
Abstract
Globular glial tauopathies (GGTs) are 4-repeat tauopathies neuropathologically characterized by tau-positive, globular glial inclusions, including both globular oligodendroglial inclusions and globular astrocytic inclusions. No mutations have been found in 25 of the 30 GGT cases reported in the literature who have been screened for mutations in microtubule associated protein tau (MAPT). In this report, six patients with GGT (four with subtype III and two with subtype I) were screened for MAPT mutations. They included 4 men and 2 women with a mean age at death of 73 years (55–83 years) and mean age at symptomatic onset of 66 years (50–77 years). Disease duration ranged from 5 to 14 years. All were homozygous for the MAPT H1 haplotype. Three patients had a positive family history of dementia, and a novel MAPT mutation (c.951G>C, p.K317N) was identified in one of them, a patient with subtype III. Recombinant tau protein bearing the lysine-to-asparagine substitution at amino acid residue 317 was used to assess functional significance of the variant on microtubule assembly and tau filament formation. Recombinant p.K317N tau had reduced ability to promote tubulin polymerization. Recombinant 3R and 4R tau bearing the p.K317N mutation showed decreased 3R tau and increased 4R tau filament assembly. These results strongly suggest that the p.K317N variant is pathogenic. Sequencing of MAPT should be considered in patients with GGT and a family history of dementia or movement disorder. Since several individuals in our series had a positive family history but no MAPT mutation, genetic factors other than MAPT may play a role in disease pathogenesis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Tauopathies are a group of neurodegenerative disorders characterized by pathologic accumulation of hyperphosphorylated and insoluble tau protein within neurons and glia [32]. Tau protein is encoded by a gene (MAPT) on chromosome 17 [37]. Mutations in MAPT cause frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17T) [19]. MAPT has 14 coding exons, and it undergoes alternative splicing of exons 2, 3 and 10 [5]. Alternative splicing of exon 10 results in tau isoforms containing either three or four imperfect and highly conserved 31-amino acid repeats in the microtubule binding domain (3R tau and 4R tau) [5]. In normal adult human brains, 3R and 4R tau are expressed at nearly the same level [21]. In primary tauopathies, the ratio of 3R to 4R tau is altered. Pick’s disease contains predominantly 3R tau [10], while 4R tau disproportionately accumulates in progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), and argyrophilic grain disease (AGD) [32, 45].
Primary tauopathies have variable and distinctive neuronal and glial pathology with phospho-tau immunohistochemistry that permits pathological classification with an imperfect fit to clinical phenotype. Pathologic tau accumulates in neurons as pretangles, neurofibrillary tangles, Pick bodies, neuropil threads, and argyrophilic grains. Tau-positive astrocytic lesions that have disease-specificity include astrocytic plaques of CBD and tufted astrocytes of PSP [27]. Less specific tau-positive astrocytic lesions are detected in AGD and Pick’s disease as well as the aging brain [31]. Oligodendroglial coiled bodies, originally reported in AGD [8], are found in many primary tauopathies [16, 50]. Unusual globular inclusions in oligodendroglia have been described in sporadic neurodegenerative disorders, often presenting with progressive aphasia [36], frontal lobe dementia [7], PSP clinical syndrome [25] or frontotemporal dementia with motor neuron disease [3, 30]. The distinctive nature of globular oligodendroglial inclusions (GOI) has led to recognition of a new 4R tauopathy, characterized by extensive white matter involvement, termed globular glial tauopathy (GGT) [2]. In addition to GOI, globular astrocytic inclusions (GAI) are also found in GGT. In some cases, GOI are accompanied by coiled bodies [2, 3]. Both coiled bodies and GOI are positive with Gallyas silver stain. GAI are gray matter astrocytes with globular or dot-like inclusions in their cell body and proximal processes. Their distinctive appearance usually permits differentiation from astrocytic plaques of CBD and tufted astrocytes of PSP, but occasionally the distinction is difficult [2]. Typical GAI in GGT show no (or only weak) staining with Gallyas silver stain, while astrocytic plaques and tufted astrocytes are strongly argyrophilic [27].
Three different clinicopathological subtypes of GGT have been proposed by an international consensus committee after round-robin review of slides of GGT cases [2]. All subtypes have significant degeneration of the white matter with GOI. Subtype I is clinically characterized by frontotemporal dementia and neuropathologically by frontotemporal distribution of pathology (“FTD type”). Subtype II cases are clinically characterized by frontotemporal dementia with pyramidal and extrapyramidal features and neuropathologically by predominant involvement of motor and dorsolateral premotor cortices (“MND type”) as well as extrapyramidal nuclei. Subtype III cases often present with a combination of FTD and MND, including lower motor neuron pathology in some cases, with pathology in frontotemporal and motor cortices, and variable subcortical or spinal cord pathology (“FTD and MND type”). Whether the subtypes represent distinctive entities or a spectrum of one disease remains to be determined. Of the 30 cases included in the consensus paper, 25 had MAPT sequencing (Supplementary material in [2]) and no mutations were found.
More than 50 different MAPT mutations have been identified to cause tauopathies (see http://www.molgen.ua.ac.be/ADMutations). MAPT mutations have been shown to impair microtubule assembly and axonal transport or to promote pathological tau filament formation [9]. They occur in exons 1, 9, 10, 11, 12, 13 and in intronic sequences flanking exon 10, with clinical presentations including Parkinsonism, dementia, personality changes, language difficulties, gaze palsy, epilepsy, myoclonus and pyramidal signs [40]. Mutations in MAPT are associated with a range of neuropathologic phenotypes, with tau pathology predominantly in neurons (exons 9, 11, 12, 13) or in both neurons and glia (exons 1, 10, introns 9 and 10), including a report with numerous glial inclusions in a patient harboring p.K317M mutation [51].
In this report, we review clinical and pathologic features and results of MAPT sequencing analysis of six pathologically confirmed GGT cases from the Mayo Clinic brain bank. A functionally significant mutation was found in one patient, and pathogenicity was confirmed by in vitro assays of microtubule assembly and tau pathologic filament formation.
Materials and methods
Subjects and samples
Brain samples from six patients with GGT were obtained from the Mayo Clinic brain bank for neurodegenerative disorders between August 2004 and August 2013. All met pathologic criteria for GGT with tau immunohistochemistry and Gallyas silver staining, although there was pathologic heterogeneity [2] (see Online Resource Fig. 1). As a crude estimate the frequency of GGT in a referral brain bank that specializes in neurodegenerative tauopathies [23], the six cases in this study represent <1 % of 850 primary tauopathy cases (mostly PSP and CBD) in this time period.
Genealogical, clinical and laboratory investigations
Genealogical and clinical information were summarized from review of available medical records, supplemented by telephone interviews of relatives of Patient 1. Patient and family interviews, as well as collection of blood samples from relatives, were ethically performed under an approved Mayo Clinic Institutional Review Board protocol. Autopsies were obtained after informed consent of the legal next-of-kin.
Genetic analysis
DNA was extracted from frozen cerebellar tissue with Gentra Puregene kit (Qiagen, Venlo, Netherlands). DNA was extracted from blood samples of two relatives of Patient 1. Polymerase chain reactions (PCR) were performed using primer sets designed to flank intronic sequences of exons 0–5, 7, and 9–13/14 and labeled with unique M13 sequences to facilitate sequencing. Prior to sequencing, PCR products were purified using the AMPure system (Agencourt Biosciences/Beckman Coulter, Brea, CA). Purified PCR products for each exon were sequenced on both strands using M13 primers and Big Dye Cycle Sequencing chemistry (Life Technologies, Carlsbad, CA). Sequence reactions were purified with CleanSEQ (Agencourt Biosciences) and then run on an ABI 3730xl DNA Analyzer (Applied Biosystems, Foster City, CA). Sequence data were analyzed using Sequencher 4.5 software (Gene Codes, Ann Arbor, MI). MAPT H1/H2 haplotype was defined by the single-nucleotide polymorphism rs1052553 in MAPT exon 9.
