
Abstract
In cerebral amyloid angiopathy (CAA), amyloid fibrils deposit in walls of arteries, arterioles and less frequently in veins and capillaries of the central nervous system, often resulting in secondary degenerative vascular changes. Although the amyloid-β peptide is by far the commonest amyloid subunit implicated in sporadic and rarely in hereditary forms of CAA, a number of other proteins may also be involved in rare familial diseases in which CAA is also a characteristic morphological feature. These latter proteins include the ABri and ADan subunits in familial British dementia and familial Danish dementia, respectively, which are also known under the umbrella term BRI2 gene-related dementias, variant cystatin C in hereditary cerebral haemorrhage with amyloidosis of Icelandic-type, variant transthyretins in meningo-vascular amyloidosis, disease-associated prion protein (PrPSc) in hereditary prion disease with premature stop codon mutations and mutated gelsolin (AGel) in familial amyloidosis of Finnish type. In this review, the characteristic morphological features of the different CAAs is described and the implication of the biochemical, genetic and transgenic animal data for the pathogenesis of CAA is discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Amyloid, which is ultrastructurally composed of highly insoluble, 8- to 10-nm wide fibrils, is the end product of a protein conformation disorder. The initial phase of amyloid formation is characterised by aggregation and polymerization of soluble, often circulating proteins, marked by the conversion of random-coil secondary structures into toxic β-sheet-rich conformations. Once such conformers have been produced and protein concentration has exceeded a critical level, protofibrillar intermediate species and subsequently high-ordered amyloid fibrils are formed [108]. A frequent precondition of amyloid formation is the proteolytic processing of a larger precursor protein. Examples of this include processing by the β- and γ-secretases of the amyloid precursor protein (APP), which releases the amyloid-β (Aβ) peptide in Alzheimer’s disease (AD) [117] or processing by furin of the mutated BRI2 precursor proteins, which releases the ABri or ADan amyloid proteins in familial British dementia (FBD) [138] and familial Danish dementia (FDD) [140], respectively. A number of mechanisms are known to facilitate the destabilization of the secondary structure of soluble native proteins and these include genetic and posttranslational modifications, increased concentrations of proteins, low pH and the presence of metal ions, among others [108]. The genetic abnormalities include missense mutations in the coding region of a gene resulting in an amino acid substitution in an amyloid protein, which can alter or influence the rate of conversion of a native protein to a fibrillar conformer. A classical example is the mutant E22Q of the Aβ peptide, which is associated with hereditary cerebral haemorrhage with amyloidosis of Dutch type (HCHWA-D) [63]. Increased dosage of amyloid proteins is exemplified by trisomy of chromosome 21 in Down’s syndrome or the duplication of the APP gene associated with early onset familial AD [83, 114]. A number of amyloid-associated proteins (AAPs) or ‘pathological chaperons’ co-deposit with different cerebral parenchymal and cerebrovascular amyloids. Such proteins are structurally and functionally diverse and their binding to the amyloid fibrils or their precursors may be additional factors influencing the formation of toxic misfolded proteins [34, 61].
The umbrella term cerebral amyloid angiopathy (CAA) describes a group of biochemically and genetically diverse disorders, which are unified by the morphological finding of amyloid fibrils deposited in the walls of small to medium-sized, mostly arterial blood vessels, and in some instances, also in capillaries of the CNS parenchyma and leptomeninges. CAA may contribute to cognitive decline due to cerebral ischaemia and microhaemorrhages [12, 41, 86, 93, 97, 121, 152]. As a result of degenerative changes secondary to amyloid deposition into blood vessel walls, they may become predisposed to rupture, which is a major cause of spontaneous, frequently recurrent, lobar cerebral haemorrhages in the elderly [91]. Data from animal models also suggest that CAA may exert a functional effect on cerebral microvasculature, leading to alterations in vessel tone and reactivity [58]. Inflammatory changes recognized as a significant clinicopathological feature of CAA could also play a potentially significant role in the pathogenesis of CAA-related ischaemia [115, 131, 144].
Out of more than 25 human proteins or their proteolytic fragments that have been identified to form amyloid fibrils in vivo, only 7 have been described in diseases of the central nervous system (CNS), in which CAA can also be a prominent feature [29, 108, 110]. The most common form of CAA is due to Aβ deposition, which occurs sporadically in the elderly or in association with AD. Aβ-CAA may also be prominent in variants of familial AD with mutations of the APP, presenilin-1 (PSEN1) or presenilin-2 (PSEN2) genes. CAAs associated with other amyloid proteins are rare hereditary conditions, which include (a) FBD with deposition of the amyloid protein ABri, (b) FDD with deposition of the amyloid subunit ADan, (c) hereditary cerebral haemorrhage with amyloidosis of Icelandic type (HCHW-I) with deposition of mutant cystatin C (ACys), (d) meningo-vascular amyloidosis with variant transthyretins (ATTR) as amyloid proteins, (e) variants of familial prion disease with vascular deposition of disease-associated prion protein (PrPSc), and (f) amyloidosis of Finnish type with mutated gelsolin (AGel) as its amyloid subunit.
In this review, we wish to discuss the major neuropathological, biochemical and genetic features of the different forms of CAAs and also their clinical significance.
Neuropathology of CAA
Leptomeningeal and cortical small and medium-sized arteries and arterioles are most frequently affected by amyloid deposition, although veins may also be involved. Blood vessels with advanced CAA show an acellular thickening with a smudgy appearance of their walls on the haematoxylin and eosin stained sections. Similar to amyloid deposits elsewhere, blood vessels with CAA appear apple green in Congo red preparations when viewed in polarized light, show green fluorescence when stained with Thioflavin S, and observed under ultraviolet light (Figs. 1, 2). Binding of both of these dyes is dependent on the high β-sheet content of amyloids and is considered specific in pathological practice [108]. The predilection sites of CAA due to Aβ deposition are the occipital, parietal, frontal and temporal lobes while the medial temporal structures and hippocampus are often spared [125]. Aβ-CAA has been reported to start in leptomeningeal or parenchymal blood vessels in the neocortex, followed by amyloid formation in blood vessels of allocortical regions and cerebellum and finally of deep grey nuclei, white matter and brainstem [124]. If capillary amyloid deposition is present, it can affect a number of areas, including neocortex, subiculum, CA1 and CA4 hippocampal subregions, amygdala, thalamus, hypothalamus, nucleus basalis of Meynert, midbrain, cerebellum and pons [126]. In some forms of familial CAAs including HCHWA-I, FBD and FDD, CAA is extensive and in addition to sites commonly affected by Aβ-CAA in most cases, it can also be found in cerebral and cerebellar white matter, deep grey nuclei, brainstem and spinal cord [47, 48, 99].

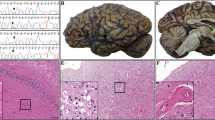
a A Congo red-positive cortical arteriole in sporadic Aβ-CAA showing (b) characteristic apple green birefringence in polarised light. c Deposition of Aβ peptide in the vascular wall is extensive and double barrelling with ‘vessel-within-vessel’ appearance is also seen. The bar on a represents 30 μm on all images

a–c Marked activated microglial reaction in relation to amyloid laden blood vessels in familial British dementia (confocal microscopy, a Thioflavin S, b Cr3/43, c combined image). d–f The C1q component of the classical complement cascade co-localises with ADan deposition in cerebrovascular amyloid in familial Danish dementia (confocal microscopy, a ADan, b C1q, c combined image). The bar on a represents 30 μm on a–c and 60 μm on d–f
Cerebrovascular amyloid deposition is a multi-step process with Aβ first appearing around smooth muscle cells in the abluminal aspect of the tunica media and the adventitia [142]. This initial phase is followed by a gradual infiltration of the intimal layers by Aβ and with further progression amyloid will gradually replace the smooth muscle cells. A similar, gradual infiltration of blood vessel walls by ABri and ADan amyloid has also been documented by immunoelectron microscopic investigations [47, 48]. Degenerative changes may accompany the amyloid deposition, including fibrous thickening with an “onion skin” appearance of the vessel wall, “double barrelling”, thinning of the degenerative vessel wall sometimes with microaneurysm formation, fibrinoid necrosis and evidence of blood breakdown products around affected blood vessels [145]. CAA grading systems commonly used in research are taken into consideration in this process of progressive amyloid deposition [92, 146]. According to one of the frequently used systems in “mild” CAA, there is amyloid in the media without significant smooth muscle cell loss, while in “moderate” CAA together with the expansion of amyloid deposition in the media, smooth muscle cell loss is conspicuous. In “severe” CAA, the smooth muscle cell layer loss is complete and this is accompanied by degenerative changes of the affected vessel walls often with evidence of leakage of blood [146].
Irrespective of the nature of the amyloid protein, a significant perivascular inflammatory response with a prominent reaction by activated microglia and astrocytes and activation of the complement cascade around amyloid-laden vessels (Fig. 2) may be found in human and experimental CAAs [46–48, 112]. Such CAA-associated angiitis due to Aβ peptide deposition has now been defined as a clinicopathological entity with patients frequently presenting with alterations in mental status, headaches, seizures and focal neurological deficits [25, 115]. Pathologically Aβ-CAA related angiitis usually consists of angiodestructive inflammation with a pronounced adventitial and perivascular infiltrate of lymphocytes and histiocytes including multinucleate giant cells with Aβ phagocytosis as well as meningeal lymphocytosis [115, 156]. The likely trigger of the vasculitic process, which has also been documented in the APP23 transgenic mouse model of AD, is vascular Aβ-peptide deposition [3, 152, 156].
In brains with CAA-related haemorrhages, there may be several lobar haemorrhages showing different stages of organization. In some instances, microhaemorrhages can be seen without evidence of a major lobar intracerebral haemorrhage. Loss of smooth muscle cells accompanied by degenerative changes, which results in weakening of the vessel walls, is thought to be the likely pathological substrate of the underlying blood vessel rupture. The possession of the ApoE ε2 allele has been reported to increase the risk of cerebral haemorrhage in patients with Aβ CAA and vasculopathic changes that result in blood vessel rupture [43, 88]. In transgenic animals with CAA, there is also a spatial and temporal relationship between microhaemorrhages and CAA. In addition, there is a positive correlation between haemorrhages and vascular amyloid load (for review see [45]). Cerebral infarctions and focal or diffuse white matter ischaemic lesions may also be the consequence of CAA irrespective of the nature of the vascular amyloid [39, 40, 66, 77, 100].
