
Abstract
Recent studies reported both positive [Thal et al. (2003) J Neuropathol Exp Neurol 62:1287–1301] and negative [Tian et al. (2003) Neurosci Lett 352:137–140] correlations between cerebral amyloid angiopathy (CAA) and Alzheimer’s disease (AD) pathology. We have recently shown high correlations between neuritic AD pathology and amyloid β peptide (Aβ) deposits in the capillary/pericapillary compartment (CapCAA) with only low correlations to general CAA (non-capillary). We have now studied the relationship between CapCAA and AD pathology with respect to the distribution of Aβ40 and 42 in the frontal cortex of 100 human postmortem brains from both male and female, demented and non-demented patients (mean age ± SD 84.3±9.3 years). Using polyclonal antibodies to Aβ40 and 42, capillary and plaques positivity were assessed semiquantiatively on a four-point scale. Aβ42 deposits in capillaries correlated highly with both Aβ42 deposits in plaques and morphological AD criteria (CERAD, Braak stages, and NIA-Reagan-Institute criteria), while only a low correlation with CAA was observed. Aβ40 deposits in capillaries differed morphologically from Aβ42 ones: they were limited to capillary walls, were significantly less frequent in both capillaries and plaques compared to Aβ42 (P<0.01), and showed a low correlation with morphological AD criteria (P<0.05) and general CAA (P<0.01). By contrast, Aβ42 deposits were seen in the glia limitans rather than in capillary walls themselves, and showed high correlation with morphological AD criteria (P<0.01). These data indicate that CapCAA is characterized by Aβ42 deposits in pericapillary spaces or in the glia limitans. A low correlation between CAA and CapCAA, but high correlations between morphological AD criteria and CapCAA suggest different pathomechanisms for both types of CAA, and a close relation between CapCAA and AD pathology (both neuritic and plaque type). These data support the concept of a neuronal origin of Aβ via drainage from interstitial fluid from the central nervous system along basement membranes to capillaries.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The pathological hallmarks of sporadic late onset Alzheimer’s disease (AD), the most common cause of dementia in the elderly, include extracellular deposition of β-amyloid peptide (Aβ) in the neuropil (senile plaques, SPs) and in the cerebral vasculature (cerebral amyloid angiopathy, CAA), and neuritic cytoskeletal lesions with deposition of hyperphosphorylated microtubule-associated tau protein in neurons and dendrites, as neurofibrillary tangles (NFTs) and neuropil threads (NTs), and around amyloid deposits as neuritic plaques (NPs, [10]). The neuropathological diagnostic criteria for AD is the (semi)quantitative assessment and topographical staging of NPs and NFTs [4, 13, 24].
CAA is defined as deposition of a congophilic material in meningeal and cerebral arteries, arterioles, capillaries and veins, representing deposition of Aβ in the vessel walls [17, 29, 46]. It is a common finding in the brains of elderly demented and non-demented individuals; its incidence and severity increase with age [8, 11, 17, 23, 30, 31, 32, 38, 44], and it is associated with cerebral hemorrhages, infarctions, and white matter lesions [5, 17, 26, 27, 28, 29, 30, 31, 32, 37a, 38] (for review see [31]). The prevalence of CAA in AD patients, according to different studies, varies from 70% to 100% [2, 3, 9, 11, 17, 37, 37a, 38, 39, 40, 45].
By immunohistochemistry, two forms of Aβ can be distinguished. Aβ terminating at amino acid position 40 (Aβ40) and at position 42 or 43 (Aβ42). In CAA, Aβ40 affects vessel walls more frequently and more severely than Aβ42 [7, 12, 14, 15, 16, 18, 20, 22]. In SPs and NPs, however, Aβ42 is predominant and Aβ40 is rarely detected [12, 14, 15, 16, 21, 34].
The relationship between CAA and AD is poorly understood and the origin of Aβ in CAA remains unclear. Basically, three different mechanisms have been proposed (see [31]): (1) derivation of Aβ from blood and/or cerebrospinal fluid [19, 30, 47]; (2) production of Aβ by smooth muscle cells within vessel walls and/or pericytes [1, 25, 30]; or (3) derivation of Aβ from the neuropil (i.e., SP, NP), in the course of its perivascular drainage [6, 29, 33, 41, 42, 43].
