworth P, Sims R et al (2009) Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet 41:1088–1093
Heilig EA, Xia W, Shen J, Kelleher RJ 3rd (2010) A presenilin-1 mutation identified in familial Alzheimer disease with cotton wool plaques causes a nearly complete loss of gamma-secretase activity. J Biol Chem 285:22350–22359
Hermey G (2009) The Vps10p-domain receptor family. Cell Mol Life Sci 66:2677–2689
Himanen JP, Saha N, Nikolov DB (2007) Cell-cell signaling via Eph receptors and ephrins. Curr Opin Cell Biol 19:534–542
Hoglinger GU, Melhem NM, Dickson DW, Sleiman PM et al (2011) Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nat Genet 43:699–705
Holen HL, Nustad K, Aasheim HC (2010) Activation of EphA receptors on CD4+ CD45RO+ memory cells stimulates migration. J Leukoc Biol 87:1059–1068
Hollingworth P, Harold D, Sims R, Gerrish A et al (2011) Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet 43:429–435
Hollingworth P, Sweet R, Sims R, Harold D et al (2011) Genome-wide association study of Alzheimer’s disease with psychotic symptoms. Mol Psychiatry. doi:10.1038/mp.2011.125
Holmes D (2011) Mind the IGAP. Lancet Neurol 10:502–503
Houlden H, Baker M, McGowan E, Lewis P et al (2000) Variant Alzheimer’s disease with spastic paraparesis and cotton wool plaques is caused by PS-1 mutations that lead to exceptionally high amyloid-beta concentrations. Ann Neurology 48:806–808
Houlden H, Crook R, Dolan RJ, McLaughlin J et al (2001) A novel presenilin mutation (M233V) causing very early onset Alzheimer’s disease with Lewy bodies. Neurosci Lett 313:93–95
Innerarity TL, Hui DY, Bersot TP, Mahley RW (1986) Type III hyperlipoproteinemia: a focus on lipoprotein receptor–apolipoprotein E2 interactions. Adv Exp Med Biol 201:273–288
Ishibashi K, Suzuki M, Sasaki S, Imai M (2001) Identification of a new multigene four-transmembrane family (MS4A) related to CD20, HTm4 and beta subunit of the high-affinity IgE receptor. Gene 264:87–93
Ishii K, Lippa C, Tomiyama T, Miyatake F et al (2001) Distinguishable effects of Presenilin-1 and APP717 mutations on amyloid plaque deposition. Neurobiol Aging 22:367–376
Ishikawa A, Piao YS, Miyashita A, Kuwano R et al (2005) A mutant PSEN1 causes dementia with Lewy bodies and variant Alzheimer’s disease. Ann Neurol 57:429–434
Ivanov AI, Romanovsky AA (2006) Putative dual role of ephrin–Eph receptor interactions in inflammation. IUBMB Life 58:389–394
Iwamoto N, Abe-Dohmae S, Sato R, Yokoyama S (2006) ABCA7 expression is regulated by cellular cholesterol through the SREBP2 pathway and associated with phagocytosis. J Lipid Res 47:1915–1927
Iwatsubo T, Odaka A, Suzuki N, Mizusawa H et al (1994) Visualization of a-beta-42(43) and a-beta-40 in senile plaques with end-specific a-beta monoclonals—evidence that an initially deposited species is a-beta-42(43). Neuron 13:45–53
Jarrett JT, Berger EP, Lansbury PT Jr (1993) The carboxy terminus of the beta amyloid protein is critical for the seeding of amyloid formation: implications for the pathogenesis of Alzheimer’s disease. Biochemistry 32:4693–4697
Jayadev S, Leverenz JB, Steinbart E, Stahl J et al (2010) Alzheimer’s disease phenotypes and genotypes associated with mutations in presenilin 2. Brain 133:1143–1154
Jones L, Harold D, Williams J (2010) Genetic evidence for the involvement of lipid metabolism in Alzheimer’s disease. Biochim Biophys Acta 1801:754–761