Tissue sampling and neuropathologic assessment
The brains submitted for neuropathologic evaluation were divided in a mid-sagittal plane with the left half fixed in 10 % formalin and the right half frozen at −80 °C. Formalin-fixed tissue was sampled with standardized dissection methods and embedded in paraffin blocks. Regions sampled included frontal, temporal, parietal, motor, visual and cingulate cortices; amygdala; two levels of hippocampus; nucleus accumbens; lentiform nucleus; thalamus at level of subthalamic nucleus; midbrain; pontomesencephalic junction; rostral pons; medulla; and two sections of cerebellum including vermis and deep nuclei. Sections were evaluated with tau immunohistochemistry as previously described [29]. Alzheimer-type pathology was assessed with thioflavin S fluorescent microscopy as previously described [48]. Hematoxylin and eosin-stained sections were used to evaluate neuronal loss and gliosis. White matter pathology and tract degeneration were assessed with Luxol fast blue for myelin and ionizing binding adapter protein 1 (IBA-1) immunohistochemistry.
Immunohistochemistry was performed using a DAKO Autostainer (Universal Staining System, Carpinteria, CA) using 3,3′-diaminobenzidine as the chromogen. Antibodies used for immunohistochemistry included IBA-1 (mouse IgG1, 1:3000; Wako Chemicals USA, Richmond, VA), phospho-tau (CP13, mouse IgG1, 1:1000; Peter Davies, Feinstein Institute for Medical Research, North Shore/Long Island Jewish Health Care System), 3R and 4R tau (RD3 and RD4, mouse IgG, Millipore, Temecula, CA), and TDP-43 (MC2085, rabbit Ig, 1:5000; Mayo Clinic, Jacksonville, FL).
A small portion of caudate nucleus from the formalin-fixed brain of Patient 1 was processed for post-embedding immunoelectron microscopy (IEM). The sample was post-fixed in 4 % paraformaldehyde in 100 mM phosphate buffer; dehydrated in 30, 50, 70, and 90 % ethanol; and embedded in LR White resin (Polysciences, Warrington, PA). Ultrathin sections collected on Formvar-coated nickel grids were incubated in PHF1 (IgG1, 1:10; from Peter Davies, Feinstein Institute for Medical Research, North Shore/Long Island Jewish Health Care System) overnight at 4 °C followed by species-specific secondary antibodies conjugated with colloidal gold particles (5 or 18 nm). Sections were examined with a Philips 208S electron microscope, and images were captured with a Gatan 831 Orius digital camera (Gatan Pleasanton, CA).
Recombinant tau purification
Recombinant tau isoforms were expressed and purified as previously described [1], including wild-type 3R tau without exon 2 or 3 inserts (3R0N), 4R0N tau, 3R0N and 4R0N with p.K317N or p.P301L. cDNA isoforms were cloned into pET30a and expressed in competent BL21 (DE3) cells. Overnight cultures were used to inoculate bulk media at 1:100. These were grown to an optical density (600 nm) of 0.5 and induced by adding 0.5 mM IPTG for 2.5 h. Cell pellets were collected, washed in PBS and stored at −80 °C. The cells were lysed with three freeze–thaw cycles, and tau proteins were purified by heating lysates for 10 min at 80 °C. Tau proteins were isolated from clarified supernatants with ion exchange chromatography. The fractions containing tau proteins were dialyzed overnight in 10 mM HEPES at pH 7.4 with two buffer changes. The purity of the tau preparations was analyzed by SDS-polyacrylamide gel electrophoresis and Coomassie blue staining. Protein concentrations were determined using the BCA protein assay kit with bovine serum albumin as a standard (Pierce, Rockford, IL).
Microtubule assembly
Microtubule assembly with recombinant tau proteins was performed in 96-well plates in a final volume of 100 µl. Ice-cold tubulin at 3.0 mg/ml (60 µM) (Cytoskeleton Inc., Denver, CO) was added to an equal volume of 0.24 mg/ml (6 µM) recombinant 4R0N tau or 0.30 mg/ml (8 µM) 3R0N tau in assembly buffer (80 mM PIPES, 2 mM MgCl2, 0.5 mM EGTA, 1 mM GTP, pH 6.8) at 37 °C. The extent of tubulin assembly was monitored by turbidity assay per the manufacturer’s recommendation. The absorbance was measured at 340 nm on SpectraMax M5 Multi-Mode Microplate Readers (Molecular Devices, Sunnyvale, CA). Reactions were run in triplicate, and both the rate and extent of microtubule polymerization were assessed.
Tau filament formation
Reactions of poly-glycosaminoglycan-induced tau aggregation were performed as previously described [1]. To assess tau filament formation, 8 µM of recombinant tau and 0.04 mg/ml of low molecular weight heparin or dextran sulfate were incubated at 37 °C (in 10 mM HEPES at pH 7.4, 100 mM NaCl) and analyzed at 2, 20, 48 and 72 h. Measurement of tau filament polymerization was assessed by adsorption of 10 µl of reaction mixture onto a 400-mesh carbon/Formvar grid (EM Sciences, Hatfield, PA) for 30 s and staining with 2 % uranyl acetate for 45 s. Electron micrographs were captured randomly from low magnification fields at 3 predetermined grid coordinates using a Philips 208S electron microscope and a Gatan digital camera.
Measurements and quantification of tau filament length were determined using NIH Image J (Version 1.47n). For each sample, 6–12 images were captured and analyzed, with respect to total number of tau filaments and lengths of individual filaments. Total polymer mass was determined for each image as the product of the two measurements. Properties of tau aggregation were confirmed independently with thioflavin S fluorescence, which provides a measure of the cross β-sheet secondary structure. Polymerized tau filaments were pelleted by ultracentrifugation at 150,000×g for 30 min to quantitate the amount of soluble and aggregated tau filaments in each fraction on SDS-PAGE.
Tau biochemical characterization
Homogenates were prepared from 150 mg of frontal cortex of Patient 1, as well as sporadic GGT, and two Alzheimer disease (AD), and PSP cases according to published procedures [42]. Briefly, samples were homogenized in 6 volumes of TBS lysis buffer (50 mM Tris–HCl, 150 mM NaCl, 1 mM PMSF, protease and phosphatase inhibitors, pH 7.6) and centrifuged at 150,000×g at 4 °C for 15 min. The supernatants (soluble fractions) were saved, and the pellets were then homogenized with 3 volumes of high-salt buffer (10 mM Tris–HCl, 800 mM NaCl, 10 % sucrose, 1 mM EGTA, 1 mM PMSF, pH 7.5). After centrifugation at 150,000×g at 4 °C for 15 min, the resulting supernatants were saved, and the previous step was repeated. The two supernatants were combined, brought to 1 % sarkosyl, and incubated for 1 h at 37 °C. The samples were then centrifuged at 150,000×g at 4 °C for 45 min, after which the supernatants (sarkosyl-soluble fractions) were collected, leaving the sarkosyl-insoluble pellets. These pellets (which contain insoluble tau protein) were re-suspended in 1:4 volumes of TBS, pH 7.6, based upon their original wet weights and used for both electron microscopy and western blotting.