Reports focusing on capillary involvement in CAA (CapCAA) showed that Aβ is present as globular deposits on the capillary wall and as linear thin layers in the pericapillary basement membrane, often with dyshoric Aβ deposits in the adjacent neuropil [29, 35, 39, 42]. CapCAA is thought to be related to AD-related Aβ deposition in the brain parenchyma, thus supporting the concept of derivation of Aβ from the neuropil in CAA (see above) [2, 35]. It is still uncertain, however, whether CapCAA represents a distinct type of CAA or is the result of coincidental CAA and AD-related parenchymal β-amyloidosis, or whether it represents an end stage of CAA [35].
Examining 19 different regions from 52 human aged brains, Thal et al. [36] reported that vascular Aβ deposition was accompanied by parenchymal plaques in most instances, and only in a few cases was CAA seen in the absence of Aβ deposits in a given area; only two demented and several non-demented individuals exhibited Aβ plaques in the complete absence of CAA. On the other hand, Tian et al. [37] recently reported a negative association between Aβ plaques and CAA in AD. In line with the latter findings, we recently showed that CAA grading (of non-capillary vasculature) did not correlate with neuritic AD pathology, while the prevalence and severity of CapCAA correlated significantly at a medium degree with the presence of high-grade AD pathology and at a high degree with CERAD, Braak, and NIA-Reagan-Institute (NIA-RI) criteria, respectively, suggesting that CapCAA has pathomechanisms different from those of CAA [2]. In this study, however, we did not discriminate between Aβ40 and Aβ42 distribution in both vessels and plaques.
Consequently, the aim of the present study was to investigate the distribution of Aβ40 and Aβ42 in leptomeningeal and intracortical arteries/arterioles, cortical capillaries and plaques to detect possible differences in the correlation between CAA, CapCAA, and AD plaque pathology with respect to different patterns of Aβ deposits.
Material and methods
We investigated 100 human brains obtained at autopsy from both genders aged 60–100 years (mean age ± SD 84.3±9.3 years), with a clinical diagnosis of dementia in 64 patients. Eight of the 36 non-demented (22.2%) and 42 of demented (65.6%) individuals showed high grade AD pathology (i.e., CERAD B, C; Braak V, VI; NIA-RI high probability [4, 13, 24]).
Our cohort consisted of 50 brains each with high- and low-grade or negative AD pathology; details were described previously [2] (Table 1).
Tissue was fixed in a 8% aqueous solution of formaldehyde, blocks were taken from the cerebral cortex (frontal, temporal, parietal, and occipital lobe), the hippocampus with adjacent entorhinal cortex and amygdala, basal ganglia, brainstem, and cerebellum. Paraffin sections, 4 μm thick, were stained with hematoxylin and eosin, Klüver-Barrera, and cresyl violet. For assessment of AD-related pathology, blocks from frontal and temporal cortex and hippocampus with adjacent entorhinal cortex were stained with modified Bielschowsky silver stain and incubated with commercially available monoclonal mouse anti-human paired helical filament-tau antibody (AT8) according to the manufacturer’s directions (Innogenetics, Ghent, Belgium). Neuropathological assessment of AD was performed using CERAD, Braak scores and NIA-RI criteria (see [2]).
For detection of Aβ40 and Aβ42 in cerebral vessels and brain parenchyma, adjacent sections from the frontal lobe were immunostained with commercially available rabbit polyclonal antibody Aβ 1–40, which is specific for the C terminus of Aβ40, and Aβ 1–42, which is specific for the C terminus of Aβ42, respectively, according to the manufacturer’s directions (Signet Laboratories, Dedham, MA).
Evaluation of Aβ40 and Aβ42
The total number of leptomeningeal vessels in a given slide differed from case to case; we therefore calculated percentages as follows. All leptomeningeal vessels were counted in each slide. All leptomeningeal vessels positive for Aβ40 were counted, discriminating between total and partial vessel wall positivity. The percentage of leptomeningeal Aβ40-positive vessels per slide was calculated (Aβ40L). To evaluate optimally the severity of Aβ40 deposits, the number of vessels with Aβ in only parts of the vessel wall was multiplied by 0.5 and added to the number of vessels with involvement of the whole vessel wall. The resulting number was used to calculate again the percentage of Aβ40 positivity (Aβ40Lsev). The same procedure was performed with Aβ42-positive leptomeningeal vessels (Aβ42L, Aβ42Lsev).