CAA with Aβ peptide deposition
Sporadic Aβ-CAA
The majority of Aβ-CAA is sporadic, mostly affecting elderly individuals, with or without morphological evidence of additional AD pathology (Fig. 1c). Sporadic CAA and AD have overlapping biology with shared risk factors [155]. The incidence of both diseases steadily increases with age with the incidence of CAA approaching 50% in elderly individuals aged over 70 years [8, 24, 27, 69, 75, 130, 143, 146]. CAA is present in over 80% of all AD cases [8, 24, 27, 69, 146] and involvement of capillaries by Aβ deposition is particularly overrepresented in advanced AD [4, 5].
Genetic risk factors for sporadic Aβ-CAA
The overlapping biology of CAA and AD is underpinned by the observation that CAA is more common and morphologically more severe in AD cases [155] than in controls and that genetic polymorphisms that have been described as risk factors for AD, such as those in ApoE, PS1, α-1-antichymotrypsin and neprilysin, which is one of the major Aβ degrading enzymes in the brain (see below), are also implicated in CAA [89]. The severity of CAA without significant AD pathology was found to correlate with the possession of the ApoE ε4 allele [105], which is also a risk factor for both sporadic CAA and CAA-related cerebral haemorrhage [42, 55, 105]. Increasing doses of ApoE ε4 have been shown to be associated with increasing amounts of Aβ40 per affected cortical vessel without increasing the proportion of amyloid-laden vessels [2]. In AD, there is a strong association between the ε4 allele frequency and the severity of CAA [14, 53] and the occurrence of CAA-related cerebral haemorrhage [55, 105]. According to a more recent study, two types of sporadic CAA can be identified and the major difference between the two is the presence (CAA type 1) or absence (CAA type 2) of capillary amyloid. In CAA type 1 cases, the frequency of the ApoE ε4 allele is reported to be more common than in CAA type 2 cases and in controls [125]. Furthermore, individuals with AD and an ApoE ε4/ε4 genotype were found to have the overall greatest Aβ deposition, more frequent arteriolar Aβ deposition, in particular in white matter [132]. Although the mechanism underlying this increased risk is not entirely clear, there is evidence to show that ApoE binds to the low density lipoprotein receptor related protein-1 (LRP-1), and interacts with soluble and aggregated Aβ both in vitro and in vivo, influencing its conformation and clearance [49].
CAA in Alzheimer’s disease treated with immunotherapy
Data are now available to indicate that following active immunisation of AD patients with Aβ42, in addition to a decrease in plaque load, there is probably a temporary manifold increase in the quantity of Aβ deposition in leptomeningeal and cerebral cortical blood vessels. This would be in keeping with observations made in immunised APP transgenic animals showing that plaque removal is accompanied by an increase in severity of CAA [102, 151]. Human studies have also demonstrated that CAA in patients treated with immunotherapy contains increased amounts of Aβ42 and Aβ40 due to solubilisation of parenchymal Aβ lesions and that, as in transgenic animals treated with passive immunisation, there is a higher density of microhaemorrhages and microvascular lesions [9, 98].
Hereditary CAAs due to Aβ peptide deposition
Missense mutations of the APP gene are within or just outside the coding region of the Aβ peptide (Fig. 3). Mutations, localized close to the β-secretase or γ-secretase cleavage sites with amino acid substitutions flanking the Aβ sequence, result in clinicopathological phenotypes of early onset AD, while those resulting in an amino acid substitution within residues 21–23 and 34 of the Aβ peptide are associated with a neuropathological phenotype, which also includes prominent CAA. The classical example of cerebrovascular disease manifestation is HCHWA-D, in which there is a glutamine for glutamic acid substitution at position 22 of Aβ (E22Q) due to a G for C nucleotide change at codon 693 of APP [63]. The clinical presentation of HCHWA-D includes strokes, including cerebral haemorrhage, although the initial presentation may be dementia [67]. Leptomeningeal and cerebral cortical blood vessels are affected by severe CAA (Fig. 4a) and, although diffuse Aβ plaques are found, classical dense core Aβ plaques are not seen and neurofibrillary degeneration is limited [67, 86]. In the Italian (E693 K), Arctic (E693G), Iowa (D694 N) and Piedmont (L705 V) variants, severe CAA has been confirmed to be a pathological feature, although it is not known why the Arctic and Iowa mutants co-exist with abundant neurofibrillary pathology while the Italian and Piedmont variants do not. The Flemish mutation (A692G) interferes with the normal processing APP and results in increased production of Aβ by the β-secretase homologue BACE-2. Affected individuals may develop cerebral haemorrhage or early onset AD. The neuropathological changes include neurofibrillary degeneration and AD-type Aβ parenchymal plaques, centered on blood vessels affected by CAA (for review see [158]). Recently, a novel mutation of the APP gene (E693Δ) was reported from Japan. This variant Aβ (E22Δ) lacking glutamate at position 22 is more resistant to proteolytic degradation and shows enhanced oligomerisation properties, but no fibrillisation. Although no neuropathological data have been reported to date, these data could be consistent with the hypothesis that the cause of dementia in this pedigree is due to enhanced formation of synaptotoxic Aβ oligomers [129]. Over-expression of wild-type APP without amino acid substitution in the protein sequence results in severe parenchymal and vascular Aβ deposition in early-onset familial AD caused by duplication of the APP gene and in Down syndrome [52, 114].

Mutations in the APP gene and their relationship to the amino acid sequence of the Aβ peptide. The Dutch and London mutations of the APP gene, shown in red, were the first described within and outside the sequence of the Aβ peptide, respectively

Different amyloid peptides in hereditary CAAs. a Deposition of Aβ peptide in blood vessels and diffuse parenchymal plaques in cerebral cortex in HCHWA-D. b Accumulation of mutated cystatin C in leptomeningeal and cerebral cortical blood vessels in HCHWA-I. c Widespread deposition of ABri in FBD d and ADan in FDD is characteristic (c and d: cerebellar cortex). e: ATTR deposition is abundant in the leptomeninges and leptomeningeal blood vessels in the Hungarian form of meningovascular amyloidosis (lumbar cord). f PrPSc deposition in blood vessels and parenchyma is a characteristic morphological feature of human prion disease with the novel Y163STOP mutation of the PRNP gene (cerebellum). g Deposition of AGel in skin blood vessels in familial amyloidosis of the Finnish type. The bar on b repsents 60 μm on a, 30 μm on b–e and g, 15 μm on f
Severe Aβ CAA has been well documented in affected members of families with mutations in the PSEN1 and presenilin-2 PSEN2 genes [23, 50, 71, 90].
Aβ in blood vessels
Aβ deposited in blood vessel walls is highly heterogeneous at both N- and C-termini. Although Aβ species ending at position 40 are usually predominant, those ending at position 42 are often present and are particularly enriched in capillaries [5]. In some animal models, the first species deposited in the vessel wall is Aβ42 and the more soluble Aβ40 is deposited subsequently [133]. Aβ42 has been demonstrated in HCHWA-D cerebrovascular lesions [87] as well as co-deposited with ADan in FDD cases [48, 127]. N-terminally truncated and post-translationally modified Aβ species, which have enhanced aggregation propensities, have also been documented to contribute to vascular amyloid [123].
Biochemical studies of Aβ in HCHWA-D [104] and in the Iowa variant [128] of FAD demonstrated that the amyloid deposits in CAA are composed of both variant (either E22Q or D23N) and wild-type Aβ in ~50:50 ratio. Compared with wild-type Aβ, both the Dutch and Iowa Aβ40 synthetic peptides rapidly assemble to form amyloid fibrils in vitro, which are toxic to cultured human cerebrovascular endothelial cells and smooth muscle cells [79, 134]. In Aβ-CAA, a number of amyloid-associated proteins including complement components, serum amyloid-P component, Apolipoprotein E (ApoE), complement inhibitors such as apolipoprotein J (ApoJ) and vitronectin, α1-antichymotrypsin, glycosaminoglycans and extracellular matrix proteins are also present [136].
Clinical and experimental studies have demonstrated that in vivo the Aβ40:Aβ42 ratio is an important determinant of amyloid formation in different cerebral compartments, i.e. whether Aβ primarily deposits in blood vessel walls or brain parenchyma. There are considerable differences between these two major classes of Aβ protein species; Aβ42 aggregates more readily perhaps because it nucleates more efficiently. In contrast, the more soluble Aβ40 is less able to initiate nucleation events capable of promoting amyloid deposition, and it may also have a possible protective role with direct inhibitory effect on Aβ42 aggregation into amyloid both in vitro and in vivo [57, 76, 122]. The relationship between the Aβ40:Aβ42 ratio and the morphological phenotype is based on the different aggregation and fibrillisation propensities of the two major classes of Aβ when they are in different compositions [45]. An increase in total cerebral Aβ with an increase in both Aβ40 and Aβ42 levels, results in increased degree of amyloid deposition in both cerebral vasculature and parenchyma. An example is the KM670/671NL Swedish double mutation, which affects the two residues located just N-terminal to the β-secretase cleavage site and results in a six to eightfold increase of both Aβ40 and Aβ42 [17, 84]. The neuropathological phenotype of both human disease and its transgenic animal model is characterised by a mixed plaque and CAA-rich picture [13, 60]. Overexpression of wild-type Aβ in familial AD with duplication of the APP gene and in Down syndrome has a similar effect with an increase of both Aβ40 and Aβ42 and amyloid deposition in both parenchyma and blood vessels [52, 114]. In contrast, mutations such as the London mutation (V717I), which are just C-terminal to the γ-secretase cleavage site of the APP gene, specifically increase the levels of the more insoluble and fibrillogenic Aβ42 and, as such mutations do not influence total Aβ production, there is a consequent decrease in the Aβ40 to Aβ42 ratio. In such human cases and their transgenic animal models there is significant parenchymal, but less vascular Aβ deposition [45]. In contrast, both human and experimental data indicate that an increased Aβ40 to Aβ42 ratio, such as that is found in affected members of families with HCHWA-D and the transgenic mouse model of this disease, significantly shifts Aβ deposition towards the cerebral vasculature resulting in prominent CAA [46]. That the high cerebral Aβ40 to Aβ42 ratio is an important marker of the morphological phenotype of HCHWA-D is further underpinned by data from experimental studies, in which the APPDutch mice were crossbred with mice overexpressing the hPS1 G384A mutation, which increases Aβ42 production. As a consequence, in the double-transgenic mice there is a decrease in the cerebral Aβ40 to Aβ42 ratio accompanied by a redistribution of the amyloid pathology from blood vessels to cerebral parenchyma [46].