Sarkosyl-insoluble protein fractions extracted from frontal cortex of all cases and human recombinant tau isoform ladder (rPeptide, Bogart, GA) were reduced/denatured and subjected to PAGE on 10 % Tris–glycine gels (Invitrogen Life Technologies, Grand Island, NY). Separated proteins were transferred to a PVDF membrane (EMD Millipore, Billerica, MA) and blocked in 5 % non-fat milk/TBS-T. Immunoblotting was performed as previously described [29] using anti-tau (human-specific, rabbit anti-E1 tau antibody, 1:20,000) and secondary anti-rabbit IgG (Jackson ImmunoResearch Labs, West Grove, PA, 1:5000). Grids for electron microscopy of extracted samples were prepared as described above.
Results
Sequencing and association studies of MAPT
Sequence analysis of the entire coding region of the MAPT gene in six neuropathologically confirmed GGT cases identified one novel coding mutation in exon 11, c.951G>C predicted to lead to p.K317N substitution (Online Resource Fig. 2) in Patient 1 and her two asymptomatic children. All six patients with GGT, including Patient 1, were homozygous for the MAPT H1 haplotype. Genetic, clinical and genealogical data for all cases are summarized in Table 1.
Patient 1 clinical features and family history
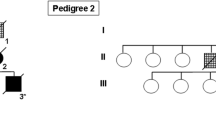
Patient 1 was a 69-year-old woman with a 5-year disease course who presented initially with difficulty expressing herself and producing the correct sounds of words, consistent with primary progressive apraxia of speech [24]. Later, she developed symptoms suggestive of PSP with vertical gaze palsy, axial rigidity, backward falls, and apraxia of eyelid opening. Subsequently, she developed limb apraxia and signs of lower motor neuron disease, including muscle fasciculations. There was no response to 1000 mg of carbidopa/levodopa therapy. The clinical presentation of Patient 1 was consistent with GGT subtype III, including features of FTD, extrapyramidal and motor neuron disease signs. Her five children were asymptomatic. Her mother and maternal aunt were clinically diagnosed with Alzheimer’s dementia (pedigree structure is presented in Fig. 1). Both had disease durations <0 years, and they died at the ages of 63 and 70 years, respectively. In neither case was an autopsy performed to confirm the diagnosis.

Pedigree of Family 1. Round symbols indicate women; squares indicate men; diamonds indicate undisclosed sex; numbers inside symbols indicate number of children; diagonal lines indicate that the individual is deceased. The arrow indicates the proband. Black symbols indicate individuals with dementia, or/and extrapyramidal and/or behavioral disorders. Asterisks indicate family members who underwent genetic testing and are p.K317N mutation carriers
Patient 1 neuropathology
The calculated brain weight was 1220 g. It had lobar atrophy, most marked in inferior frontal gyrus, but also affecting premotor and motor cortices (Fig. 2). The parietal lobe was affected to a lesser degree. Histologically, there was marked neuronal loss and gliosis in affected cortices. The subjacent white matter, especially beneath the motor cortex, was pale, vacuolated, and gliotic with depletion of myelinated fibers.

Macroscopic findings in Patient 1. circumscribed cortical atrophy is most marked in the frontal lobe, especially the inferior frontal gyrus and the precentral gyrus, as well as mild atrophy of frontal pole and superior parietal lobule. There is loss of neuromelanin pigmentation in the substantia nigra (arrow), but visible pigmentation in the locus ceruleus (arrowhead). The cerebellar peduncles showed no unusual features
Tau immunohistochemistry showed many neuronal and glial lesions (Fig. 3a), including GAI, GOI and inclusions that resembled coiled bodies. The affected cortices had tau-positive neuronal inclusions that resembled pretangles and pleomorphic NFTs. Astrocytic lesions had dense granular or globular inclusions in proximal cell processes that were immunoreactive for phospho-tau (Fig. 3b) and 4R tau (Fig. 3c). They were positive with Gallyas silver stain (Fig. 3d, Online Resource Fig. 1). In the white matter were many GOI and coiled bodies that had immunoreactivity for phospho-tau (Fig. 3e) and 4R tau (Fig. 3f), and weak staining with Gallyas silver stain (Fig. 3g, Online Resource Fig. 1). All GAI and GOI were negative for 3R tau (not shown). Distribution of tau pathology in Patient 1 was consistent with GGT subtype III.

Tau pathology in Patient 1. The motor cortex has numerous GAI (a) that show immunoreactivity with phospho-tau (CP13) (b) and 4R tau (c) with variable staining with Gallyas silver stain (d). In the white matter are GOI and coiled bodies, which have immunoreactivity with phospho-tau (CP13) (e) and 4R tau (f) and variable staining with Gallyas silver stain (g). Immunoelectron microscopy of a GOI with phospho-tau (PHF-1; arrows point to gold particles) has circumscribed, but not membrane-bound, randomly orientated straight filaments (open arrow heads), about 18 nm diameter, that displace cytoplasmic organelles (h, i). Bar in a = 50 μm; bar in g = 20 μm (applies for b–g); bars in h = 0.5 μm; bar in j = 50 nm
Immunoelectron microscopy of GOI with phospho-tau showed circumscribed, but non-membrane-bound, randomly orientated straight filaments of about 18 nm diameter that displaced cytoplasmic organelles (Fig. 3h, i). Alzheimer-type paired helical filaments and long periodicity paired helical filaments were not detected.
Alzheimer-type neurofibrillary pathology assessed with thioflavin S fluorescent microscopy was limited to NFTs in anteromedial temporal lobe structures, consistent with Braak NFT stage III. Neither senile plaques nor Lewy bodies were identified with thioflavin S fluorescent microscopy or α-synuclein immunohistochemistry, respectively. Macroscopic examination showed decreased pigmentation in the substantia nigra (Fig. 2) associated with mild-to-moderate neuronal loss and tau-positive neuronal inclusions on microscopic study. There was mild pathology in the corticospinal tract with Luxol fast blue and IBA-1 immunohistochemistry, but there were many tau-positive GOI and threads in the corticospinal tract and the medullary pyramid. The hypoglossal nucleus had no neuronal loss or neuronal inclusions. The cerebellum had pretangles in the dentate nucleus, but no grumose degeneration, and there was no degeneration in the superior cerebellar peduncle.
Summary of clinicopathologic features of other GGT cases
The five other GGT patients included 4 men and 1 woman. Their average age at death was 73.4 years (55–83); average disease duration 7.6 years (5–14); and average age at symptomatic disease onset was 63 years (50–77). Two patients had a positive history of dementia in first- or second-degree relatives or both. None of the patients responded to treatments with anti-dementia, antioxidants or dopaminergic drugs. Parkinsonian signs were noted in four of the five patients. Clinical, genealogical and genetic data are summarized in Table 1.