The total number of cortical vessels in a given slide depends on the size of the specimen, which was approximately the same in every case. Therefore, we counted affected vessels as follows. Cortical arteries/arterioles (not capillaries) positive for Aβ40 were counted in each slide, discriminating between vessels with total and partial Aβ deposits. The total sum of Aβ40-positive vessels was calculated (Aβ40C), and, to evaluate the severity of Aβ40 affection, the number of vessels with partial vessel wall positivity was multiplied with 0.5 and added to the number of vessels with total vessel wall positivity (Aβ40Csev). The same procedure was performed with Aβ42 positive cortical vessels (Aβ42C, Aβ42Csev).
To combine Aβ40Lsev, Aβ40Csev, Aβ42Lsev, and Aβ42Csev the following gradings were used: Aβ40Lsev and Aβ42Lsev were graded on a four-point scale, with 0, absent; 1, 1−20%; 2, 1–40%; 3, >40%, leading to an Aβ40 leptomeningeal score (Aβ40LS) and an Aβ42 leptomeningeal score (Aβ42LS), respectively. Aβ40Csev and Aβ42Csev were also graded on a four-point scale, with 0, absent; 1, 1–5; 2, 6–20; 3, >20, leading to an Aβ40 cortical score (Aβ40CS) and an Aβ42 cortical score (Aβ42CS), respectively. Mean values of Aβ40LS+Aβ40CS, Aβ42LS+Aβ42CS, Aβ40LS+Aβ42LS, and Aβ40CS+Aβ42SC lead to Aβ40, Aβ42, leptomeningeal, and cortical total scores (Aβ40TS, Aβ42TS, AβLTS, AβCTS). Mean values of Aβ40TS+Aβ42TS and AβLTS+AβCTS were equal and each lead to CAA total score (CAATS).
Aβ40 and Aβ42 capillary positivity were assessed semiquantitatively, by two of the authors independently, by counting affected vessels in ten high-power fields with ×400 magnification (HPF). The average of both counts for each section was calculated, and the severity of CapCAA was graded as follows: 0, no affected capillary; 1, less than 1 affected capillary/HPF; 2, one to two affected capillaries; 3, more than two affected capillaries/HPF. This resulted in an Aβ40 and an Aβ42 capillary score (Aβ40CapS, Aβ42CapS).
Aβ40 and Aβ42 plaque positivity were assessed semiquantitatively on a four-point scale: 0, absent; 1, mild; 2, moderate; 3, severe, resulting in an Aβ40 and an Aβ42 plaque score (Aβ40PS, Aβ42PS).
Apolipoprotein E genotyping in this sample is in progress (Attems and Jellinger, in preparation).
Statistics
The Mann-Whitney U test was used to test for differences in the prevalence and severity of each Aβ40 and Aβ42 between cases with high-grade AD pathology/low-grade or no AD pathology, and between clinically demented and non-demented individuals, and the Wilcoxon test for differences between the scores. The Spearman’s rank correlation coefficient was calculated for all correlations.
Results
Morphology of Aβ40 and Aβ42 deposits
No qualitative differences in the morphology of either Aβ40 or Aβ42 deposits in leptomeningeal and cortical arterial vessels was observed (Fig. 1a, b), but Aβ40 deposition was much heavier in the vessels. The overall morphology was in concordance with published criteria of CAA morphology [31, 35, 38, 40, 46] (Fig. 2a–c). Severe CAA expressing both Aβ40 and 42 was observed in some brains without any detectable plaques (Fig. 2a, b). Partial vessel wall positivity ranged from only focal to almost total. Therefore, multiplying the number of these vessels with 0.5, to achieve a better evaluation of overall severity seemed appropriate.