Jun G, Naj AC, Beecham GW, Wang LS et al (2010) Meta-analysis confirms CR1, CLU, and PICALM as Alzheimer disease risk loci and reveals interactions with APOE genotypes. Arch Neurol 67:1473–1484
Kamino K, Orr HT, Payami H, Wijsamn EM et al (1992) Linkage and mutational analysis of familial Alzheimer disease kindreds for the APP gene region. Am J Hum Genet 51:998–1014
Kaneko H, Kakita A, Kasuga K, Nozaki H et al (2007) Enhanced accumulation of phosphorylated alpha-synuclein and elevated beta-amyloid 42/40 ratio caused by expression of the presenilin-1 deltaT440 mutant associated with familial Lewy body disease and variant Alzheimer’s disease. J Neurosci 27:13092–13097
Kang J, Lemaire H-G, Unterbeck A, Salbaum JM et al (1987) The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature 325:733–736
Kasuga K, Shimohata T, Nishimura A, Shiga A et al (2009) Identification of independent APP locus duplication in Japanese patients with early-onset Alzheimer disease. J Neurol Neurosurg Psychiat 80:1050–1052
Kauwe JS, Jacquart S, Chakraverty S, Wang J et al (2007) Extreme cerebrospinal fluid amyloid beta levels identify family with late-onset Alzheimer’s disease presenilin 1 mutation. Ann Neurol 61:446–453
Keene CD, Cudaback E, Li X, Montine KS et al (2011) Apolipoprotein E isoforms and regulation of the innate immune response in brain of patients with Alzheimer’s disease. Curr Opin Neurobiol 21:920–928
Kim J, Basak JM, Holtzman DM (2009) The role of apolipoprotein E in Alzheimer’s disease. Neuron 63:287–303
Kim JM, Wu H, Green G, Winkler CA et al (2003) CD2-associated protein haploinsufficiency is linked to glomerular disease susceptibility. Science 300:1298–1300
Kim WS, Weickert CS, Garner B (2008) Role of ATP-binding cassette transporters in brain lipid transport and neurological disease. J Neurochem 104:1145–1166
Kimura R, Yamamoto M, Morihara T, Akatsu H et al (2009) SORL1 is genetically associated with Alzheimer disease in a Japanese population. Neurosci Lett 461:177–180
Kolsch H, Jessen F, Wiltfang J, Lewczuk P et al (2009) Association of SORL1 gene variants with Alzheimer’s disease. Brain Res 1264:1–6
Kumar-Singh S, Cras P, Wang R, Kros JM et al (2002) Dense-core senile plaques in the Flemish variant of Alzheimer’s disease are vasocentric. Am J Pathol 161:507–520
Kwok JBJ, Halliday GM, Brooks WS, Dolios G et al (2003) Presenilin-1 mutation L271V results in altered exon 8 splicing and Alzheimer’s disease with non-cored plaques and no neuritic dystrophy. J Biol Chem 278:6748–6754
Lai KO, Ip NY (2009) Synapse development and plasticity: roles of ephrin/Eph receptor signaling. Curr Opin Neurobiol 19:275–283
Lambert JC, Heath S, Even G, Campion D et al (2009) Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet 41:1094–1099
Lambert JC, Sleegers K, Gonzalez-Perez A, Ingelsson M et al (2010) The CALHM1 P86L polymorphism is a genetic modifier of age at onset in Alzheimer’s disease: a meta-analysis study. J Alzheimers Dis 22:247–255
Lantos PL, Ovenstone IM, Johnson J, Clelland CA et al (1994) Lewy bodies in the brain of two members of a family with the 717 (Val to Ile) mutation of the amyloid precursor protein gene. Neurosci Lett 172:77–79