The average calculated whole brain weight was 1156 g (1000–1260 g). Cortical atrophy affected the frontal lobe in cases 1, 3, 5 and 6; with temporal lobe atrophy notable in cases 2 and 4. There was a range of Alzheimer-type neurofibrillary pathology (Braak stage I–V). The patient with Braak Stage V had cortical tangles with thioflavin S fluorescent microscopy, but no amyloid deposits in blood vessels or brain parenchyma. In Patients 2 and 4, there was severe neuronal loss and gliosis of the subthalamic nucleus, and in Patient 3 midbrain atrophy was noted. The substantia nigra had varying degrees of neuronal loss in all cases, but none had Lewy bodies. Patients 2 and 4 had marked neuronal loss and gliosis in hippocampus, consistent with hippocampal sclerosis, and both had sparse TDP-43-positive neuronal inclusions in the anteromedial temporal lobe, including some inclusions in white matter. Distribution of TDP-43 pathology in these two patients corresponded with stage II of the TDP-43 staging in AD proposed by Josephs et al. [26]. There was no TDP-43 pathology in frontal or precentral gyri or in the basal ganglia. None of the other GGT cases had TDP-43 immunoreactive neuronal or glial inclusions. Atrophy of the basal ganglia and pyramidal tract degeneration were not significant in any of the cases. Patient 2 had grains, pretangles and ballooned neurons in the medial temporal lobe, especially the amygdala, consistent with AGD. Neuropathologic data are summarized in Table 2.
Neuronal lesions, GOI, and GAI were strongly positive for phospho-tau and 4R tau, but completely negative for 3R tau. GAI were completely negative on Gallyas in Patient 2, Patient 3 and Patient 4; in the remaining patients, GAI had weak or positive staining. GOI were more consistently detected with Gallyas silver stain than were GAI, being darkly positive in Patients 2 and 4, who had GGT subtype I with predominant GOI in white matter of temporal cortex (Online Resource Fig. 1). Patients 1, 3, 5 and 6 had GGT subtype III with GOI and GAI distribution in the frontotemporal and motor cortex. Anatomic distribution and severity of tau pathology as well as staining profile of lesions are summarized in Table 3.
Tau biochemistry and electron microscopy
Biochemical analysis was performed on sarkosyl-insoluble fractions extracted from frontal cortices of Patient 1, as well as two of the sporadic GGT patients, two patients with AD and two patients with PSP (Fig. 4a). Immunoblotting with the E1 polyclonal antibody, which is specific to human tau epitope in exon 1, demonstrated bands at ~64 and ~68 kDa, which is characteristic of 4R tauopathies such as PSP [10]. Lower molecular weight bands, consistent with tau cleavage fragments [6], were detected in Patient 1 and the two sporadic GGT patients, as well as in PSP. This contrasts with AD, which showed strong bands at 60, as well as 64 and ~68 kDa. Reprobing of the blots with the 3R tau specific antibody demonstrated that the p.K317N MAPT carrier had no detectable 3R tau at the 64 and 68 kDa position (data not shown). Together the results indicate that Patient 1 and sporadic GGT and PSP patients preferentially deposit 4R tau, confirming the results with immunohistochemistry using 3R and 4R specific monoclonal antibodies presented earlier (see Table 3). Electron microscopy of the purified samples showed sarkosyl-insoluble tau filaments from Patient 1 that were indistinguishable from those in sporadic GGT and composed of straight filaments that were clearly different from paired helical filaments isolated from an AD brain (Fig. 4b).

Biochemical and ultrastructural analysis of sarkosyl-insoluble tau in GGT. Comparison of insoluble tau aggregates from AD, GGT and PSP cases indicates that Patient 1 (lane 4) has similar, but not identical, tau isoform composition and degradation patterns as in sporadic GGT and PSP, but clear differences from AD. The GGT and PSP samples have bands are 64 and 68 kDa with little of the 60 kDa band observed in AD. They also have lower molecular weight fragments seen best on the dark exposure (a). Electron microscopy of insoluble straight tau filaments (b) from Patient 1 are structurally similar to the straight filaments from the sporadic GGT cases and structurally distinct from paired helical filaments prepared the same way from Alzheimer brain. (Scale bar 50 nm)
Effects of recombinant tau with p.K317N mutation on microtubule assembly
Many pathogenic MAPT missense mutations lead to decreased ability of tau protein to promote tubulin polymerization into microtubules. To test this with the new MAPT mutation, recombinant p.K317N tau on either a 3R0N or 4R0N isoform sequences was purified (Fig. 5a) and mixed on ice with purified tubulin and then transferred to a warm cuvette in the presence of GTP to assess tubulin polymerization. Wild-type 3R0N, 4R0N, and 4R0N with p.P301L mutation were used as controls. The rate and final extent of tubulin assembly were both significantly decreased with 3R0N with p.K317N mutation (Fig. 5b). These effects were slightly more robust with 4R0N p.K317N than 3R0N p.K317N, with more substantial decreases in both the rate and extent of tubulin assembly compared to 3R0N (Fig. 5c). These results indicate that the p.K317N MAPT mutation is likely pathogenic, as it has a similar inhibitory effect on microtubule assembly as the most common causative FTDP-17 mutation, p.P301L.

Effects of p.K317N on microtubule assembly. a Coomassie stained recombinant wild-type 3R0N (3R0N-WT), 3R0N p.K317N (3R-K317N), wild-type 4R0N (4R0N-WT), 4R0N p.K317N (4R-K317N), and 4R0N p.P301L (4R-P301L) gels show purity of preparations used for in vitro studies. b Assembly with 3R0N p.K317N shows 20 % decrease in rate and 6 % decrease in final extent compared to 3R0N (*p < 0.01). c Assembly with 4R0N p.K317N shows 24 % decrease in rate and 8 % decrease in final extent compared to 4R0N. The effects of the p.K317N mutation were almost as large as the 29 % decrease in rate and 9 % decrease in extent as p.P301L tau. (*p < 0.01; error bars at time points = standard deviations)
Effects of recombinant 3R0N tau with p.K317N mutation on filament assembly
In addition to a loss of normal function, many tau mutants increase the aggregation properties of tau. This can be assessed with filament pelleting by ultracentrifugation or by fluorescence assays using thioflavin S. These assays are complementary in that not all aggregated tau is polymerized into filaments and similarly not all misfolded tau is insoluble or filamentous. As shown in Fig. 6a, the amount of aggregated 3R0N began to accumulate within 2 h of incubation with dextran sulfate, and by day 1, the majority of 3R0N had aggregated. At both time points, the amount of pelletable 3R0N p.K317N was significantly reduced, representing a 43 % decrease at 2 h and a 31 % decrease at 20 h. By 48 h essentially all the tau protein was aggregated in both samples (data not shown). Similar results were observed with thioflavin S binding as a marker for misfolded tau. As shown in Fig. 6b, misfolded 3R0N was detected at 2 h, and the signal was increased as more of the tau became aggregated. Again, the 3R0N p.K317N mutant had reduced tau misfolding, with a 17 % decrease at 2 h and a 37 % decrease at 20 h compared to 3R wild-type tau.

Effects of p.K317N on 3R0N tau aggregation. Recombinant 3R0N p.K317N has reduced dextran sulfate-induced aggregation compared with 3R0N at both 2 and 20 h (a). Thioflavin S fluorescence shows decreased misfolding of 3R0N p.K317N compared with 3R0N at both 2 and 20 h (b). The number of filaments observed per field was no different at 2 h, but at 20 h there were fewer filaments with 3R0N p.K317N compared with 3R0N (c). The total filament length (nm per field) was decreased for 3R0N p.K317N compared to 3R0N at both 2 and 20 h (d). (*p < 0.01; bars with black fill 3R0N; bars with gray fill 3R0N p.K317N; error bars show standard deviation)
When applicable, quantitative electron microscopy provides a means to directly assess the polymerization properties of tau, and it can provide insights into the relative nucleation rates of tau proteins. Fortunately, both 3R0N and 3R0N with p.K317N mutation had filaments that were evenly dispersed with little clumping or bundling. As shown in Fig. 6c, the number of filaments per field was unchanged at 2 h, but at 20 h the number of filaments was significantly decreased by 42 % for 3R0N p.K317N compared to 3R0N. Interestingly, as shown in Fig. 6d, at 2 h the total filament length per field was significantly decreased by 29 % for 3R0N p.K317N since the mutant filaments were shorter, and at 20 h the total filament length per field was significantly decreased by 36 % for 3R0N p.K317N, though the average filament length was statistically the same.