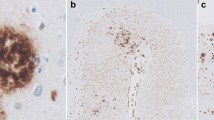
a Aβ40-positive leptomeningeal and cortical arteries; no decoration of plaques. b Aβ42-positive meningeal and cortical arteries and multiple plaques. a, b Frontal cortex, ×100

CAA. a, b CAA of meningeal and cortical vessels without detectable plaques. c Higher magnification of meningeal and cortical CAA. Immunostaining with Aβ against Aβ40 (a, c) and Aβ42 (b) (CAA cerebral amyloid angiopathy)
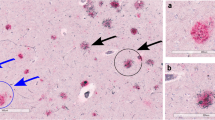
In capillaries, Aβ40 was present as strong, continuous deposits in the capillary wall with little or no association with dyshoric changes or plaques, whereas Aβ42 was present as globular deposits and linear thin layers particularly in the glia limitans and in the perivascular basement membrane often with dyshoric changes (Fig. 3a–e).

CapCAA and amyloid plaques. Immunostaining with Aβ against Aβ40 (a) and Aβ42 (b–e) (CapCAA capillary CAA). a, e ×1,000; b, d ×400; e ×600
The morphology of Aβ40 and Aβ42 deposits in plaques was in concordance with published data on the range of Aβ plaque morphology [10] (Fig. 3b, d).
Distribution of Aβ40 and Aβ42 in vessels and plaques
The distribution of Aβ40 and Aβ42 in vessels and plaques is shown in Table 2. To validate the reliability of Aβ40LS, Aβ40CS, Aβ42LS, and Aβ42CS, the corresponding variables Aβ40L/Aβ40Lsev/Aβ40LS, Aβ40C/Aβ40Csev/Aβ40CS, Aβ42L/Aβ42Lsev/Aβ42LS, and Aβ42C/Aβ42Csev/Aβ42CS were correlated, and all showed very high positive correlations (ρ>0.985, P<0.01). We thus used these scores for all further analysis.
Aβ40LS and Aβ42LS showed no significant difference (mean Aβ40LS 0.91±1.093, mean Aβ42LS 0.71±0.924; P>0.01) and highly correlated with each other (ρ=0.828, P<0.01), Aβ40CS was significantly higher than Aβ42CS (mean Aβ40CS 0.85±1.132, mean Aβ42CS 0.85±0.966; P<0.01) and highly correlated with Aβ42CS (ρ=0.725, P<0.01). No significant difference and high correlations were seen between Aβ40LS and Aβ40CS/Aβ42LS and Aβ42CS (ρ=0.728, ρ=0.712, P<0.01).
Aβ40TS was significantly higher than Aβ42TS (mean Aβ40TS 0.88±1.035, mean Aβ42TS 0.65±0.871; P<0.01) and highly correlated with Aβ42TS (ρ=0.819, P<0.01). AβLTS and AβCTS showed no significant differences (mean AβLTS 0.81±0.942, mean AβCTS 0.715±0.975) and highly correlated with each other (ρ=0.755, P<0.01).
Aβ42CapS and Aβ42PS were significantly higher than Aβ40CapS and Aβ40PS, respectively (mean Aβ40CapS 0.15±359, mean Aβ42CapS 2.02±1.279; P<0.01; mean Aβ40PS 0.19±0.456, mean Aβ42PS 2.12±1.241; P<0.01). Low correlations were seen between Aβ40CapS and Aβ42CapS/Aβ40PS and Aβ42PS (ρ=0.260/ρ=0.275, P<0.01), a high correlation between Aβ40CapS and Aβ40PS (ρ=0.855, P<0.01), and a very high correlation between Aβ42CapS and Aβ42PS (ρ=0.946, P<0.01).
Aβ40CapS, Aβ42CapS, Aβ40PS, and Aβ42PS showed no to low positive correlations with each Aβ40TS and Aβ42TS respectively (ρ<0.5, P<0.05), with the exception of a medium correlation between Aβ40PS and Aβ40TS (ρ=0.511, P<0.01).