Laumet G, Chouraki V, Grenier-Boley B, Legry V et al (2010) Systematic analysis of candidate genes for Alzheimer’s disease in a French, genome-wide association study. J Alzheimers Dis 20:1181–1188
Le TV, Crook R, Hardy J, Dickson DW (2001) Cotton wool plaques in non-familial late-onset Alzheimer disease. J Neuropathol Exp Neurol 60:1051–1061
Lee JH, Cheng R, Barral S, Reitz C et al (2011) Identification of novel loci for Alzheimer disease and replication of CLU, PICALM, and BIN1 in Caribbean Hispanic individuals. Arch Neurol 68:320–328
Lee JH, Cheng R, Honig LS, Vonsattel JPG et al (2008) Association between genetic variants in SORL1 and autopsy-confirmed Alzheimer disease. Neurology 70:887–889
Lee JH, Cheng R, Schupf N, Manly J et al (2007) The association between genetic variants in SORL1 and Alzheimer disease in an urban, multiethnic, community-based cohort. Arch Neurol 64:501–506
Lehtonen S, Zhao F, Lehtonen E (2002) CD2-associated protein directly interacts with the actin cytoskeleton. Am J Physiol Renal Physiol 283:F734–F743
Leverenz JB, Fishel MA, Peskind ER, Montine TJ et al (2006) Lewy body pathology in familial Alzheimer disease: evidence for disease- and mutation-specific pathologic phenotype. Arch Neurol 63:370–376
Leverenz JB, Raskind MA (1998) Early amyloid deposition in the medial temporal lobe of young Down syndrome patients: a regional quantitative analysis. Exp Neurol 150:296–304
Levy-Lahad E, Wasco W, Poorkaj P, Romano DM et al (1995) Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science 269:973–977
Li H, Wetten S, Li L, St Jean PL et al (2008) Candidate single-nucleotide polymorphisms from a genomewide association study of Alzheimer disease. Arch Neurol 65:45–53
Li Y, Rowland C, Catanese J, Morris J et al (2008) SORL1 variants and risk of late-onset Alzheimer’s disease. Neurobiol Dis 29:293–296
Liang Y, Tedder TF (2001) Identification of a CD20-, FcepsilonRIbeta-, and HTm4-related gene family: sixteen new MS4A family members expressed in human and mouse. Genomics 72:119–127
Lippa CF, Fujiwara H, Mann DM, Giasson B et al (1998) Lewy bodies contain altered alpha-synuclein in brains of many familial Alzheimer’s disease patients with mutations in presenilin and amyloid precursor protein genes. Am J Pathol 153:1365–1370
Lippa CF, Saunders AM, Smith TW, Swearer JM et al (1996) Familial and sporadic Alzheimer’s disease: neuropathology cannot exclude a final common pathway. Neurology 46:406–412
Lipsky LPE, Abramson SB, Crofford L, Dubois RN et al (1998) The classification of cyclooxygenase inhibitors. J Rheumatol 25:2298–2303
Liu F, AriasVasquez A, Sleegers K, Aulchenko YS et al (2007) A genomewide screen for late-onset Alzheimer disease in a genetically isolated Dutch population. Am J Hum Genet 81:17–31
Lowik MM, Groenen PJ, Levtchenko EN, Monnens LA et al (2009) Molecular genetic analysis of podocyte genes in focal segmental glomerulosclerosis—a review. Eur J Pediatr 168:1291–1304
Mahley RW, Rall SC Jr (1999) Is epsilon4 the ancestral human apoE allele? Neurobiol Aging 20:429–430
Mahley RW, Rall SC Jr (2000) Apolipoprotein E: far more than a lipid transport protein. Annu Rev Genomics Hum Genet 1:507–537
Mahley RW, Weisgraber KH, Huang Y (2009) Apolipoprotein E: structure determines function, from atherosclerosis to Alzheimer’s disease to AIDS. J Lipid Res 50(Suppl):S183–S188