Effects of recombinant 4R0N tau with p.K317N mutation on filament assembly
Assessment of 4R0N tau filament assembly at 2 and 20 h demonstrated 4R0N aggregated and misfolded much more rapidly than 3R0N (Online Resource Fig. 3). The 4R0N p.K317N was found to be significantly more insoluble at 2 h than 4R0N WT, but at 20 h the samples were essentially all polymerized and indistinguishable (Online Resource Fig. 3a). Similar results were obtained using thioflavin S binding to assess misfolding, though at 2 h there was a small and statistically significant reduction in the fluorescence (Online Resource Fig. 3b). As reactions with 4R0N were faster than 3R0N, additional time points were assessed at 1 h for 4R0N, 4R0N with p.K317N or p.P301L mutations. As shown in Fig. 7a, detectable and significant increases were still observed for 4R0N p.K317N aggregation, and these were similar in magnitude to those observed for 4R0N p.P301L. Likewise, as shown in Fig. 7b, detectable and significant decreases were still observed for 4R0N p.K317N misfolding by thioflavin S fluorescence, but 4R0N p.P301L demonstrated an increase in misfolding compared to 4R0N. Together these data suggest the 4R0N p.K317N might be increasing the filament assembly of 4R tau since the aggregation is increased.

Effects of 4R0N p.K317N (KN), 4R0N p.P301L (PL) and 4R0N (WT) on aggregation. Recombinant 4R0N p.K317N and 4R0N p.P301L have increased dextran sulfate-induced aggregation compared with 4R0N (a). Thioflavin S fluorescence shows decreased misfolding of 4R0N p.K317N compared to both 4R0N and 4R0N p.P301L at 1 h (b). The number of filaments observed per field is increased for both 4R0N p.K317N and 4R0N p.P301L compared with 4R0N (c). Increased total filament length (nm per field) for both 4R0N p.K317N and 4R0N p.P301L compared to 4R0N (d). (*p < 0.01, # p < 0.05; error bars show standard deviation)
Analysis of the electron micrographs was used to clarify the differences observed between the aggregation and misfolding data, which indicated WT tau misfolded more rapidly, but the newly formed assembly-competent WT monomer was not as aggregation prone as the misfolded K317N tau. As shown in representative electron micrographs in Online Resource Fig. 4a, b, at 1 h there was a pronounced increase in length of the filaments and total filament length per field for both 4R0N p.K317N and 4R0N p.P301L (not shown) tau compared to 4R0N WT. As quantitated in Fig. 7c at 1 h, there were significantly more filaments for the 4R0N p.K317N and 4R0N p.P301L mutants compared to 4R0N WT, and the mutant filaments were significantly longer (WT = 179 ± 25 nm, p.K317N = 265 ± 34 nm, p.P301L = 215 ± 16 nm, p < 0.01). As shown in Fig. 7d, this led to a substantial increase in the amount of tau that assembled into filaments as represented by the total filament length per field for both the 4R0N p.K317N and 4R0N p.P301L tau compared to 4R0N. Together these results indicate that the p.K317N mutation accelerates tau filament assembly in 4R tau isoforms, while decreasing it in 3R isoforms.
Discussion
The six patients in this study met neuropathologic criteria for GGT [2]. While GGT is usually considered a sporadic condition, three patients had a positive family history of dementia in first- or second-degree relatives or both, including Patient 1 in whom we identified a MAPT mutation. This patient carried the novel substitution p.K317N (c.951G>C in exon 11). Functional analysis using recombinant wild-type tau and mutant p.K317N tau on both 3R0N and 4R0N isoforms demonstrated that the p.K317N reduces normal tau function, and that the effect of this mutation on microtubule polymerization was nearly as great as that seen in the most common pathogenic MAPT mutation, p.P301L. Furthermore, the p.K317N mutation had a strong isoform effect on assembly properties of recombinant tau. When expressed in the context of 3R0N, the p.K317N mutation decreased tau aggregation, misfolding, and filament assembly compared to 3R0N, and this is understandable as GGT is a 4R tauopathy. In contrast, when expressed in the context of 4R0N, the p.K317N mutation increased tau aggregation and filament assembly. The effects of p.K317N were nearly as large in magnitude as that observed for the pathogenic p.P301L. The results with the thioflavin S assay highlighted different capabilities of tau filaments and soluble species to bind thioflavin S. The lack of thioflavin S staining of neuronal and glial lesions in GGT, as well as other 4R tauopathies, is well known [47]. Increased aggregation and filamentous 4R K317N tau observed with thioflavin S fluorescence at 1 h indicated that although the wild-type and mutant 4R tau misfolded at similar rates, the K317N 4R monomer was more assembly-competent at 1 h than wild-type tau. Although not observed with the 3R tau, this suggests that the mutation may affect the structure necessary for 4R tau filament assembly more than that required for thioflavin S binding. The limitation of thioflavin S binding to differentiate misfolded soluble and aggregated tau in stoichiometrically equivalent amounts has been reported previously [33, 43].
The effects of tau mutations on microtubule polymerization are typically modest in size compared to their ability to increase tau filament assembly [12, 28]. The p.K317N tau mutation is directly upstream of a KXIGS motif, which is thought to regulate microtubule polymerization through phosphorylation by MARK kinase [34]. The lysine-to-asparagine substitution is predicted to result in loss of positive charge in the microtubule binding domain, which is thought to interact electrostatically with the anionic carboxyl terminus of tubulin. This may explain similar effects on microtubule polymerization when p.K317N is expressed in either a 3R or 4R context.
The p.K317N mutation might be predicted to affect tau misfolding required for self-assembly into tau filaments. The in vitro studies suggest a more prominent gain of abnormal function for 4R tau compared to 3R tau filament assembly. This effect may also be charge-dependent since tau is induced to assemble into filaments by anionic inducers or phosphorylation, which also lowers the isoelectric point of tau. Since wild-type 4R tau assembles into filaments more rapidly than 3R tau, it is not surprising that the mutation would exert a larger gain of function on 4R tau.
The effect of the mutation on abolishing one of the predicted tau acetylation sites is unknown [11], but lysine-317 lies in the middle of the microtubule binding domain and is <10 amino acids from the most prominent amyloidogenic VQIVYK sequence in tau, and the mutation changes both charge and acetylation sites [11]. Taken together, the in vitro data strongly suggest that the p.K317N mutation is pathogenic. Definitive proof that p.K317N is a causative mutation would come from evidence that the genetic variant segregates with disease in Patient 1’s family. Two of her children carry the mutation, but are both currently asymptomatic.
Comparison of Patient 1 to FTDP-17T
To our knowledge, this is the first explicit mention of GGT pathology in FTDP-17T. Patient 1 shared clinical, genealogical and neuropathologic features with sporadic GGT and is tentatively referred to as “GGT caused by MAPT mutation” rather than FTDP-17T, but we admit that the distinction is largely semantic. Most descriptions of neuropathology of FTDP-17T were made before nosologic separation of this disorder from other 4R tauopathies [2]. Indeed, some of the reported pathologic and biochemical features of FTDP-17T are not easily distinguished from GGT [19]. The current criteria of GGT emphasize differential staining of glial lesions with Gallyas silver stains—with weak or no staining of GAI, which contrasts with the strong staining of astrocytic lesions in other 4R tauopathies, such as PSP and CBD [2]. Most oligodendroglial lesions in GGT are dense globular inclusions that are strongly positive with Gallyas, but in a subset of cases, particularly subtype II, oligodendroglial lesions may be more similar to coiled bodies seen in PSP [2, 22]. Results of Gallyas staining are frequently missing in published reports of FTDP-17T.