Hereditary CAAs in FBD and FDD (BRI2 gene-related dementias)
Two distinct mutations of the BRI2 gene are associated with neurodegenerative diseases with striking morphological resemblance to AD. The neuropathological hallmarks of FBD include parenchymal ABri amyloid and preamyloid plaques, widespread ABri-CAA (Fig. 4c) and neurofibrillary tangle pathology [47, 100, 109, 153, 154]. In addition, white matter ischaemic change thought to be secondary to the severe CAA is also a characteristic feature [100]. The anatomical distribution of ADan deposition in FDD is rather similar to that seen in FBD (Fig. 4d), although the parenchymal lesions are primarily of preamyloid nature (defined ultrastructurally as granular, sparsely fibrillar protein deposits) [48]. In the Danish pedigree, there is also frequent co-deposition of Aβ, mostly Aβ42, with vascular and perivascular ADan amyloid [48, 127]. The BRI2 gene is located on the long arm of chromosome 13 and is broadly expressed in a number of peripheral organs and neurons and glial cells of the CNS [62, 113, 138]. The BRI2 gene encodes a 266-amino acid-long type II transmembrane protein, and furin-like proteolysis between peptide bonds 243 and 244 results in the release of a short 23-amino acid-long C-terminal peptide [138]. The BRI2 protein with a still largely unknown biological function, is widely expressed in the CNS and transported along axons [1, 62, 113]. BRI2 protein interacts with APP and is able to modify its processing. It may also act as a tumour suppressor and has been suggested to have pro-apoptotic properties [38].
A point mutation (T to A) of the normal stop codon of the BRI2 gene is the genetic abnormality underlying FBD, while a 10-nt duplication insertion mutation between codons 265 and 266 is associated with FDD [38, 138, 140]. As both mutations abolish the normal stop codon, they result in extended precursor proteins, which possess 277 amino acids instead of the normal 266. In both diseases, 34-amino-acid-long C-terminal peptides, ABri in FBD and ADan in FDD, are cleaved from the mutated precursor proteins, and readily form amyloid fibrils in vitro. Both ABri and ADan are neurotoxic, which may partly be due to their ability to form ion channel-like structures in cell membranes [107]. ABri and ADan species with post-translationally modified N-termini, are the main components of the amyloid, preamyloid parenchymal deposits and vascular amyloid in the CNS and systemic organs in FBD and FDD, respectively [36, 127, 138, 140]. A constant feature of both FBD and FDD is that CAA is extensive and involves not only the blood vessels of the leptomeninges and cerebral cortex, but also of the white matter, deep grey nuclei, brainstem, cerebellum and spinal cord [47, 48, 100]. There is a wide range of amyloid-associated proteins in both ABri and ADan parenchymal lesions and CAA in a pattern similar to that seen in association with vascular and parenchymal Aβ deposits [61]. A marked astrocytic and activated microglial response together with complement activation of both the classical and alternative pathways have been documented in relation to ABri and ADan amyloid lesions, including CAA [47, 48, 62, 112]. As reported for other non-Aβ cerebral amyloidosis (see below), mostly vascular systemic deposits of ABri and ADan can be found in a variety of peripheral tissues [36].
CAA due to mutated cystatin C in hereditary cerebral haemorrhage with amyloidosis of Icelandic-type (HCHWA-I)
HCHWA-I is an autosomal dominant disorder with often fatal, early onset cerebral haemorrhage while dementia may develop in those surviving the initial episode of haemorrhagic stroke. HCHWA-I is associated with a glutamine for leucine amino acid substitution due to an A to T point mutation at codon 68 of the cystatin C gene located on chromosome 20 [37, 64]. The cystatin C protein is a member of the type II family of cysteine protease inhibitors and is produced by many cell types including neurons of the CNS. The protein species widely deposited as vascular amyloid in the leptomeninges, cerebral cortex, basal ganglia, brainstem and cerebellum is an N-terminal degradation product of the mutated cystatin C protein. A characteristic feature of HCHWA-I is that in addition to CAA (Fig. 4b), as in FBD and FDD, amyloid deposits can also be found in peripheral tissues including lymphoid organs, skin, salivary glands, and testes [29].
CAAs due to deposition of variant transthyretins
Multiple mutations of the TTR gene, located on chromosome 18 are the most common cause of familial amyloid polyneuropathy. The amyloid subunit is composed of one of more than 60 known variants of the protein transthyretin, a molecule involved in the transport of the retinol and the thyroid hormone [51]. Although the major component of these amyloid lesions is mutated transthyretin (ATTR), wild-type transthyretin species have been also found incorporated in the amyloid fibrils [65, 103, 157]. As in other cerebral amyloidosis, amyloid fibrils are often composed of both full-length TTR as well as C- and N-terminal degradation fragments of various sizes.
The clinical manifestations of the disease are rather protean; phenotypic heterogeneity was not only found between mutations but among patients with the same mutation [18]. The most common neurological phenotype is familial amyloid sensorimotor polyneuropathy with or without associated autonomic neuropathy [7]. In some of the variants deposition of ATTR in the vitreous, leptomeninges and meningeal blood vessels is a feature. In the Hungarian (D18G) (Fig. 4e) and Ohio (V30G) pedigrees there is severe amyloid deposition in the leptomeninges and cerebral parenchyma and CAA is also documented [96, 139].
CAA in human prion diseases
A central feature of prion diseases is that disease-associated prions recruit normal cellular prion protein (PrPC), encoded by a chromosomal gene (PRPN) localized on chromosome 20, and facilitate the conversion of the cellular isoform into a disease-associated prion protein isoform (PrPSc) [106]. PrPC and PrPSc have an identical amino acid sequence, albeit with different conformations. While PrPC is rich in α-helical regions, PrPSc is characterised by a β-pleated sheet-rich secondary structure. Limited proteolysis of PrPSc of about 142 amino acids length produces shorter, protease-resistant protein species (PrP 27-30), which is capable of polymerization and forming amyloid fibrils [106]. As a general rule, PrPSc-CAA is usually not a feature of human prion diseases, which include Creutzfeldt–Jakob disease (CJD), the Gerstmann–Sträussler–Scheinker’s syndrome, fatal familial insomnia, kuru and variant CJD. However, rare hereditary disease forms, which are characterised by a premature stop codon mutation of the PRPN gene, seem to emerge as a noticeable exception to this general rule. Detailed neuropathological data are available from a family, in which a T to G mutation occurring at codon 145 results in an early stop codon (Y145STOP) and the production of an N- and C-terminally truncated, 70 amino acid-long PrP. In this pedigree, there is extensive PrP-positive CAA together with parenchymal perivascular PrP deposition and neurofibrillary tangle pathology [31]. One of us (JLH) recently had the opportunity to observe a case from a family with a novel Y163STOP mutation with a neuropathological phenotype characterised by vascular (Fig. 4f) and parenchymal disease-associated PrP deposition and extensive neurofibrillary tangle pathology (unpublished data). Both the Y145STOP and Y163STOP mutations result in truncated C-termini with loss of the glycosylphosphatidylinositol (GPI) anchor, which is added post-translationally to the C-terminus of PrP and is required to attach it to the outer leaflet of the plasma membrane [95]. As in the human disease due to Y145STOP or the Y163STOP mutation, in transgenic mice lacking the GPI anchor and infected with scrapie there are PrPSc-positive amyloid plaques and CAA. Data indicate that the GPI moiety might interfere with the ability of PrP to form amyloid fibrils and when it is absent, PrP readily forms amyloid fibrils also resulting in cerebrovascular amyloid deposition [15].
In a recently reported CJD case of an elderly individual, the accompanying CAA was mainly due to deposition of Aβ, although some additional PrP immunoreactivity was also observed [94]. It remains to be proven that this observation represents true vascular deposition of disease-associated PrP in a sporadic CJD case or the presence of PrP in the Aβ-CAA is more analogous to frequently observed co-deposition of PrPC in Aβ amyloid plaques in AD [26].
Gelsolin-related-familial amyloidosis of the Finnish type
In this form of systemic amyloidosis, the clinical presentation includes ophthalmological, dermatological and neurological symptoms and signs. Both the G654A and the G654T mutations of the gelsolin gene located on chromosome 9 have been described in a number of countries, the G654A mutation is the characteristic genetic abnormality in the Finnish pedigrees. The actin-binding protein, gelsolin has two isoforms, one is cytoplasmic with a molecular weight of 80 kDa while the other is present in the plasma and has a molecular weight of 83 kDa. The disease-associated variant AGel composing amyloid, consists of proteolytic fragments of the secretary form of gelsolin spanning positions 173–243 or 173–225 [35, 73]. Due to nucleotide changes noted above, AGel is characterised by a single amino acid substitution at residue 187, D187 N in the Finnish families and D187Y in the Danish/Czech kindreds [35, 73, 74]. AGel deposits in basement membranes and as amyloid angiopathy in systemic organs (Fig. 4g) and the CNS.
Pathogenesis of CAA
Studies of the mechanisms of disease pathogenesis in CAA are currently centered on the structural changes that affect the various amyloid subunits. Conformational transitions occurring in native soluble amyloid molecules increase their content in β-sheet structures favouring the formation of more insoluble oligomeric structures that are not physiologically catabolised and accumulate in the form of intra- and extra-cellular amorphous aggregates and fibrillar deposits. In turn, they trigger a secondary cascade of events that include, among others, release of inflammatory components, activation of the complement system, oxidative stress, alteration of the blood-brain barrier (BBB) permeability, formation of ion-like channels and cell toxicity [38, 111]. Although this information has been primarily obtained with the Aβ peptide it is clear that all known amyloid subunits share many aspects of these pathogenic mechanisms [111].
Aβ is a normal soluble component (sAβ) of biological fluids and brain interstitial fluid, in which its concentration appears to directly correlate with neuronal activity—being decreased under conditions of depressed neuronal function [10, 33, 85, 118, 120, 141]. Both deposited and sAβ molecules are identical in their primary structure, but exhibit completely different solubility and tinctorial properties. It is believed that the sAβ forms are immediate precursors of the deposited species, which through mechanisms, not completely understood, change their conformation into a predominantly β-sheet structure, highly prone to oligomerization and fibrillization. The identification of sAβ species in circulation, brain interstitial fluid and cerebrospinal fluid (CSF), together with the ability of the BBB to regulate Aβ transport in both directions, originally pointed out to the potential importance of plasma sAβ as the precursor of the deposited species [159]. However, the lack of brain lesions in a transgenic model with several fold increased plasma sAβ [56] strongly argues against the sole contribution of circulating species to brain deposition and draws attention to the brain itself as the source of Aβ. Since smooth muscle cells, pericytes and endothelial cells all express APP [11] and isolated cerebral microvessels and meningeal blood vessels are able to produce Aβ [54] the cerebral vasculature itself was proposed as a possible source of cerebral Aβ. This was supported by the close association of Aβ CAA with smooth-muscle cells [28]. Nevertheless, the sole contribution of smooth-muscle cells to Aβ-CAA is made less likely by the existence of amyloid deposits in capillaries (which are devoid of smooth muscle), a frequent finding in Aβ- as well as in ABri- and ADan- associated disorders [47, 48, 149]. Another argument against this hypothesis is that larger arteries with more abundant smooth muscle are usually less affected by amyloid deposition than small arteries and arterioles. In recent years, the notion of neuronal origin of Aβ and other amyloid proteins has been strengthened and is supported by the observation that APP transgenic models and more recently a transgenic model of FDD, all driven by neuronal promoters, develop CAA [11, 15, 46, 133, 137]. It has been proposed that the amyloid protein produced by neurons is drained along the perivascular interstitial fluid pathways of the brain parenchyma and leptomeninges and that under specific pathologic conditions it deposits along the vessels [149, 150].