Mann DMA, Iwatsubo T, Cairns NJ, Lantos PL et al (1996) Amyloid beta protein (A beta) deposition in chromosome 14-linked Alzheimer’s disease: predominance of A beta(42(43)). Ann Neurol 40:149–156
Mann DMA, Iwatsubo T, Ihara Y, Cairns NJ et al (1996) Predominant deposition of amyloid-beta(42(43)) in plaques in cases of Alzheimer’s disease and hereditary cerebral hemorrhage associated with mutations in the amyloid precursor protein gene. Am J Pathol 148:1257–1266
Mann DMA, Iwatsubo T, Nochlin D, Sumi SM et al (1997) Amyloid (A beta) deposition in chromosome 1-linked Alzheimer’s disease: the Volga German families. Ann Neurol 41:52–57
Mann DMA, Pickering-Brown SM, Takeuchi A, Iwatsubo T (2001) Amyloid angiopathy and variability in amyloid beta deposition is determined by mutation position in presenilin-1-linked Alzheimer’s disease. Am J Pathol 158:2165–2175
Masters CL, Simms G, Weinman NA, Multhaup G et al (1985) Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci USA 82:4245–4249
McGeer PL, Rogers J (1992) Anti-inflammatory agents as a therapeutic approach to Alzheimer’s disease. Neurology 42:447–449
Meng Y, Lee JH, Cheng R, GeorgeHyslop P et al (2007) Association between SORLI and Alzheimer’s disease in a genome-wide study. NeuroReport 18:1761–1764
Miklossy J, Taddei K, Suva D, Verdile G et al (2003) Two novel presenilin-1 mutations (Y256S and Q222H) are associated with early-onset Alzheimer’s disease. Neurobiol Aging 24:655–662
Minster RL, DeKosky ST, Kamboh MI (2008) No association of SORL1 SNPs with Alzheimer’s disease. Neurosci Lett 440:190–192
Musunuru K, Strong A, Frank-Kamenetsky M, Lee NE et al (2010) From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature 466:714–719
Naj AC, Beecham GW, Martin ER, Gallins PJ et al (2010) Dementia revealed: novel chromosome 6 locus for late-onset Alzheimer disease provides genetic evidence for folate-pathway abnormalities. PLoS Genet 6:e1001130
Naj AC, Jun G, Beecham GW, Wang L-S et al (2011) Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet 43:436–441
Namba Y, Tomonaga M, Kawasaki H, Otomo E et al (1991) Apolipoprotien E immunoreactivity in cerebral amyloid deposits and neurofibrillary tangles in Alzheimer’s disease and kuru plaque amyloid in Creutzfeldt–Jakob disease. Brain Res 541:163–166
Naslund J, Schierhorn A, Hellman U, Lannfelt L et al (1994) Relative abundance of Alzheimer a-beta amyloid peptide variants in Alzheimer-disease and normal aging. Proc Natl Acad Sci USA 91:8378–8382
Natte R, Maat-Schieman MLC, Haan J, Bornebroek M et al (2001) Dementia in hereditary cerebral hemorrhage with amyloidosis-Dutch type is associated with cerebral amyloid angiopathy but is independent of plaques and neurofibrillary tangles. Ann Neurol 50:765–772
Nilsberth C, Westlinddanielsson A, Eckman CB, Condron MM et al (2001) The ‘Arctic’ APP mutation (E693G) causes Alzheimer’s disease by enhanced A beta protofibril formation. Nat Neurosci 4:887–893
Nuutinen T, Suuronen T, Kauppinen A, Salminen A (2009) Clusterin: a forgotten player in Alzheimer’s disease. Brain Res Rev 61:89–104
O’Brien RJ, Wong PC (2011) Amyloid precursor protein processing and Alzheimer’s disease. Annu Rev Neurosci 34:185–204
Pasquale EB (2005) Eph receptor signalling casts a wide net on cell behaviour. Nat Rev Mol Cell Biol 6:462–475