Comparison of Aβ40 and Aβ42 with AD pathology and clinical data
Aβ40 deposits were present in leptomeningeal vessels/cortical arteries-arterioles/capillaries/plaques in 32 (64.0%)/30 (60.0%)/12 (24%)/14 (28%) of cases with high-grade AD pathology, and in 32 (50.0%)/31 (48.4%)/12 (18.8%)/15 (23.4%) individuals with clinically overt dementia, with prevalence of Aβ40 deposits in leptomeningeal vessels and cortical arteries/arterioles (Fig. 2c). The number of plaques was significantly higher in cases with high-grade AD pathology compared to those with no or low-grade AD pathology (P<0.01).
Aβ42 deposits were present in leptomeningeal vessels/cortical arteries-arterioles/capillaries/plaques in 26 (52.0%)/17 (34.0%)/50 (100%)/50 (100%) of cases with high-grade neuritic AD pathology and in 27 (42.2%)/19 (29.7%)/56 (87%)/57 (89%) individuals with clinically overt dementia. There was a prevalence of Aβ42 deposits in capillaries and plaques, being significantly higher in both cases with high-grade AD pathology than in those with no or low-grade AD lesions, and in subjects with clinically overt dementia compared to non-demented ones (P<0.01). The presence of Aβ42 in/at capillaries and in plaques showed medium positive correlations with high-grade neuritic AD pathology (ρ=0.531/ρ=0.562, P<0.01).
Aβ40LS, Aβ40CS, Aβ40TS, Aβ40PS, Aβ42CapS, and Aβ42PS were significantly higher in cases with high-grade AD pathology compared to those with no or low-grade AD lesions (P<0.01). Aβ42CapS and Aβ42PS were significantly higher in individuals with clinically overt dementia compared to non-demented subjects (P<0.01).
Correlation with CERAD, Braak stages, and NIA-RI criteria
We correlated all CAA/CapCAA scores with CERAD, Braak stages, and NIA-RI criteria (Fig. 4a–c); high positive correlations were seen between each Aβ42CapS and Aβ42PS and each of the morphological AD criteria (ρ>0.76, P<0.01), and low positive correlations between each Aβ40LS, Aβ40CS, Aβ40TS, AβLTS, AβCTS, CAATS, Aβ40CapS, and Aβ40PS and all three AD criteria (0.2<ρ<0.5, P<0.05). When controlling the positive correlation between each AβLTS, AβCTS, and CAATS and the three morphological AD criteria for Aβ40LS and Aβ40CS, no positive correlation was seen, reflecting the influence of Aβ40LS and Aβ40CS on each AβLTS, AβCTS, and CAATS. This was also true for Aβ40TS as it represents the mean value of Aβ40LS+Aβ40CS.

Correlations between CAA and CapCAA with CERAD and Braak scores, and NIA-RI criteria for Aβ. Italic figures: number of cases
Discussion
The present study clearly indicates that the severity of Aβ42 deposits in/at capillaries (i.e., Aβ42CapS) significantly correlates with the severity of neuritic AD pathology using CERAD, Braak stages, and NIA-RI criteria, and to a very high degree with the severity of Aβ42 deposits in plaques (i.e., Aβ42PS). The severity of Aβ40 in both leptomeningeal and cortical arterial vessels (i.e., Aβ40LS, Aβ40CS, Aβ40TS) increased significantly with increasing CERAD, Braak stages, and NIA-RI criteria; however, only low positive correlations between non-capillary CAA and AD pathology were observed. The very high correlation between Aβ42CapS and Aβ42PS in the present study, supports the concept of perivascular drainage of Aβ, with Aβ entering the perivascular pathways at the level of capillaries, leading to (peri)capillary Aβ deposition and consequently to CapCAA [2, 29, 41, 42, 43]. Although the flow of interstitial fluid within the perivascular space occurs in the opposite direction to that of the arterial blood flow, it may be enhanced by the pulsatile arterial distension. A failure of this propulsive mechanism has been proposed to explain the association of capillary CAA with thrombosis of overlying cortical arteries. It is widely accepted, however, that CapCAA only occurs in the presence of CAA, and thus may be a part of CAA [6, 35]. This assumption, however, is contrasted by our findings, since Aβ42CapS showed no correlation with Aβ42TS and only low correlation with Aβ40TS. This is reflected by the fact that in 17 cases of the present cohort with high Aβ42CapS, no Aβ deposits were detected in leptomeningeal and cortical arterial vessels (Fig. 5a, b), whereas severe CAA was present in a few brains without any plaques detectable by either Aβ40 or 42 immunohistochemistry (Fig. 2a, b). The low correlation of Aβ42PS with each Aβ40TS and Aβ42LS, and the lack of a correlation with Aβ42CS indicates that general CAA is not a result of perivascular draining of Aβ. This is in line with a previously observed inverse relation between the overall severity of CAA and parenchymal Aβ load in patients with moderate to severe AD [41].