With the exception of a small number (<5%) of AD familial cases with inherited mutations in APP or PSEN genes, no increased Aβ production has been demonstrated, suggesting an imbalance between Aβ production and clearance as a major element in the formation of amyloid deposits. The amphyphilic nature of Aβ precludes its crossing through the BBB unless mediated by specialised carriers and/or receptor transport mechanisms. In fact, the BBB has both the capability to control the uptake of circulating Aβ (free or complexed to carrier lipoproteins) into the CNS [20, 32, 68, 70, 72, 101, 160, 161] and regulate brain clearance via transport-mediated mechanisms [6, 21, 22, 30, 80, 119, 162]. Of the receptors involved, RAGE (the receptor for advance glycation end-products) actively participates in brain uptake of free Aβ at the vessel wall level [20] whereas other receptors are more relevant for the transport of Aβ complexed with other molecules, which, in turn, are ligands of specific receptors. In this sense, LRP-1 mediates transcytosis of Aβ-ApoE complexes contributing to rapid CNS clearance [119] whereas megalin mediates in the cellular uptake and transport of Aβ-ApoJ complexes across both the blood–brain and the blood–CSF barriers [161]. Also involved in Aβ efflux at the BBB is p-glycoprotein, highly expressed on the brain capillary endothelial cells, and the expression of which appears to correlate inversely with Aβ deposits [16, 59].
The notion of defective Aβ degradation as a contributing mechanism to brain accumulation should not be overlooked. Although the pathways by which Aβ is generated from its precursor are largely known (Fig. 3), Aβ catabolism under physiological and pathological conditions is only starting to be unveiled. Neprilysin (NEP), endothelin-converting enzyme (ECE), insulin-degrading enzyme (IDE), beta-amyloid-converting enzyme 1 (BACE-1), plasmin and matrix metalloproteases (MMPs) are among the major enzymes known to participate in brain Aβ catabolic pathways (Fig. 5) (for review see [78, 81, 116, 148]). Reduced levels and/or catalytic activity of Aβ degrading enzymes as a result of age and genetic factors as well as specific disease conditions favour Aβ accumulation, an issue well documented in murine models in which gene deletion of different proteases translate into increased levels of Aβ deposition (for review see [116, 135, 148]). The specific association of many of these enzymes with vascular components points to their active participation in CAA pathogenesis [44, 82].

Schematic representation of Aβ proteolysis by major Aβ-degrading enzymes. Thick arrows indicate major cleavage sites
Experimental models of CAA
CAA due to deposition of Aβ has been reported in aged dogs and primates [147]. A number of transgenic mouse models with Aβ-CAA has been described, including the Tg2576 mice overexpressing human APP containing the Swedish double mutation under the control of a hamster PrP promoter. In the APP23 transgenic mouse model of AD, in which the same mutation is utilized under another neuron-specific promoter (murine Thy1), there is CAA with vasculopathic alterations and CAA-related cerebral haemorrhages, enhanced by either passive anti-Aβ immunotherapy or thrombolytic treatment [98, 152]. A significant degree of CAA has also been documented in aged mice with human APP transgene harbouring the London mutation. As already described above in detail there is severe CAA in the APPDutch mice, over-expressing E693Q-mutated human APP under the control of the neuron-specific murine Thy1 promoter [46]. In another transgenic mouse model (Tg-SwDI) expressing human APP possessing the Swedish double mutation and Dutch/Iowa (E693Q/D694 N) mutations at levels below those of endogenous mouse APP, the animals develop CAA in capillaries and occasional microhaemorrhages [19]. Recently an animal model of FDD has been described, which recapitulates major morphological features of the human disease including ADan parenchymal deposits and ADan-CAA [137].
Conclusions
The mechanisms of amyloid formation and deposition in cerebral blood vessels are certainly complex. Histopathological, biochemical, genetic and physicochemical studies in conjunction with data obtained from genetically engineered transgenic animal models support the notion that different amyloid subunits undergo common abnormal folding pathways rendering similar endpoint oligomeric and fibrillar structures that deposit in the vessels and eventually replace the whole vessel wall. Once established, these deposits alter the BBB permeability and affect the normal supply of oxygen and nutrients, triggering a cascade of secondary events that include inflammation, oxidative stress and cell toxicity, key elements in the development of neurodegeneration.
References
Akiyama H, Kondo H, Arai T, Ikeda K, Kato M, Iseki E, Schwab C, McGeer PL (2004) Expression of BRI, the normal precursor of the amyloid protein of familial British dementia, in human brain. Acta Neuropathol 107:53–58. doi:10.1007/s00401-003-0783-1
Alonzo NC, Hyman BT, Rebeck GW, Greenberg SM (1998) Progression of cerebral amyloid angiopathy: accumulation of amyloid-beta40 in affected vessels. J Neuropathol Exp Neurol 57:353–359. doi:10.1097/00005072-199804000-00008
Anders KH, Wang ZZ, Kornfeld M, Gray F, Soontornniyomkij V, Reed LA, Hart MN, Menchine M, Secor DL, Vinters HV (1997) Giant cell arteritis in association with cerebral amyloid angiopathy: immunohistochemical and molecular studies. Hum Pathol 28:1237–1246. doi:10.1016/S0046-8177(97)90196-9
Attems J, Jellinger KA (2004) Only cerebral capillary amyloid angiopathy correlates with Alzheimer pathology—a pilot study. Acta Neuropathol 107:83–90. doi:10.1007/s00401-003-0796-9
Attems J, Lintner F, Jellinger KA (2004) Amyloid beta peptide 1–42 highly correlates with capillary cerebral amyloid angiopathy and Alzheimer disease pathology. Acta Neuropathol 107:283–291. doi:10.1007/s00401-004-0822-6
Bading JR, Yamada S, Mackic JB, Kirkman L, Miller C, Calero M, Ghiso J, Frangione B, Zlokovic BV (2002) Brain clearance of Alzheimer’s amyloid-beta40 in the squirrel monkey: a SPECT study in a primate model of cerebral amyloid angiopathy. J Drug Target 10:359–368. doi:10.1080/10611860290031831
Benson MD (1996) Leptomeningeal amyloid and variant transthyretins. Am J Pathol 148:351–354
Bergeron C, Ranalli PJ, Miceli PN (1987) Amyloid angiopathy in Alzheimer’s disease. Can J Neurol Sci 14:564–569
Boche D, Zotova E, Weller RO, Love S, Neal JW, Pickering RM, Wilkinson D, Holmes C, Nicoll JA (2008) Consequence of Abeta immunization on the vasculature of human Alzheimer’s disease brain. Brain 131:3299–3310. doi:10.1093/brain/awn261
Brody DL, Magnoni S, Schwetye KE, Spinner ML, Esparza TJ, Stocchetti N, Zipfel GJ, Holtzman DM (2008) Amyloid-beta dynamics correlate with neurological status in the injured human brain. Science 321:1221–1224. doi:10.1126/science.1161591
Burgermeister P, Calhoun ME, Winkler DT, Jucker M (2000) Mechanisms of cerebrovascular amyloid deposition. Lessons from mouse models. Ann N Y Acad Sci 903:307–316. doi:10.1111/j.1749-6632.2000.tb06381.x
Cadavid D, Mena H, Koeller K, Frommelt RA (2000) Cerebral beta amyloid angiopathy is a risk factor for cerebral ischemic infarction. A case control study in human brain biopsies. J Neuropathol Exp Neurol 59:768–773
Calhoun ME, Burgermeister P, Phinney AL, Stalder M, Tolnay M, Wiederhold KH, Abramowski D, Sturchler-Pierrat C, Sommer B, Staufenbiel M, Jucker M (1999) Neuronal overexpression of mutant amyloid precursor protein results in prominent deposition of cerebrovascular amyloid. Proc Natl Acad Sci USA 96:14088–14093. doi:10.1073/pnas.96.24.14088
Chalmers K, Wilcock GK, Love S (2003) APOE epsilon 4 influences the pathological phenotype of Alzheimer’s disease by favouring cerebrovascular over parenchymal accumulation of A beta protein. Neuropathol Appl Neurobiol 29:231–238. doi:10.1046/j.1365-2990.2003.00457.x
Chesebro B, Trifilo M, Race R, Meade-White K, Teng C, LaCasse R, Raymond L, Favara C, Baron G, Priola S, Caughey B, Masliah E, Oldstone M (2005) Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science 308:1435–1439. doi:10.1126/science.1110837
Cirrito JR, Deane R, Fagan AM, Spinner ML, Parsadanian M, Finn MB, Jiang H, Prior JL, Sagare A, Bales KR, Paul SM, Zlokovic BV, Piwnica-Worms D, Holtzman DM (2005) P-glycoprotein deficiency at the blood-brain barrier increases amyloid-beta deposition in an Alzheimer disease mouse model. J Clin Invest 115:3285–3290. doi:10.1172/JCI25247