Pastor P, Roe CM, Villegas A, Bedoya G et al (2003) Apolipoprotein E epsilon 4 modifies Alzheimer’s disease onset in an E280A PS1 kindred. Ann Neurol 54:163–169
Pennisi E (2011) The biology of genomes. Disease risk links to gene regulation. Science 332:1031
Pericak-Vance MA, Bebout JL, Gaskell PC, Yamaoka LH et al (1991) Linkage studies in familial Alzheimer disease: evidence for chromosome 19 linkage. Am J Hum Genet 48:1034–1050
Poliakov A, Cotrina M, Wilkinson DG (2004) Diverse roles of eph receptors and ephrins in the regulation of cell migration and tissue assembly. Dev Cell 7:465–480
Prendergast GC, Muller AJ, Ramalingam A, Chang MY (2009) BAR the door: cancer suppression by amphiphysin-like genes. Biochim Biophys Acta 1795:25–36
Rao Y, Haucke V (2011) Membrane shaping by the Bin/amphiphysin/Rvs (BAR) domain protein superfamily. Cell Mol Life Sci 68:3983–3993
Reiman EM, Webster JA, Myers AJ, Hardy J et al (2007) GAB2 alleles modify Alzheimer’s risk in APOE epsilon 4 carriers. Neuron 54:713–720
Reitz C, Cheng R, Rogaeva E, Lee JH et al (2011) Meta-analysis of the association between variants in SORL1 and Alzheimer disease. Arch Neurol 68:99–106
Ren G, Vajjhala P, Lee JS, Winsor B et al (2006) The BAR domain proteins: molding membranes in fission, fusion, and phagy. Microbiol Mol Biol Rev 70:37–120
Revesz T, McLaughlin JL, Rossor MN, Lantos PL (1997) Pathology of familial Alzheimer’s disease with Lewy bodies. J Neural Transm Suppl 51:121–135
Revuz J (2009) Hidradenitis suppurativa. J Eur Acad Dermatol Venereol 23:985–998
Rogaeva E, Meng Y, Lee JH, Gu Y et al (2007) The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat Genet 39:168–177
Roses AD, Lutz MW, Amrine-Madsen H, Saunders AM et al (2010) A TOMM40 variable-length polymorphism predicts the age of late-onset Alzheimer’s disease. Pharmacogenomics J 10:375–384
Rossi G, Giaccone G, Maletta R, Morbin M et al (2004) A family with Alzheimer disease and strokes associated with A713T mutation of the APP gene. Neurology 63:910–912
Rovelet-ecruz A, Hannequin D, Raux G, LeMeur N et al (2006) APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat Genet 38:24–26
Sakamoto A, Sugamoto Y, Tokunaga Y, Yoshimuta T et al (2011) Expression profiling of the ephrin (EFN) and Eph receptor (EPH) family of genes in atherosclerosis-related human cells. J Int Med Res 39:522–527
Scheuner D, Eckman C, Jensen M, Song X et al (1996) Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat Med 2:864–870
Schjeide BM, McQueen MB, Mullin K, DiVito J et al (2009) Assessment of Alzheimer’s disease case–control associations using family-based methods. Neurogenetics 10:19–25
Seshadri S, DeStefano AL, Au R, Massaro JM et al (2007) Genetic correlates of brain aging on MRI and cognitive test measures: a genome-wide association and linkage analysis in the Framingham Study. BMC Med Genet 8(Suppl 1):S15
Seshadri S, Fitzpatrick AL, Ikram MA, DeStefano AL et al (2010) Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA 303:1832–1840
Shen J, Kelleher RJ 3rd (2007) The presenilin hypothesis of Alzheimer’s disease: evidence for a loss-of-function pathogenic mechanism. Proc Natl Acad Sci USA 104:403–409
Shen L, Kim S, Risacher SL, Nho K et al (2010) Whole genome association study of brain-wide imaging phenotypes for identifying quantitative trait loci in MCI and AD: a study of the ADNI cohort. Neuroimage 53:1051–1063