A different missense mutation at the same codon (c.950A>T; p.K317M) has been reported in 13 patients with FTDP-17T from a founder in the Basque region of Spain [51]. The clinical presentation of patients with p.K317M mutation included Parkinsonism, upper motor neuron signs, and dysarthria. Behavioral and personality changes were not prominent features. Lower motor neuron signs were detected in half of them. The mean age of symptomatic onset was 48 years, and the mean disease duration was 6 years. Eight patients came to autopsy, and neuropathologic findings revealed degeneration of the substantia nigra without Lewy bodies as the most consistent finding, followed by degeneration of corticospinal tracts, frontotemporal cortices, and motor neurons in anterior horns of spinal cord. Macroscopically, four brains had no significant atrophy, and four had atrophy in frontal lobe and less frequently in temporal lobe. Immunocytochemistry showed widespread tau pathology in neurons and glia. Oligodendroglial and astrocytic inclusions were abundant. Some of the illustrated glial lesions resembled GOI and GAI (see Figure 4 in [51]). Gallyas silver stains were not described, and the final diagnostic classification, therefore, remains uncertain based upon the criteria for GGT proposed by Ahmed et al. [2]. On the other hand, biochemical analysis of tau revealed two bands of ~64 and ~68 kDa consistent with a 4R tauopathy and electron microscopy of the purified sarkosyl-insoluble tau showed straight filaments similar to Patient 1. The similarities of our Patient 1 to in this Basque family with p.K317M mutation suggest that this may be the first report of GGT due to MAPT mutation.
Interestingly, clinicopathologic presentations of two other mutations in exon 11 (p.L315R and p.S320F) were different from Patient 1. Both mutations had Pick body-like neuronal inclusions, which is not characteristic of GGT. While they had extensive tau-positive astrocytic pathology, numerous oligodendroglial coiled bodies and significant white matter pathology were not reported [41, 49]. Unfortunately, glial lesions were not characterized with Gallyas silver stain. The biochemical profiles of both p.L315R and p.S320F were similar to Pick’s disease, with immunoblots showing prominent bands at ~60 and ~64 kDa rather than ~64 and ~68 kDa [32].
Although most missense mutations located outside exon 10 of MAPT are thought to lead to neuronal more than glial tau pathology [40], the three reported mutations in exon 11—p.K317M and p.L315R, as well as our new mutation, p.K317N—all had neuronal and glial tau pathology. In our case, oligodendroglia and astrocytic pathology were striking, leading to the diagnosis of GGT.
Comparison of Patient 1 to other GGT patients in the Mayo series and in the literature
Patient 1 was clinically and demographically very similar to five sporadic GGT in our series and to GGT patients reported in the literature. Previously reported individuals with GGT showed no sex differences (four women and three men) and an age of death that ranged from 63 to 81 years (mean 76 years) [30]. The most frequent clinical feature in the seven previously reported cases [30] was behavioral changes (n = 6), followed by speech disorder (n = 4) and upper motor neuron signs (n = 4). While vertical gaze palsy, corticobasal syndrome, and parkinsonism have not been previously emphasized in reports of GGT [2, 3], they were common in our series, possibly reflecting a referral bias, as our brain bank is a major referral site in North America for PSP and CBD [23]. The MAPT haplotype has been studied previously in GGT [3, 30], and all individuals have been homozygous for the H1 haplotype, as were all of our GGT cases.
Patient 1 had no significant pathologic differences from the five sporadic GGT cases in our series or from other GGT cases reported in the literature. The brain weights of patients with GGT have ranged from 960 to 1300 g (mean 1112 g) [2], compared to 1000–1260 g (median 1156 g) in our series. Macroscopically, all cases have had frontotemporal lobar atrophy. The defining lesions of GGT are GOI and GAI, which are variably argyrophilic and positive for phospho-tau. Although GOI have been reported to stain weakly for 3R [2, 16], none of our GGT cases had 3R tau immunoreactivity, but all were intensely 4R positive. The staining profile of glial lesions with Gallyas silver stain has been suggested to be one of the most important histopathologic features in differentiating GGT from other 4R tauopathies [2]. On the other hand, there are several different protocols for Gallyas silver stains, and experience from multicenter reliability studies clearly shows that silver staining methods are not ideal for inter-laboratory consensus diagnoses [4, 13, 35]. A more specific biomarker is needed to assist in diagnosis of GGT.
Four patients from this study, including Patient 1, had neurodegeneration in frontotemporal cortices, including motor cortex, with clinical presentations of FTD with extrapyramidal signs consistent with GGT subtype III. Two of the four had evidence of motor neuron disease. In contrast, Patients 2 and 4 had prominent neurodegeneration in the temporal lobe with severe white matter degeneration, as well as clinical presentations fitting with FTD, although Patient 2 was reported to show some rigidity in the final stage of disease, but no features of motor neuron disease. These clinicopathologic findings fit with GGT subtype I.
The four previously reported patients (two women and two men) with GGT subtype III [17, 38] all had extrapyramidal features, as well as symptoms of upper and lower motor neuron disease. Their disease duration ranged from 4 to 6 years, with age at death ranging from 44 to 76 years. Two patients had no significant cognitive impairment [17]. Brain weights ranged from 1090 to 1310 g and all had cortical atrophy in a predominantly frontotemporal distribution. Importantly, neuronal loss and gliosis was noted in premotor and motor cortices as well as anterior horn cells in all four cases. The glial and neuronal lesions were positive for 4R tau. MAPT haplotype, reported in one patient, was H1 homozygous [38].
Thirteen patients have been reported with features of GGT subtype I, including six men and seven women [3, 7, 15, 18, 20, 30, 39]. The age of death ranged from 63 to 85 years, and disease duration from over 2 to 12 years. All presented with clinical features of FTD, with no significant Parkinsonism or features of motor neuron disease. Reported MAPT haplotypes included six H1 homozygous and one H1 H2 heterozygous. All had prominent neurodegeneration in temporal and frontal lobes with brain weights ranging from 960 to 1305 g. When reported, GOI were 4R positive and were prominent in medial temporal lobe, as well as frontal white matter. When noted, GOI were positive with Gallyas stain [30].
Biochemical studies of Patient 1 as well as other GGT in the Mayo Clinic series and those reported in the literature are similar to other 4R tauopathies, with species at 64 and 68 kDa on western blots of sarkosyl-insoluble brain homogenates. Low molecular weight cleavage fragments, which have been useful in differentiating PSP and CBD [6], are also present in GGT, but they have not shown a distinctive signature [3], with GGT cases showing variable (sometimes PSP-like) amino-terminal cleavage fragments (see Fig. 4a).
Comparison of Mayo Clinic GGT series to 4R tauopathies
Our six GGT cases had overlapping neuropathological features with other 4R tauopathies. Marked neuronal loss and gliosis in the subthalamic nucleus in two cases and midbrain atrophy with reduced pigmentation of the substantia nigra are frequent in PSP. A major distinction is the morphology and staining profile of glial lesions in GGT compared to PSP. Astrocytic lesions in our series were negative, or at most weakly positive, on Gallyas silver stain, while tufted astrocytes of PSP are strongly positive with Gallyas stains [27].