Citron M, Oltersdorf T, Haass C, McConlogue L, Hung AY, Seubert P, Vigo-Pelfrey C, Lieberburg I, Selkoe DJ (1992) Mutation of the beta-amyloid precursor protein in familial Alzheimer’s disease increases beta-protein production. Nature 360:672–674. doi:10.1038/360672a0
Connors LH, Lim A, Prokaeva T, Roskens VA, Costello CE (2003) Tabulation of human transthyretin (TTR) variants, 2003. Amyloid 10:160–184
Davis J, Xu F, Deane R, Romanov G, Previti ML, Zeigler K, Zlokovic BV, Van Nostrand WE (2004) Early-onset and robust cerebral microvascular accumulation of amyloid beta-protein in transgenic mice expressing low levels of a vasculotropic Dutch/Iowa mutant form of amyloid beta-protein precursor. J Biol Chem 279:20296–20306. doi:10.1074/jbc.M312946200
Deane R, Du YS, Submamaryan RK, LaRue B, Jovanovic S, Hogg E, Welch D, Manness L, Lin C, Yu J, Zhu H, Ghiso J, Frangione B, Stern A, Schmidt AM, Armstrong DL, Arnold B, Liliensiek B, Nawroth P, Hofman F, Kindy M, Stern D, Zlokovic B (2003) RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat Med 9:907–913. doi:10.1038/nm890
DeMattos RB, Bales KR, Cummins DJ, Paul SM, Holtzman DM (2002) Brain to plasma amyloid-beta efflux: a measure of brain amyloid burden in a mouse model of Alzheimer’s disease. Science 295:2264–2267. doi:10.1126/science.1067568
DeMattos RB, Bales KR, Parsadanian M, O’dell MA, Foss EM, Paul SM, Holtzman DM (2002) Plaque-associated disruption of CSF and plasma amyloid-beta (Aβ) equilibrium in a mouse model of Alzheimer’s disease. J Neurochem 81:229–236. doi:10.1046/j.1471-4159.2002.00889.x
Dermaut B, Kumar-Singh S, De Jonghe C, Cruts M, Lofgren A, Lubke U, Cras P, Dom R, De Deyn PP, Martin JJ, Van Broeckhoven C (2001) Cerebral amyloid angiopathy is a pathogenic lesion in Alzheimer’s disease due to a novel presenilin 1 mutation. Brain 124:2383–2392. doi:10.1093/brain/124.12.2383
Ellis RJ, Olichney JM, Thal LJ, Mirra SS, Morris JC, Beekly D, Heyman A (1996) Cerebral amyloid angiopathy in the brains of patients with Alzheimer’s disease: the CERAD experience, Part XV. Neurology 46:1592–1596
Eng JA, Frosch MP, Choi K, Rebeck GW, Greenberg SM (2004) Clinical manifestations of cerebral amyloid angiopathy-related inflammation. Ann Neurol 55:250–256. doi:10.1002/ana.10810
Esiri MM, Carter J, Ironside JW (2000) Prion protein immunoreactivity in brain samples from an unselected autopsy population: findings in 200 consecutive cases. Neuropathol Appl Neurobiol 26:273–284. doi:10.1046/j.1365-2990.2000.00239.x
Esiri MM, Wilcock GK (1986) Cerebral amyloid angiopathy in dementia and old age. J Neurol Neurosurg Psychiatry 49:1221–1226. doi:10.1136/jnnp.49.11.1221
Frackowiak J, Zoltowska A, Wisniewski HM (1994) Non-fibrillar beta-amyloid protein is associated with smooth muscle cells of vessel walls in Alzheimer disease. J Neuropathol Exp Neurol 53:637–645. doi:10.1097/00005072-199411000-00011
Frangione B, Revesz T, Vidal R, Holton J, Lashley T, Houlden H, Wood N, Rostagno A, Plant G, Ghiso J (2001) Familial cerebral amyloid angiopathy related to stroke and dementia. Amyloid 8(Suppl 1):36–42
Ghersi-Egea JF, Gorevic PD, Ghiso J, Frangione B, Patlak CS, Fenstermacher JD (1996) Fate of cerebrospinal fluid-borne amyloid beta-peptide: rapid clearance into blood and appreciable accumulation by cerebral arteries. J Neurochem 67:880–883
Ghetti B, Piccardo P, Spillantini MG, Ichimiya Y, Porro M, Perini F, Kitamoto T, Tateishi J, Seiler C, Frangione B, Bugiani O, Giaccone G, Prelli F, Goedert M, Dlouhy SR, Tagliavini F (1996) Vascular variant of prion protein cerebral amyloidosis with tau- positive neurofibrillary tangles: the phenotype of the stop codon 145 mutation in PRNP. Proc Natl Acad Sci USA 93:744–748. doi:10.1073/pnas.93.2.744
Ghilardi JR, Catton M, Stimson ER, Rogers S, Walker LC, Maggio JE, Mantyh PW (1996) Intra-arterial infusion of [125I]A beta 1–40 labels amyloid deposits in the aged primate brain in vivo. NeuroReport 7:2607–2611. doi:10.1097/00001756-199611040-00040
Ghiso J, Calero M, Matsubara E, Governale S, Chuba J, Beavis R, Wisniewski T, Frangione B (1997) Alzheimer’s soluble amyloid beta is a normal component of human urine. FEBS Lett 408:105–108. doi:10.1016/S0014-5793(97)00400-6
Ghiso J, Frangione B (2002) Amyloidosis and Alzheimer’s disease. Adv Drug Deliv Rev 54:1539–1551. doi:10.1016/S0169-409X(02)00149-7
Ghiso J, Haltia M, Prelli F, Novello J, Frangione B (1990) Gelsolin variant (Asn-187) in familial amyloidosis, Finnish type. Biochem J 272:827–830
Ghiso J, Holton J, Miravalle L, Calero M, Lashley T, Vidal R, Houlden H, Wood N, Neubert T, Rostagno A, Plant G, Revesz T, Frangione B (2001) Systemic amyloid deposits in familial British dementia. J Biol Chem 276:43909–43914. doi:10.1074/jbc.M105956200
Ghiso J, Jensson O, Frangione B (1986) Amyloid fibrils in hereditary cerebral hemorrhage with amyloidosis of Icelandic type is a variant of gamma-trace basic protein (cystatin C). Proc Natl Acad Sci USA 83:2974–2978. doi:10.1073/pnas.83.9.2974
Ghiso J, Rostagno A, Tomidokoro Y, Lashley T, Bojsen-Moller M, Braendgaard H, Plant G, Holton J, Lal R, Revesz T, Frangione B (2006) Genetic alterations of the BRI2 gene: familial British and Danish dementias. Brain Pathol 16:71–79. doi:10.1111/j.1750-3639.2006.tb00563.x
Grabowski TJ, Cho HS, Vonsattel JP, Rebeck GW, Greenberg SM (2001) Novel amyloid precursor protein mutation in an Iowa family with dementia and severe cerebral amyloid angiopathy. Ann Neurol 49:697–705. doi:10.1002/ana.1009
Gray F, Dubas F, Roullet E, Escourolle R (1985) Leukoencephalopathy in diffuse hemorrhagic cerebral amyloid angiopathy. Ann Neurol 18:54–59. doi:10.1002/ana.410180110
Greenberg SM (2002) Cerebral amyloid angiopathy and dementia: two amyloids are worse than one. Neurology 58:1587–1588
Greenberg SM, Rebeck GW, Vonsattel JP, Gomez-Isla T, Hyman BT (1995) Apolipoprotein E epsilon 4 and cerebral hemorrhage associated with amyloid angiopathy. Ann Neurol 38:254–259. doi:10.1002/ana.410380219
Greenberg SM, Vonsattel JP, Segal AZ, Chiu RI, Clatworthy AE, Liao A, Hyman BT, Rebeck GW (1998) Association of apolipoprotein E epsilon2 and vasculopathy in cerebral amyloid angiopathy. Neurology 50:961–965
Hersh LB (2003) Peptidases, proteases and amyloid beta-peptide catabolism. Curr Pharm Des 9:449–454. doi:10.2174/1381612033391676
Herzig MC, Van Nostrand WE, Jucker M (2006) Mechanism of cerebral beta-amyloid angiopathy: murine and cellular models. Brain Pathol 16:40–54. doi:10.1111/j.1750-3639.2006.tb00560.x
Herzig MC, Winkler DT, Burgermeister P, Pfeifer M, Kohler E, Schmidt SD, Danner S, Abramowski D, Sturchler-Pierrat C, Burki K, van Duinen SG, Maat-Schieman ML, Staufenbiel M, Mathews PM, Jucker M (2004) Abeta is targeted to the vasculature in a mouse model of hereditary cerebral hemorrhage with amyloidosis. Nat Neurosci 7:954–960. doi:10.1038/nn1302
Holton JL, Ghiso J, Lashley T, Rostagno A, Guerin CJ, Gibb G, Houlden H, Ayling H, Martinian L, Anderton BH, Wood NW, Vidal R, Plant G, Frangione B, Revesz T (2001) Regional distribution of amyloid-BRI deposition and its association with neurofibrillary degeneration in familial British dementia. Am J Pathol 158:515–526
Holton JL, Lashley T, Ghiso J, Braendgaard H, Vidal R, Guerin CJ, Gibb G, Hanger DP, Rostagno A, Anderton BH, Strand C, Ayling H, Plant G, Frangione B, Bojsen-Moller M, Revesz T (2002) Familial Danish dementia: a novel form of cerebral amyloidosis associated with deposition of both amyloid-Dan and amyloid-beta. J Neuropathol Exp Neurol 61:254–267
Holtzman DM (2004) In vivo effects of ApoE and clusterin on amyloid-beta metabolism and neuropathology. J Mol Neurosci 23:247–254. doi:10.1385/JMN:23:3:247
Houlden H, Baker M, McGowan E, Lewis P, Hutton M, Crook R, Wood NW, Kumar-Singh S, Geddes J, Swash M, Scaravilli F, Holton JL, Lashley T, Tomita T, Hashimoto T, Verkkoniemi A, Kalimo H, Somer M, Paetau A, Martin JJ, Van Broeckhoven C, Golde T, Hardy J, Haltia M, Revesz T (2000) Variant Alzheimer’s disease with spastic paraparesis and cotton wool plaques is caused by PS-1 mutations that lead to exceptionally high amyloid-beta concentrations. Ann Neurol 48:806–808. doi:10.1002/1531-8249(200011)48:5<806::AID-ANA18>3.0.CO;2-F
Ihse E, Stangou AJ, Heaton ND, O’Grady J, Ybo A, Hellman U, Edvinsson S, Westermark P (2008) Proportion of wild-type transthyretin in truncated compared to full-length ATTR: an analysis on transplanted transthyretin T60A amyloidosis patients. Biochem Biophys Res Commun 379:846–850
Iwatsubo T, Mann DM, Odaka A, Suzuki N, Ihara Y (1995) Amyloid beta protein (A beta) deposition: a beta 42(43) precedes A beta 40 in Down syndrome. Ann Neurol 37:294–299. doi:10.1002/ana.410370305