Cortical plaques without detectable CAA in meningeal and cortical vessels. Immunostaining with Aβ against Aβ40 (a) and Aβ42 (b). a, b ×100
Aβ40 deposits in capillary walls were rarely detected and morphologically differed from capillary Aβ42 deposits (see results, Fig. 3), and, albeit low correlation with Aβ40TS (ρ=0.490, P<0.01), was almost exclusively seen in cases with high Aβ40TS. These findings indicate that Aβ40 deposits in capillaries are a sign of extensive CAA and are not associated with Aβ42CapCAA.
It was previously reported that Aβ40 is more frequent and more severe than Aβ42 in CAA [7, 12, 14, 15, 16, 20, 22]. This is in concordance with our findings. In addition, we observed high correlations between Aβ40TS and Aβ42TS, suggesting that both Aβ40 and Aβ42 contribute to Aβ deposition in CAA.
Our findings of low/no correlations between Aβ40TS/Aβ42TS and neuritic AD pathology argue against the possibility of one common pathomechanism for both AD pathology and non-capillary CAA. Since we only used sections from to frontal cortex to evaluate CAA, this could have led to the lower prevalence and severity of CAA in our cohort, thus biasing the results. On the other hand, we observed high CAATS in the complete absence of AD pathology, suggesting different pathomechanisms for AD and CAA. The presence of CAA could, under yet unknown additional influences, have a promoting effect on AD pathology, which is reflected by the significant increase of Aβ40TS with increasing neuritic AD lesions in our cohort and the significant association between CAA and AD pathology in previous studies [35, 36]. Support for this comes from the occasional finding of tau immunopositive neurites clustered around larger arteries with dyshoric CAA (angiopathy in which amyloid extends from the affected blood vessels into the surrounding brain parenchyma) [18].
In conclusion our results suggest that:
-
CAA is characterized by Aβ40/Aβ42 deposits in leptomeningeal and cortical arterial vessels, with Aβ40 being more frequent and more severe. Involvement of capillaries is very rare and is considered to represent an indicator of high-grade CAA. CAA (together with other influences) possibly promotes AD pathology.
-
By contrast, CapCAA is characterized by globular Aβ42 deposits entrapped in the glia limitans of cortical capillaries and in pericapillary compartments, often in conjunction with parenchymal Aβ42 deposits. It is presumably a result of Aβ42 drainage from SPs and NPs along basement membranes, i.e. perivascular drainage, and, thus, closely related to both plaques and neurofibrillary AD pathology, but not to general (non-capillary) CAA.