Corticospinal tract degeneration is reported in some cases of GGT [3], and it was detected in about half of our cases. Patient 1 had mild corticospinal tract degeneration as assessed with myelin and microglial stains, as well as GOI and threads in the corticospinal tract. Marked corticospinal tract degeneration is also a feature of “atypical PSP with corticospinal tract degeneration (PSP–CST)” [25]. PSP–CST can have clinical presentations similar to the GGT, especially GGT subtype II, including atypical Parkinsonism, vertical gaze palsy, and corticobasal syndrome [25].
Patient 6 had a clinical presentation of corticobasal syndrome; however, astrocytic plaques, the most specific histopathologic lesion in the neuropathologic diagnosis of CBD [14], were not present. Patient 2 had characteristic features of AGD, an age-associated 4R tauopathy preferentially affecting the medial temporal lobe [46]. It is increasingly recognized that AGD may coexist with other degenerative disorders, most often 4R tauopathies, including CBD and PSP [44]. It remains to be determined the frequency of argyrophilic grains in GGT.
Concluding comments
The major finding of this study is that pathologically confirmed GGT may be caused by MAPT mutations. Whether one lumps such cases as FTDP-17T or splits them into a distinct category is purely semantic. Nevertheless, sequencing of MAPT should be considered in patients with GGT if there is a positive family history of dementia, motor neuron disease, or parkinsonism. Given that some GGT patients have a positive family history of neurodegenerative disease but lack MAPT mutations, additional genetic studies are warranted. Other genetic factors may play a role in disease pathogenesis. Finally, although it is difficult to make firm statements based upon small series, GGT subtype III was more frequent than subtype I. Further studies are needed to determine if subtype III is more likely to be associated with MAPT mutations.
References
Adams SJ, DeTure MA, McBride M, Dickson DW, Petrucelli L (2010) Three repeat isoforms of tau inhibit assembly of four repeat tau filaments. PLoS One 5:e10810
Ahmed Z, Bigio EH, Budka H et al (2013) Globular glial tauopathies (GGT): consensus recommendations. Acta Neuropathol 126:537–544
Ahmed Z, Doherty KM, Silveira-Moriyama L et al (2011) Globular glial tauopathies (GGT) presenting with motor neuron disease or frontotemporal dementia: an emerging group of 4-repeat tauopathies. Acta Neuropathol 122:415–428
Alafuzoff I, Pikkarainen M, Al-Sarraj S et al (2006) Interlaboratory comparison of assessments of Alzheimer disease-related lesions: a study of the BrainNet Europe Consortium. J Neuropathol Exp Neurol 65:740–757
Andreadis A, Brown WM, Kosik KS (1992) Structure and novel exons of the human tau gene. Biochemistry 31:10626–10633
Arai T, Ikeda K, Akiyama H et al (2004) Identification of amino-terminally cleaved tau fragments that distinguish progressive supranuclear palsy from corticobasal degeneration. Ann Neurol 55:72–79
Bigio EH, Lipton AM, Yen SH et al (2001) Frontal lobe dementia with novel tauopathy: sporadic multiple system tauopathy with dementia. J Neuropathol Exp Neurol 60:328–341
Braak H, Braak E (1987) Argyrophilic grains: characteristic pathology of cerebral cortex in cases of adult onset dementia without Alzheimer changes. Neurosci Lett 76:124–127
Brandt R, Hundelt M, Shahani N (2005) Tau alteration and neuronal degeneration in tauopathies: mechanisms and models. Biochim Biophys Acta 1739:331–354
Buee L, Delacourte A (1999) Comparative biochemistry of tau in progressive supranuclear palsy, corticobasal degeneration, FTDP-17 and Pick’s disease. Brain Pathol 9:681–693
Cook C, Carlomagno Y, Gendron TF et al (2014) Acetylation of the KXGS motifs in tau is a critical determinant in modulation of tau aggregation and clearance. Hum Mol Genet 23:104–116
DeTure M, Ko LW, Yen S et al (2000) Missense tau mutations identified in FTDP-17 have a small effect on tau–microtubule interactions. Brain Res 853:5–14
Duyckaerts C, Delaere P, Hauw JJ et al (1990) Rating of the lesions in senile dementia of the Alzheimer type: concordance between laboratories. A European multicenter study under the auspices of EURAGE. J Neurol Sci 97:295–323
Feany MB, Dickson DW (1995) Widespread cytoskeletal pathology characterizes corticobasal degeneration. Am J Pathol 146:1388–1396
Ferrer I, Hernandez I, Boada M et al (2003) Primary progressive aphasia as the initial manifestation of corticobasal degeneration and unusual tauopathies. Acta Neuropathol 106:419–435
Ferrer I, Lopez-Gonzalez I, Carmona M et al (2014) Glial and neuronal tau pathology in tauopathies: characterization of disease-specific phenotypes and tau pathology progression. J Neuropathol Exp Neurol 73:81–97
Fu YJ, Nishihira Y, Kuroda S et al (2010) Sporadic four-repeat tauopathy with frontotemporal lobar degeneration, Parkinsonism, and motor neuron disease: a distinct clinicopathological and biochemical disease entity. Acta Neuropathol 120:21–32
Gelpi E, Cullel F, Navarro-Otano J, Llado A (2013) Globular glial-like inclusions in a patient with advanced Alzheimer’s disease. Acta Neuropathol 126:155–157
Ghetti B, Wszolek ZK, Boeve BF, Spina S, Goedert M (2011) Frontotemporal dementia and parkinsonism linked to chromosome 17. In: Dickson DW, Weller RO (eds) Neurodegeneration: the molecular pathology of dementia and movement disorders, 2nd edn. Wiley-Blackwell, West Sussex, pp 110–134
Giaccone G, Marcon G, Mangieri M et al (2008) Atypical tauopathy with massive involvement of the white matter. Neuropathol Appl Neurobiol 34:468–472
Goedert M, Spillantini MG, Potier MC, Ulrich J, Crowther RA (1989) Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats: differential expression of tau protein mRNAs in human brain. EMBO J 8:393–399
Hayashi S, Toyoshima Y, Hasegawa M et al (2002) Late-onset frontotemporal dementia with a novel exon 1 (Arg5His) tau gene mutation. Ann Neurol 51:525–530
Josephs KA, Dickson DW (2003) Diagnostic accuracy of progressive supranuclear palsy in the Society for Progressive Supranuclear Palsy brain bank. Mov Disord 18:1018–1026
Josephs KA, Duffy JR, Strand EA et al (2012) Characterizing a neurodegenerative syndrome: primary progressive apraxia of speech. Brain 135:1522–1536
Josephs KA, Katsuse O, Beccano-Kelly DA et al (2006) Atypical progressive supranuclear palsy with corticospinal tract degeneration. J Neuropathol Exp Neurol 65:396–405
Josephs KA, Murray ME, Whitwell JL et al (2014) Staging TDP-43 pathology in Alzheimer’s disease. Acta Neuropathol 127:441–450
Komori T, Arai N, Oda M et al (1998) Astrocytic plaques and tufts of abnormal fibers do not coexist in corticobasal degeneration and progressive supranuclear palsy. Acta Neuropathol 96:401–408