Shepherd CE, Gregory GC, Vickers JC, Brooks WS et al (2004) Positional effects of presenilin-1 mutations on tau phosphorylation in cortical plaques. Neurobiol Dis 15:115–119
Shih NY, Li J, Cotran R, Mundel P et al (2001) CD2AP localizes to the slit diaphragm and binds to nephrin via a novel C-terminal domain. Am J Pathol 159:2303–2308
Sleegers K, Brouwers N, Gijselinck I, Theuns J et al (2006) APP duplication is sufficient to cause early onset Alzheimer’s dementia with cerebral amyloid angiopathy. Brain 129:2977–2983
Sleegers K, Lambert JC, Bertram L, Cruts M et al (2010) The pursuit of susceptibility genes for Alzheimer’s disease: progress and prospects. Trends Genet 26:84–93
Snider BJ, Norton J, Coats MA, Chakraverty S et al (2005) Novel presenilin 1 mutation (S170F) causing Alzheimer disease with Lewy bodies in the third decade of life. Arch Neurol 62:1821–1830
Spoelgen R, vonArnim CAF, Thomas AV, Peltan ID et al (2006) Interaction of the cytosolic domains of sorLA/LR11 with the amyloid precursor protein (APP) and beta-secretase beta-site APP-cleaving enzyme. J Neurosci 26:418–428
St George-Hyslop P, Crapper McLachlan D, Tuda T, Rogaev E et al (1994) Alzheimer’s disease and possible gene interaction. Science 263:537
Steiner H, Revesz T, Neumann M, Romig H et al (2001) A pathogenic presenilin-1 deletion causes abberrant A beta 42 production in the absence of congophilic amyloid plaques. J Biol Chem 276:7233–7239
Strittmatter WJ, Saunders AM, Schmechel D, Pericak Vance M et al (1993) Apolipoprotein-E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci USA 90:1977–1981
Sudo S, Shiozawa M, Cairns NJ, Wada Y (2005) Aberrant accentuation of neurofibrillary degeneration in the hippocampus of Alzheimer’s disease with amyloid precursor protein 717 and presenilin-1 gene mutations. J Neurol Sci 234:55–65
Taira K, Bujo H, Hirayama S, Yamazaki H et al (2001) LR11, a mosaic LDL receptor family member, mediates the uptake of ApoE-rich lipoproteins in vitro. Arterioscler Thromb Vasc Biol 21:1501–1506
Takao M, Ghetti B, Hayakawa I, Ikeda E et al (2002) A novel mutation (G217D) in the Presenilin 1 gene (PSEN1) in a Japanese family: presenile dementia and parkinsonism are associated with cotton wool plaques in the cortex and striatum. Acta Neuropathol 104:155–170
Tamaoka A, Fraser PE, Ishii K, Sahara N et al (1998) Amyloid-β-protein isoforms in brain of subjects with PS1-linked, βAPP-linked and sporadic Alzheimer disease. Mol Brain Res 56:178–185
Tamaoka A, Odaka A, Ishibashi Y, Usami M et al (1994) App717 missense mutation affects the ratio of amyloid-beta protein species (a-beta-1–42/43 and a-beta-1–40) in familial Alzheimers-disease brain. J Biol Chem 269:32721–32724
Tamaoka A, Sawamura N, Odaka A, Suzuki N et al (1995) Amyloid-beta protein-1–42/43 (a-beta-1–42/43) in cerebellar diffuse plaques—enzyme-linked-immunosorbent-assay and immunocytochemical study. Brain Res 679:151–156
Tan EK, Lee J, Chen CP, Teo YY et al (2009) SORL1 haplotypes modulate risk of Alzheimer’s disease in Chinese. Neurobiol Aging 30:1048–1051
Tebar F, Bohlander SK, Sorkin A (1999) Clathrin assembly lymphoid myeloid leukemia (CALM) protein: localization in endocytic-coated pits, interactions with clathrin, and the impact of overexpression on clathrin-mediated traffic. Mol Biol Cell 10:2687–2702