Jellinger KA, Lauda F, Attems J (2007) Sporadic cerebral amyloid angiopathy is not a frequent cause of spontaneous brain hemorrhage. Eur J Neurol 14:923–928. doi:10.1111/j.1468-1331.2007.01880.x
Kalaria RN, Premkumar DR, Pax AB, Cohen DL, Lieberburg I (1996) Production and increased detection of amyloid beta protein and amyloidogenic fragments in brain microvessels, meningeal vessels and choroid plexus in Alzheimer’s disease. Brain Res Mol Brain Res 35:58–68. doi:10.1016/0169-328X(95)00180-Z
Kalaria RN, Thomas A, Oakley A, Ince P, Tamaoka A, Mori H, Kenny RA, Ballard C (2003) Cerebrovascular amyloidosis and dementia. Curr Med Chem Immun Endocr Metab Agents 3:317–327
Kawarabayashi T, Shoji M, Sato M, Sasaki A, Ho L, Eckman CB, Prada CM, Younkin SG, Kobayashi T, Tada N, Matsubara E, Iizuka T, Harigaya Y, Kasai K, Hirai S (1996) Accumulation of beta-amyloid fibrils in pancreas of transgenic mice. Neurobiol Aging 17:215–222. doi:10.1016/0197-4580(95)02061-6
Kim J, Onstead L, Randle S, Price R, Smithson L, Zwizinski C, Dickson DW, Golde T, McGowan E (2007) Abeta40 inhibits amyloid deposition in vivo. J Neurosci 27:627–633. doi:10.1523/JNEUROSCI.4849-06.2007
Kimchi EY, Kajdasz S, Bacskai BJ, Hyman BT (2001) Analysis of cerebral amyloid angiopathy in a transgenic mouse model of Alzheimer disease using in vivo multiphoton microscopy. J Neuropathol Exp Neurol 60:274–279
Lam FC, Liu R, Lu P, Shapiro AB, Renoir JM, Sharom FJ, Reiner PB (2001) beta-Amyloid efflux mediated by p-glycoprotein. J Neurochem 76:1121–1128. doi:10.1046/j.1471-4159.2001.00113.x
Lannfelt L, Bogdanovic N, Appelgren H, Axelman K, Lilius L, Hansson G, Schenk D, Hardy J, Winblad B (1994) Amyloid precursor protein mutation causes Alzheimer’s disease in a Swedish family. Neurosci Lett 168:254–256. doi:10.1016/0304-3940(94)90463-4
Lashley T, Holton JL, Verbeek MM, Rostagno A, Bojsen-Moller M, David G, Van HJ, Braendgaard H, Plant G, Frangione B, Ghiso J, Revesz T (2006) Molecular chaperons, amyloid and preamyloid lesions in the BRI2 gene-related dementias: a morphological study. Neuropathol Appl Neurobiol 32:492–504. doi:10.1111/j.1365-2990.2006.00747.x
Lashley T, Revesz T, Plant G, Bandopadhyay R, Lees AJ, Frangione B, Wood NW, de Silva R, Ghiso J, Rostagno A, Holton JL (2008) Expression of BRI2 mRNA and protein in normal human brain and familial British dementia: its relevance to the pathogenesis of disease. Neuropathol Appl Neurobiol 34:492–505
Levy E, Carman MD, Fernandez-Madrid IJ, Power MD, Lieberburg I, van Duinen SG, Bots GT, Luyendijk W, Frangione B (1990) Mutation of the Alzheimer’s disease amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science 248:1124–1126. doi:10.1126/science.2111584
Levy E, Lopez-Otin C, Ghiso J, Geltner D, Frangione B (1989) Stroke in Icelandic patients with hereditary amyloid angiopathy is related to a mutation in the cystatin C gene, an inhibitor of cysteine proteases. J Exp Med 169:1771–1778. doi:10.1084/jem.169.5.1771
Liepnieks JJ, Wilson DL, Benson MD (2006) Biochemical characterization of vitreous and cardiac amyloid in Ile84Ser transthyretin amyloidosis. Amyloid 13:170–177. doi:10.1080/13506120600877003
Love S (2004) Contribution of cerebral amyloid angiopathy to Alzheimer’s disease. J Neurol Neurosurg Psychiatry 75:1–4. doi:10.1136/jnnp.2003.034249
Maat-Schieman ML, van Duinen SG, Bornebroek M, Haan J, Roos RA (1996) Hereditary cerebral hemorrhage with amyloidosis-Dutch type (HCHWA-D): II—a review of histopathological aspects. Brain Pathol 6:115–120. doi:10.1111/j.1750-3639.1996.tb00794.x
Mackic JB, Weiss MH, Miao W, Kirkman E, Ghiso J, Calero M, Bading J, Frangione B, Zlokovic BV (1998) Cerebrovascular accumulation and increased blood-brain barrier permeability to circulating Alzheimer’s amyloid beta peptide in aged squirrel monkey with cerebral amyloid angiopathy. J Neurochem 70:210–215
Mandybur TI (1975) The incidence of cerebral amyloid angiopathy in Alzheimer’s disease. Neurology 25:120–126
Maness LM, Banks WA, Podlisny MB, Selkoe DJ, Kastin AJ (1994) Passage of human amyloid beta-protein 1–40 across the murine blood-brain barrier. Life Sci 55:1643–1650. doi:10.1016/0024-3205(94)00331-9
Mann DM, Pickering-Brown SM, Takeuchi A, Iwatsubo T (2001) Amyloid angiopathy and variability in amyloid beta deposition is determined by mutation position in presenilin-1-linked Alzheimer’s disease. Am J Pathol 158:2165–2175
Martel CL, Mackic JB, Matsubara E, Governale S, Miguel C, Miao W, McComb JG, Frangione B, Ghiso J, Zlokovic BV (1997) Isoform-specific effects of apolipoproteins E2, E3, and E4 on cerebral capillary sequestration and blood-brain barrier transport of circulating Alzheimer’s amyloid beta. J Neurochem 69:1995–2004
Maury CP (1991) Gelsolin-related amyloidosis. Identification of the amyloid protein in Finnish hereditary amyloidosis as a fragment of variant gelsolin. J Clin Invest 87:1195–1199. doi:10.1172/JCI115118
Maury CP, Nurmiaho-Lassila EL, Rossi H (1994) Amyloid fibril formation in gelsolin-derived amyloidosis. Definition of the amyloidogenic region and evidence of accelerated amyloid formation of mutant Asn-187 and Tyr-187 gelsolin peptides. Lab Invest 70:558–564
McCarron MO, Nicoll JAR (2000) ApoE genotype ain relation to sporadic and Alzheimer-related CAA. In: Verbeek MM, de Waal RMW, Vinters HV (eds) Cerebral amyloid angiopathy in Alzheimer’s disease and related disorders. Kluwer, Dordrecht, pp 81–102
McGowan E, Pickford F, Kim J, Onstead L, Eriksen J, Yu C, Skipper L, Murphy MP, Beard J, Das P, Jansen K, Delucia M, Lin WL, Dolios G, Wang R, Eckman CB, Dickson DW, Hutton M, Hardy J, Golde T (2005) Abeta42 is essential for parenchymal and vascular amyloid deposition in mice. Neuron 47:191–199. doi:10.1016/j.neuron.2005.06.030
Mead S, James-Galton M, Revesz T, Doshi RB, Harwood G, Pan EL, Ghiso J, Frangione B, Plant G (2000) Familial British dementia with amyloid angiopathy: early clinical, neuropsychological and imaging findings. Brain 123:975–991. doi:10.1093/brain/123.5.975
Miners JS, Baig S, Palmer J, Palmer LE, Kehoe PG, Love S (2008) Abeta-degrading enzymes in Alzheimer’s disease. Brain Pathol 18:240–252. doi:10.1111/j.1750-3639.2008.00132.x
Miravalle L, Tokuda T, Chiarle R, Giaccone G, Bugiani O, Tagliavini F, Frangione B, Ghiso J (2000) Substitutions at codon 22 of Alzheimer’s Aβ peptide induce diverse conformational changes and apoptotic effects in human cerebral endothelial cells. J Biol Chem 275:27110–27116
Monro OR, Mackic JB, Yamada S, Segal MB, Ghiso J, Maurer C, Calero M, Frangione B, Zlokovic BV (2002) Substitution at codon 22 reduces clearance of Alzheimer’s amyloid-beta peptide from the cerebrospinal fluid and prevents its transport from the central nervous system into blood. Neurobiol Aging 23:405–412. doi:10.1016/S0197-4580(01)00317-7
Morelli L, Llovera R, Ibendahl S, Castano EM (2002) The degradation of amyloid beta as a therapeutic strategy in Alzheimer’s disease and cerebrovascular amyloidoses. Neurochem Res 27:1387–1399. doi:10.1023/A:1021679817756
Morelli L, Llovera RE, Mathov I, Lue LF, Frangione B, Ghiso J, Castano EM (2004) Insulin-degrading enzyme in brain microvessels: proteolysis of amyloid-β vasculotropic variants and reduced activity in cerebral amyloid angiopathy. J Biol Chem 279:56004–56013. doi:10.1074/jbc.M407283200
Mrak RE, Griffin WS (2004) Trisomy 21 and the brain. J Neuropathol Exp Neurol 63:679–685
Mullan M, Crawford F, Axelman K, Houlden H, Lilius L, Winblad B, Lannfelt L (1992) A pathogenic mutation for probable Alzheimer’s disease in the APP gene at the N-terminus of beta-amyloid. Nat Genet 1:345–347. doi:10.1038/ng0892-345
Naslund J, Schierhorn A, Hellman U, Lannfelt L, Roses AD, Tjernberg LO, Silberring J, Gandy SE, Winblad B, Greengard P (1994) Relative abundance of Alzheimer Aβ amyloid peptide variants in Alzheimer disease and normal aging. Proc Natl Acad Sci USA 91:8378–8382. doi:10.1073/pnas.91.18.8378
Natte R, Maat-Schieman ML, Haan J, Bornebroek M, Roos RA, van Duinen SG (2001) Dementia in hereditary cerebral hemorrhage with amyloidosis-Dutch type is associated with cerebral amyloid angiopathy but is independent of plaques and neurofibrillary tangles. Ann Neurol 50:765–772. doi:10.1002/ana.10040
Natte R, Yamaguchi H, Maat-Schieman ML, Prins FA, Neeskens P, Roos RA, van Duinen SG (1999) Ultrastructural evidence of early non-fibrillar Aβ42 in the capillary basement membrane of patients with hereditary cerebral hemorrhage with amyloidosis, Dutch type. Acta Neuropathol 98:577–582. doi:10.1007/s004010051121