Kouri N, Carlomagno Y, Baker M et al (2014) Novel mutation in MAPT exon 13 (p. N410H) causes corticobasal degeneration. Acta Neuropathol 127:271–282
Kouri N, Murray ME, Hassan A et al (2011) Neuropathological features of corticobasal degeneration presenting as corticobasal syndrome or Richardson syndrome. Brain 134:3264–3275
Kovacs GG, Majtenyi K, Spina S et al (2008) White matter tauopathy with globular glial inclusions: a distinct sporadic frontotemporal lobar degeneration. J Neuropathol Exp Neurol 67:963–975
Kovacs GG, Milenkovic I, Wohrer A et al (2013) Non-Alzheimer neurodegenerative pathologies and their combinations are more frequent than commonly believed in the elderly brain: a community-based autopsy series. Acta Neuropathol 126:365–384
Lee VM, Goedert M, Trojanowski JQ (2001) Neurodegenerative tauopathies. Annu Rev Neurosci 24:1121–1159
Maeda S, Sahara N, Saito Y et al (2007) Granular tau oligomers as intermediates of tau filaments. Biochemistry 46:3856–3861
Mandelkow E, Thies E, Trinczek B, Biernat J, Mandelkow E (2004) MARK/PAR1 kinase is a regulator of microtubule-dependent transport in axons. J Cell Biol 167:99–110
Mirra SS, Gearing M, McKeel DW Jr et al (1994) Interlaboratory comparison of neuropathology assessments in Alzheimer’s disease: a study of the Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). J Neuropathol Exp Neurol 53:303–315
Molina JA, Probst A, Villanueva C et al (1998) Primary progressive aphasia with glial cytoplasmic inclusions. Eur Neurol 40:71–77
Neve RL, Harris P, Kosik KS, Kurnit DM, Donlon TA (1986) Identification of cDNA clones for the human microtubule-associated protein tau and chromosomal localization of the genes for tau and microtubule-associated protein 2. Brain Res 387:271–280
Piao YS, Tan CF, Iwanaga K et al (2005) Sporadic four-repeat tauopathy with frontotemporal degeneration, parkinsonism and motor neuron disease. Acta Neuropathol 110:600–609
Powers JM, Byrne NP, Ito M et al (2003) A novel leukoencephalopathy associated with tau deposits primarily in white matter glia. Acta Neuropathol 106:181–187
Rademakers R, Cruts M, van Broeckhoven C (2004) The role of tau (MAPT) in frontotemporal dementia and related tauopathies. Hum Mutat 24:277–295
Rosso SM, van Herpen E, Deelen W et al (2002) A novel tau mutation, S320F, causes a tauopathy with inclusions similar to those in Pick’s disease. Ann Neurol 51:373–376
Sahara N, Lewis J, DeTure M et al (2002) Assembly of tau in transgenic animals expressing P301L tau: alteration of phosphorylation and solubility. J Neurochem 83:1498–1508
Santa-Maria I, Perez M, Hernandez F, Avila J, Moreno FJ (2006) Characteristics of the binding of thioflavin S to tau paired helical filaments. J Alzheimer’s Dis: JAD 9:279–285
Togo T, Cookson N, Dickson DW (2002) Argyrophilic grain disease: neuropathology, frequency in a dementia brain bank and lack of relationship with apolipoprotein E. Brain Pathol 12:45–52
Togo T, Sahara N, Yen SH et al (2002) Argyrophilic grain disease is a sporadic 4-repeat tauopathy. J Neuropathol Exp Neurol 61:547–556
Tolnay M, Schwietert M, Monsch AU, Staehelin HB, Langui D, Probst A (1997) Argyrophilic grain disease: distribution of grains in patients with and without dementia. Acta Neuropathol 94:353–358
Uchihara T (2007) Silver diagnosis in neuropathology: principles, practice and revised interpretation. Acta Neuropathol 113:483–499
Uchikado H, Lin WL, DeLucia MW, Dickson DW (2006) Alzheimer disease with amygdala Lewy bodies: a distinct form of alpha-synucleinopathy. J Neuropathol Exp Neurol 65:685–697
van Herpen E, Rosso SM, Serverijnen LA et al (2003) Variable phenotypic expression and extensive tau pathology in two families with the novel tau mutation L315R. Ann Neurol 54:573–581
Yoshida M (2006) Cellular tau pathology and immunohistochemical study of tau isoforms in sporadic tauopathies. Neuropathology 26:457–470
Zarranz JJ, Ferrer I, Lezcano E et al (2005) A novel mutation (K317M) in the MAPT gene causes FTDP and motor neuron disease. Neurology 64:1578–1585
Acknowledgments
This study was supported by the NIH P50 NS072187 (DWD, ZKW, OAR, RR, AJS), the Max Kade Foundation (PT), NIH R01 NS078086 (OAR), NIH R01 NS080882 (RR), NIH R01 NS065782 (RR), NIH R01 AG026251 (RR), NIH P50 AG16574 (RR), the ALS Therapy Alliance (RR), the Consortium for Frontotemporal Degeneration Research (RR), the NIH R01 DC010367 (KAJ), the NIH R01 AG037491 (KAJ), the NIH R01 DC012519 (KAJ), and the Alzheimer’s Association (KAJ), the Mayo Clinic Foundation (LP), the NIH P50 AG016574 (LP), the NIH R01 NS089544-1 (LP), the BrightFocus Foundation (LP), the gift from Carl Edward Bolch, Jr., and Susan Bass Bolch Foundation (SF, ZKW). We are grateful to all patients, family members, and caregivers who agreed to brain donation; without their donation these studies would have been impossible. We also acknowledge expert technical assistance of Linda Rousseau and Virginia Phillips for histology and Monica Castanedes-Casey for immunohistochemistry.
Author information
Authors and Affiliations
Corresponding author
Additional information
P. Tacik and M. DeTure have contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
401_2015_1425_MOESM1_ESM.tif
Supplementary material 1 (TIFF 7578 kb) Fig. 1 Pathologic characterization of 6 GGT cases with of phospho-tau (CP13) and 4R tau (RD4) tau immunohistochemistry as well as Gallyas sliver stain of the most severely affected areas of the white and gray matter, including motor cortex (case 1, subtype III; case 5, subtype III; case 6, subtype III), temporal lobe (case 2, subtype I; case 4, subtype I), and superior frontal gyrus (case 3, subtype III). Scale bar = 20 µm
401_2015_1425_MOESM2_ESM.tif
Supplementary material 2 (TIFF 4841 kb) Fig. 2 Chromatogram of novel MAPT p.K317N mutation. The arrow indicates the mutation site within exon 11 of MAPT
401_2015_1425_MOESM3_ESM.tif
Supplementary material 3 (TIFF 464 kb) Fig 3 (a) Effects of 4R0N p.K317N on aggregation. Tau aggregation induced by incubation of recombinant tau with dextran sulfate. Aggregation is 10% greater in 4R0N p.K317N (gray bars) than 4R0N (black bars) at 2 hours, but not different at 20 hours. (*p < 0.01); (b) Effects of 4R0N p.K317N on misfolding assessed by thioflavin S fluorescence. Tau misfolding is less in 4R0N p.K317N (gray bars), compared to 4R0N (black bars) at both 2 and 20 hours. (*p < 0.05)
401_2015_1425_MOESM4_ESM.tif
Supplementary material 4 (TIFF 23686 kb) Fig 4 Electron micrographs of recombinant 4R0N filaments (4R WT) and 4R0N p.K317N (4R K317N) at 1 hour. The scale bar = 200 nm
Rights and permissions
About this article

Cite this article
Tacik, P., DeTure, M., Lin, WL. et al. A novel tau mutation, p.K317N, causes globular glial tauopathy. Acta Neuropathol 130, 199–214 (2015). https://doi.org/10.1007/s00401-015-1425-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-015-1425-0