Teslovich TM, Musunuru K, Smith AV, Edmondson AC et al (2010) Biological, clinical and population relevance of 95 loci for blood lipids. Nature 466:707–713
Thaker U, McDonagh AM, Iwatsubo T, Lendon CL et al (2003) Tau load is associated with apolipoprotein E genotype and the amount of amyloid beta protein, A beta(40), in sporadic and familial Alzheimer’s disease. Neuropathol Appl Neurobiol 29:35–44
Tyrrell J, Cosgrave M, McCarron M, McPherson J et al (2001) Dementia in people with Down’s syndrome. Int J Geriatr Psychiatry 16:1168–1174
Ungewickell EJ, Hinrichsen L (2007) Endocytosis: clathrin-mediated membrane budding. Curr Opin Cell Biol 19:417–425
Verkkoniemi A, Kalimo H, Paetau A, Somer M et al (2001) Variant Alzheimer disease with spastic paraparesis: neuropathological phenotype. J Neuropathol Exp Neurol 60:483–492
Visscher PM, Brown MA, McCarthy MI, Yang J (2012) Five years of GWAS discovery. Am J Hum Genet 90:7–24
Vitale C, Romagnani C, Falco M, Ponte M et al (1999) Engagement of p75/AIRM1 or CD33 inhibits the proliferation of normal or leukemic myeloid cells. Proc Natl Acad Sci USA 96:15091–15096
von Gunten S, Bochner BS (2008) Basic and clinical immunology of Siglecs. Ann N Y Acad Sci 1143:61–82
Wakabayashi T, De Strooper B (2008) Presenilins: members of the gamma-secretase quartets, but part-time soloists too. Physiology (Bethesda) 23:194–204
Wang B, Yang W, Wen W, Sun J et al (2010) Gamma-secretase gene mutations in familial acne inversa. Science 330:1065
Wijsman EM, Daw EW, Yu XS, Steinbart EJ et al (2005) APOE and other loci affect age-at-onset in Alzheimer’s disease families with PS2 mutation. Am J Med Genet 132B:14–20
Wijsman EM, Pankratz ND, Choi Y, Rothstein JH et al (2011) Genome-wide association of familial late-onset Alzheimer’s disease replicates BIN1 and CLU and nominates CUGBP2 in interaction with APOE. PLoS Genet 7:e1001308
Wisiewski KE, Wisniewski HM, Wen GY (1985) Occurance of neuropathologic changes and dementia of Alzheimer’s disease in Down syndrome. Ann Neurol 17:278–282
Wollmer MA (2010) Cholesterol-related genes in Alzheimer’s disease. Biochim Biophys Acta 1801:762–773
Yamazaki T, Masuda J, Omori T, Usui R et al (2009) EphA1 interacts with integrin-linked kinase and regulates cell morphology and motility. J Cell Sci 122:243–255
Yeganeh MZ, Mirabzadeh A, Khorshid HRK, Kamali K et al (2010) Novel extreme homozygote haplotypes at the human Caveolin 1 gene upstream purine complex in sporadic Alzheimer’s disease. Am J Med Genet Part B 153B:347–349
Yokota O, Terada S, Ishizu H, Ujike H et al (2003) Variability and heterogeneity in Alzheimer’s disease with cotton wool plaques: a clinicopathological study of four autopsy cases. Acta Neuropathol 106:348–356
Acknowledgments
This work was supported by Grants from the NIH (U01AG032984, RC2AG036528, and AG05136) and the Nancy and Buster Alvord Endowment. We thank Dr. Kathleen Montine and Dr. Diane Durnam for their editorial assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Schellenberg, G.D., Montine, T.J. The genetics and neuropathology of Alzheimer’s disease. Acta Neuropathol 124, 305–323 (2012). https://doi.org/10.1007/s00401-012-0996-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-012-0996-2