Nicoll JA, Burnett C, Love S, Graham DI, Dewar D, Ironside JW, Stewart J, Vinters HV (1997) High frequency of apolipoprotein E epsilon 2 allele in hemorrhage due to cerebral amyloid angiopathy. Ann Neurol 41:716–721. doi:10.1002/ana.410410607
Nicoll JA, Yamada M, Frackowiak J, Mazur-Kolecka B, Weller RO (2004) Cerebral amyloid angiopathy plays a direct role in the pathogenesis of Alzheimer’s disease. Pro-CAA position statement. Neurobiol Aging 25:589–597. doi:10.1016/j.neurobiolaging.2004.02.003
Nochlin D, Bird TD, Nemens EJ, Ball MJ, Sumi SM (1998) Amyloid angiopathy in a Volga German family with Alzheimer’s disease and a presenilin-2 mutation (N141I). Ann Neurol 43:131–135. doi:10.1002/ana.410430124
O’Donnell HC, Rosand J, Knudsen KA, Furie KL, Segal AZ, Chiu RI, Ikeda D, Greenberg SM (2000) Apolipoprotein E genotype and the risk of recurrent lobar intracerebral hemorrhage. N Engl J Med 342:240–245. doi:10.1056/NEJM200001273420403
Olichney JM, Hansen LA, Galasko D, Saitoh T, Hofstetter CR, Katzman R, Thal LJ (1996) The apolipoprotein E epsilon 4 allele is associated with increased neuritic plaques and cerebral amyloid angiopathy in Alzheimer’s disease and Lewy body variant. Neurology 47:190–196
Olichney JM, Hansen LA, Hofstetter CR, Grundman M, Katzman R, Thal LJ (1995) Cerebral infarction in Alzheimer’s disease is associated with severe amyloid angiopathy and hypertension. Arch Neurol 52:702–708
Paquet C, Privat N, Kaci R, Polivka M, Dupont O, Haik S, Laplanche JL, Hauw JJ, Gray F (2008) Cerebral amyloid angiopathy with co-localization of prion protein and beta-amyloid in an 85-year-old patient with sporadic Creutzfeldt-Jakob disease. Acta Neuropathol 116:567–573. doi:10.1007/s00401-008-0394-y
Paulick MG, Bertozzi CR (2008) The glycosylphosphatidylinositol anchor: a complex membrane-anchoring structure for proteins. Biochemistry 47:6991–7000. doi:10.1021/bi8006324
Petersen RB, Goren H, Cohen M, Richardson SL, Tresser N, Lynn A, Gali M, Estes M, Gambetti P (1997) Transthyretin amyloidosis: a new mutation associated with dementia. Ann Neurol 41:307–313. doi:10.1002/ana.410410305
Pfeifer LA, White LR, Ross GW, Petrovitch H, Launer LJ (2002) Cerebral amyloid angiopathy and cognitive function: the HAAS autopsy study. Neurology 58:1629–1634
Pfeifer M, Boncristiano S, Bondolfi L, Stalder A, Deller T, Staufenbiel M, Mathews PM, Jucker M (2002) Cerebral hemorrhage after passive anti-Abeta immunotherapy. Science 298:1379. doi:10.1126/science.1078259
Plant GT, Ghiso J, Holton JL, Frangione B, Revesz T (2004) Familial and sporadic cerebral amyloid angiopathies associated with dementia and the BRI dementias. In: Esiri MM, Lee VMY, Trojanowski JQ (eds) The neuropathology of dementia. Cambridge University Press, Cambridge, pp 330–352
Plant GT, Revesz T, Barnard RO, Harding AE, Gautier-Smith PC (1990) Familial cerebral amyloid angiopathy with nonneuritic amyloid plaque formation. Brain 113:721–747. doi:10.1093/brain/113.3.721
Poduslo JF, Curran GL, Sanyal B, Selkoe DJ (1999) Receptor-mediated transport of human amyloid beta-protein 1–40 and 1–42 at the blood-brain barrier. Neurobiol Dis 6:190–199. doi:10.1006/nbdi.1999.0238
Prada CM, Garcia-Alloza M, Betensky RA, Zhang-Nunes SX, Greenberg SM, Bacskai BJ, Frosch MP (2007) Antibody-mediated clearance of amyloid-beta peptide from cerebral amyloid angiopathy revealed by quantitative in vivo imaging. J Neurosci 27:1973–1980. doi:10.1523/JNEUROSCI.5426-06.2007
Pras M, Prelli F, Franklin EC, Frangione B (1983) Primary structure of an amyloid prealbumin variant in familial polyneuropathy of Jewish origin. Proc Natl Acad Sci USA 80:539–542. doi:10.1073/pnas.80.2.539
Prelli F, Levy E, van Duinen SG, Bots GT, Luyendijk W, Frangione B (1990) Expression of a normal and variant Alzheimer’s beta-protein gene in amyloid of hereditary cerebral hemorrhage, Dutch type: DNA and protein diagnostic assays. Biochem Biophys Res Commun 170:301–307. doi:10.1016/0006-291X(90)91274-V
Premkumar DR, Cohen DL, Hedera P, Friedland RP, Kalaria RN (1996) Apolipoprotein E-epsilon4 alleles in cerebral amyloid angiopathy and cerebrovascular pathology associated with Alzheimer’s disease. Am J Pathol 148:2083–2095
Prusiner SB (2001) Shattuck lecture—neurodegenerative diseases and prions. N Engl J Med 344:1516–1526. doi:10.1056/NEJM200105173442006
Quist A, Doudevski I, Lin H, Azimova R, Ng D, Frangione B, Kagan B, Ghiso J, Lal R (2005) Amyloid ion channels: a common structural link for protein-misfolding disease. Proc Natl Acad Sci USA 102:10427–10432. doi:10.1073/pnas.0502066102
Revesz T, Ghiso J, Lashley T, Plant G, Rostagno A, Frangione B, Holton JL (2003) Cerebral amyloid angiopathies: a pathologic, biochemical, and genetic view. J Neuropathol Exp Neurol 62:885–898
Revesz T, Holton JL, Doshi B, Anderton BH, Scaravilli F, Plant GT (1999) Cytoskeletal pathology in familial cerebral amyloid angiopathy (British type) with non-neuritic amyloid plaque formation. Acta Neuropathol 97:170–176. doi:10.1007/s004010050970
Revesz T, Holton JL, Lashley T, Plant G, Rostagno A, Ghiso J, Frangione B (2002) Sporadic and familial cerebral amyloid angiopathies. Brain Pathol 12:343–357
Rostagno A, Lal R, Ghiso J (2007) Protein misfolding, aggregation, and fibril formation: common features of cerebral and non-cerebral amyloid diseases. In: Dawbarn D, Allen S (eds) The neurobiology of Alzheimer’s disease. Oxford University Press, Oxford, pp 133–160
Rostagno A, Revesz T, Lashley T, Tomidokoro Y, Magnotti L, Braendgaard H, Plant G, Bojsen-Moller M, Holton J, Frangione B, Ghiso J (2002) Complement activation in chromosome 13 dementias. Similarities with Alzheimer’s disease. J Biol Chem 277:49782–49790. doi:10.1074/jbc.M206448200
Rostagno A, Tomidokoro Y, Lashley T, Ng D, Plant G, Holton J, Frangione B, Revesz T, Ghiso J (2005) Chromosome 13 dementias. Cell Mol Life Sci 62:1814–1825. doi:10.1007/s00018-005-5092-5
Rovelet-Lecrux A, Hannequin D, Raux G, Le MN, Laquerriere A, Vital A, Dumanchin C, Feuillette S, Brice A, Vercelletto M, Dubas F, FREBOURG T, Campion D (2006) APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat Genet 38:24–26. doi:10.1038/ng1718
Scolding NJ, Joseph F, Kirby PA, Mazanti I, Gray F, Mikol J, Ellison D, Hilton DA, Williams TL, MacKenzie JM, Xuereb JH, Love S (2005) Aβ-related angiitis: primary angiitis of the central nervous system associated with cerebral amyloid angiopathy. Brain 128:500–515. doi:10.1093/brain/awh379
Selkoe DJ (2001) Clearing the brain’s amyloid cobwebs. Neuron 32:177–180. doi:10.1016/S0896-6273(01)00475-5
Selkoe DJ (2001) Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev 81:741–766
Seubert P, Vigo-Pelfrey C, Esch F, Lee M, Dovey H, Davis D, Sinha S, Schlossmacher M, Whaley J, Swindlehurst C (1992) Isolation and quantification of soluble Alzheimer’s beta-peptide from biological fluids. Nature 359:325–327. doi:10.1038/359325a0
Shibata M, Yamada S, Kumar SR, Calero M, Bading J, Frangione B, Holtzman DM, Miller CA, Strickland DK, Ghiso J, Zlokovic BV (2000) Clearance of Alzheimer’s amyloid-β (1–40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J Clin Invest 106:1489–1499. doi:10.1172/JCI10498
Shoji M, Golde TE, Ghiso J, Cheung TT, Estus S, Shaffer LM, Cai XD, McKay DM, Tintner R, Frangione B (1992) Production of the Alzheimer amyloid beta protein by normal proteolytic processing. Science 258:126–129. doi:10.1126/science.1439760
Snowdon DA, Greiner LH, Mortimer JA, Riley KP, Greiner PA, Markesbery WR (1997) Brain infarction and the clinical expression of Alzheimer disease. The Nun Study. JAMA 277:813–817. doi:10.1001/jama.277.10.813
Snyder SW, Ladror US, Wade WS, Wang GT, Barrett LW, Matayoshi ED, Huffaker HJ, Krafft GA, Holzman TF (1994) Amyloid-beta aggregation: selective inhibition of aggregation in mixtures of amyloid with different chain lengths. Biophys J 67:1216–1228. doi:10.1016/S0006-3495(94)80591-0
Tekirian TL, Saido TC, Markesbery WR, Russell MJ, Wekstein DR, Patel E, Geddes JW (1998) N-terminal heterogeneity of parenchymal and cerebrovascular A beta deposits. J Neuropathol Exp Neurol 57:76–94. doi:10.1097/00005072-199801000-00009
Thal DR, Ghebremedhin E, Orantes M, Wiestler OD (2003) Vascular pathology in Alzheimer disease: correlation of cerebral amyloid angiopathy and arteriosclerosis/lipohyalinosis with cognitive decline. J Neuropathol Exp Neurol 62:1287–1301
Thal DR, Ghebremedhin E, Rub U, Yamaguchi H, Del Tredici K, Braak H (2002) Two types of sporadic cerebral amyloid angiopathy. J Neuropathol Exp Neurol 61:282–293
Thal DR, Griffin WS, de Vos RA, Ghebremedhin E (2008) Cerebral amyloid angiopathy and its relationship to Alzheimer’s disease. Acta Neuropathol 115:599–609. doi:10.1007/s00401-008-0366-2
Tomidokoro Y, Lashley T, Rostagno A, Neubert TA, Bojsen-Moller M, Braendgaard H, Plant G, Holton J, Frangione B, Revesz T, Ghiso J (2005) Familial Danish dementia: co-existence of Danish and Alzheimer amyloid subunits (ADan and Aβ) in the absence of compact plaques. J Biol Chem 280:36883–36894. doi:10.1074/jbc.M504038200
Tomidokoro Y, Rostagno A, Greenberg SM, Frangione B, Rebeck WG, Ghiso J (2004) Biochemical analysis of A beta amyloid deposits in the Iowa variant of Alzheimer’s disease. Neurobiol Aging 25:S38. doi:10.1016/S0197-4580(04)80125-8
Tomiyama T, Nagata T, Shimada H, Teraoka R, Fukushima A, Kanemitsu H, Takuma H, Kuwano R, Imagawa M, Ataka S, Wada Y, Yoshioka E, Nishizaki T, Watanabe Y, Mori H (2008) A new amyloid beta variant favoring oligomerization in Alzheimer’s-type dementia. Ann Neurol 63:377–387. doi:10.1002/ana.21321
Tomonaga M (1981) Cerebral amyloid angiopathy in the elderly. J Am Geriatr Soc 29:151–157
Townsend KP, Obregon D, Quadros A, Patel N, Volmar C, Paris D, Mullan M (2002) Proinflammatory and vasoactive effects of Abeta in the cerebrovasculature. Ann N Y Acad Sci 977:65–76
Trembath D, Ervin JF, Broom L, Szymanski M, Welsh-Bohmer K, Pieper C, Hulette CM (2007) The distribution of cerebrovascular amyloid in Alzheimer’s disease varies with ApoE genotype. Acta Neuropathol 113:23–31. doi:10.1007/s00401-006-0162-9
Van Dorpe J, Smeijers L, Dewachter I, Nuyens D, Spittaels K, Van Den HC MERCKENM, Moechars D, Laenen I, Kuiperi C, Bruynseels K, Tesseur I, Loos R, Vanderstichele H, Checler F, Sciot R, Van Leuven F (2000) Prominent cerebral amyloid angiopathy in transgenic mice overexpressing the London mutant of human APP in neurons. Am J Pathol 157:1283–1298
Van Nostrand WE, Melchor JP, Cho HS, Greenberg SM, Rebeck GW (2001) Pathogenic effects of D23 N Iowa mutant amyloid beta -protein. J Biol Chem 276:32860–32866. doi:10.1074/jbc.M104135200
Vardy ER, Catto AJ, Hooper NM (2005) Proteolytic mechanisms in amyloid-beta metabolism: therapeutic implications for Alzheimer’s disease. Trends Mol Med 11:464–472. doi:10.1016/j.molmed.2005.08.004
Verbeek MM, Otte-Holler I, Veerhuis R, Ruiter DJ, de Waal RM (1998) Distribution of A beta-associated proteins in cerebrovascular amyloid of Alzheimer’s disease. Acta Neuropathol 96:628–636. doi:10.1007/s004010050944
Vidal R, Barbeito AG, Miravalle L, Ghetti B (2009) Cerebral amyloid angiopathy and parenchymal amyloid deposition in transgenic mice expressing the Danish mutant form of human BRI2. Brain Pathol 19:58–68. doi:10.1111/j.1750-3639.2008.00164.x
Vidal R, Frangione B, Rostagno A, Mead S, Revesz T, Plant G, Ghiso J (1999) A stop-codon mutation in the BRI gene associated with familial British dementia. Nature 399:776–781. doi:10.1038/21637
Vidal R, Garzuly F, Budka H, Lalowski M, Linke RP, Brittig F, Frangione B, Wisniewski T (1996) Meningocerebrovascular amyloidosis associated with a novel transthyretin mis-sense mutation at codon 18 (TTRD 18G). Am J Pathol 148:361–366
Vidal R, Revesz T, Rostagno A, Kim E, Holton JL, Bek T, Bojsen-Moller M, Braendgaard H, Plant G, Ghiso J, Frangione B (2000) A decamer duplication in the 3’ region of the BRI gene originates an amyloid peptide that is associated with dementia in a Danish kindred. Proc Natl Acad Sci USA 97:4920–4925. doi:10.1073/pnas.080076097
Vigo-Pelfrey C, Lee D, Keim P, Lieberburg I, Schenk DB (1993) Characterization of beta-amyloid peptide from human cerebrospinal fluid. J Neurochem 61:1965–1968. doi:10.1111/j.1471-4159.1993.tb09841.x
Vinters HV (1987) Cerebral amyloid angiopathy. A critical review. Stroke 18:311–324
Vinters HV, Gilbert JJ (1983) Cerebral amyloid angiopathy: incidence and complications in the aging brain. II. The distribution of amyloid vascular changes. Stroke 14:924–928
Vinters HV, Natte R, Maat-Schieman ML, van Duinen SG, Hegeman-Kleinn I, Welling-Graafland C, Haan J, Roos RA (1998) Secondary microvascular degeneration in amyloid angiopathy of patients with hereditary cerebral hemorrhage with amyloidosis, Dutch type (HCHWA-D). Acta Neuropathol 95:235–244. doi:10.1007/s004010050793
Vinters HV, Vonsattel JP (2000) Neuropathologic features and grading of Alzheimer-related and sporadic CAA. In: Verbeek MM, de Waal RMW, Vinters HV (eds) Cerebral Amyloid Angiopathy in Alzheimer’s Disease and Related Disorders. Kluwer, Dordrecht, pp 137–155
Vonsattel JP, Myers RH, Hedley-Whyte ET, Ropper AH, Bird ED, Richardson EP Jr (1991) Cerebral amyloid angiopathy without and with cerebral hemorrhages: a comparative histological study. Ann Neurol 30:637–649. doi:10.1002/ana.410300503
Walker LC (1997) Animal models of cerebral beta-amyloid angiopathy. Brain Res Brain Res Rev 25:70–84. doi:10.1016/S0165-0173(97)00017-9
Wang YJ, Zhou HD, Zhou XF (2006) Clearance of amyloid-beta in Alzheimer’s disease: progress, problems and perspectives. Drug Discov Today 11:931–938. doi:10.1016/j.drudis.2006.08.004
Weller RO, Massey A, Newman TA, Hutchings M, Kuo YM, Roher AE (1998) Cerebral amyloid angiopathy—amyloid beta accumulates in putative interstitial fluid drainage pathways in Alzheimer’s disease. Am J Pathol 153:725–733
Weller RO, Subash M, Preston SD, Mazanti I, Carare RO (2008) Perivascular drainage of amyloid-beta peptides from the brain and its failure in cerebral amyloid angiopathy and Alzheimer’s disease. Brain Pathol 18:253–266. doi:10.1111/j.1750-3639.2008.00133.x
Wilcock DM, Rojiani A, Rosenthal A, Subbarao S, Freeman MJ, Gordon MN, Morgan D (2004) Passive immunotherapy against Abeta in aged APP-transgenic mice reverses cognitive deficits and depletes parenchymal amyloid deposits in spite of increased vascular amyloid and microhemorrhage. J Neuroinflammation 1:24. doi:10.1186/1742-2094-1-24
Winkler DT, Bondolfi L, Herzig MC, Jann L, Calhoun ME, Wiederhold KH, Tolnay M, Staufenbiel M, Jucker M (2001) Spontaneous hemorrhagic stroke in a mouse model of cerebral amyloid angiopathy. J Neurosci 21:1619–1627
Worster-Drought C, Greenfield JG, McMenemey WH (1940) A form of familial presenile dementia with spastic paralysis (including the pathological examination of a case). Brain 63:237–254. doi:10.1093/brain/63.3.237
Worster-Drought C, Greenfield JG, McMenemey WH (1944) A form of familial presenile dementia with spastic paralysis. Brain 67:38–43. doi:10.1093/brain/67.1.38
Yamada M (2002) Risk factors for cerebral amyloid angiopathy in the elderly. Ann N Y Acad Sci 977:37–44
Yamada M, Itoh Y, Shintaku M, Kawamura J, Jensson O, Thorsteinsson L, Suematsu N, Matsushita M, Otomo E (1996) Immune reactions associated with cerebral amyloid angiopathy. Stroke 27:1155–1162
Yazaki M, Liepnieks JJ, Kincaid JC, Benson MD (2003) Contribution of wild-type transthyretin to hereditary peripheral nerve amyloid. Muscle Nerve 28:438–442. doi:10.1002/mus.10452
Zhang-Nunes SX, Maat-Schieman ML, van Duinen SG, Roos RA, Frosch MP, Greenberg SM (2006) The cerebral beta-amyloid angiopathies: hereditary and sporadic. Brain Pathol 16:30–39. doi:10.1111/j.1750-3639.2006.tb00559.x
Zlokovic BV (2002) Vascular disorder in Alzheimer’s disease: role in pathogenesis of dementia and therapeutic targets. Adv Drug Deliv Rev 54:1553–1559. doi:10.1016/S0169-409X(02)00150-3
Zlokovic BV, Ghiso J, Mackic JB, McComb JG, Weiss MH, Frangione B (1993) Blood-brain barrier transport of circulating Alzheimer’s amyloid beta. Biochem Biophys Res Commun 197:1034–1040. doi:10.1006/bbrc.1993.2582
Zlokovic BV, Martel CL, Matsubara E, McComb JG, Zheng G, McCluskey RT, Frangione B, Ghiso J (1996) Glycoprotein 330/megalin: probable role in receptor-mediated transport of apolipoprotein J alone and in a complex with Alzheimer disease amyloid beta at the blood-brain and blood-cerebrospinal fluid barriers. Proc Natl Acad Sci USA 93:4229–4234. doi:10.1073/pnas.93.9.4229
Zlokovic BV, Yamada S, Holtzman D, Ghiso J, Frangione B (2000) Clearance of amyloid beta-peptide from brain: transport or metabolism? Nat Med 6:718–719. doi:10.1038/77397
Author information
Authors and Affiliations
Corresponding author
Additional information
T. Revesz and J. L. Holton have contributed equally to this work.
An erratum to this article can be found at http://dx.doi.org/10.1007/s00401-009-0555-7
Rights and permissions
About this article
Cite this article
Revesz, T., Holton, J.L., Lashley, T. et al. Genetics and molecular pathogenesis of sporadic and hereditary cerebral amyloid angiopathies. Acta Neuropathol 118, 115–130 (2009). https://doi.org/10.1007/s00401-009-0501-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-009-0501-8