Abbreviations
- AD:
-
Alzheimer disease
- Aβ:
-
beta amyloid peptide
- Aβ 40/42 CapS:
-
score of deposits of Aβ 1–40/42 in capillaries
- Aβ 40/42 C:
-
number of Aβ 1–40/42 positive cortical vessels
- Aβ 40/42 PS:
-
score of deposits of Aβ 1–40/42 in plaques
- Aβ 40/42 TS:
-
total score of Aβ 1–40/42 deposits
- Aβ 40/42 Csev:
-
severity of Aβ 1–40/42 affection of cortical vessels
- Aβ 40/42 CS:
-
Aβ 1–40/42 cortical score
- Aβ 40/42 L:
-
percentage of Aβ 40/42 positive leptomeningeal vessels
- Aβ 40/42 Lsev:
-
severity of Aβ 40/42 affection of leptomeningeal vessels
- Aβ 40/42 LS:
-
Aβ 40/42 leptomeningeal score
- AβCTS:
-
Aβ cortical total score
- AβLTS:
-
Aβ leptomeningeal total score
- CAA:
-
cerebral amyloid angiopathy
- CAATS:
-
CAA total score
- CapCAA:
-
capillary CAA
- CERAD:
-
Consortium to Establish a Registry of Alzheimer’s Disease
- NFT:
-
neurofibrillary tangle
- NIA:
-
National Institute of Aging
- NIA-RI:
-
National Institute of Aging and Reagan Institute
- NP:
-
neuritic plaque
- SP:
-
senile plaque
- TS:
-
total score
References
Alonzo NC, Hyman BT, Rebeck GW, Greenberg SM (1998) Progression of cerebral amyloid angiopathy: accumulation of amyloid-beta40 in affected vessels. J Neuropathol Exp Neurol 57:353–359
Attems J, Jellinger KA (2004) Only cerebral capillary amyloid angiopathy correlates with Alzheimer pathology—a pilot study. Acta Neuropathol 107:83–90
Bergeron C, Ranalli PJ, Miceli PN (1987) Amyloid angiopathy in Alzheimer’s disease. Can J Neurol Sci 14:564–569
Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82:239–259
Cadavid D, Mena H, Koeller K, Frommelt RA (2000) Cerebral beta amyloid angiopathy is a risk factor for cerebral ischemic infarction. A case control study in human brain biopsies. J Neuropathol Exp Neurol 59:768–773
Calhoun ME, Burgermeister P, Phinney AL, Stalder M, Tolnay M, Wiederhold KH, Abramowski D, Sturchler-Pierrat C, Sommer B, Staufenbiel M, Jucker M (1999) Neuronal overexpression of mutant amyloid precursor protein results in prominent deposition of cerebrovascular amyloid. Proc Natl Acad Sci USA 96:14088–14093
Castano EM, Prelli F, Soto C, Beavis R, Matsubara E, Shoji M, Frangione B (1996) The length of amyloid-beta in hereditary cerebral hemorrhage with amyloidosis, Dutch type. Implications for the role of amyloid-beta 1–42 in Alzheimer’s disease. J Biol Chem 271:32185–32191
Chalmers K, Wilcock GK, Love S (2003) APOE epsilon 4 influences the pathological phenotype of Alzheimer’s disease by favouring cerebrovascular over parenchymal accumulation of A beta protein. Neuropathol Appl Neurobiol 29:231–238
Dermaut B, Kumar-Singh S, De Jonghe C, Cruts M, Lofgren A, Lubke U, Cras P, Dom R, De Deyn PP, Martin JJ, Van Broeckhoven C (2001) Cerebral amyloid angiopathy is a pathogenic lesion in Alzheimer’s disease due to a novel presenilin 1 mutation. Brain 124:2383–2392
Duyckaerts C, Dickson DW (2003) Neuropathology of Alzheimer’s disease. In: Dickson DW (ed) Neurodegeneration. The molecular pathology of dementia and movement disorders. ISN Neuropath Press, Basel, pp 47–65
Ellis RJ, Olichney JM, Thal LJ, Mirra SS, Morris JC, Beekly D, Heyman A (1996) Cerebral amyloid angiopathy in the brains of patients with Alzheimer’s disease: the CERAD experience, Part XV. Neurology 46:1592–1596
Gravina SA, Ho L, Eckman CB, Long KE, Otvos L Jr, Younkin LH, Suzuki N, Younkin SG (1995) Amyloid beta protein (A beta) in Alzheimer’s disease brain. Biochemical and immunocytochemical analysis with antibodies specific for forms ending at A beta 40 or A beta 42(43). J Biol Chem 270:7013–7016
Hyman BT (1998) New neuropathological criteria for Alzheimer disease. Arch Neurol 55:1174–1176
Iwatsubo T, Odaka A, Suzuki N, Mizusawa H, Nukina N, Ihara Y (1994) Visualization of A beta 42(43) and A beta 40 in senile plaques with end-specific A beta monoclonals: evidence that an initially deposited species is A beta 42(43). Neuron 13:45–53
Iwatsubo T, Mann DM, Odaka A, Suzuki N, Ihara Y (1995) Amyloid beta protein (A beta) deposition: A beta 42(43) precedes A beta 40 in Down syndrome. Ann Neurol 37:294–299
Iwatsubo T, Saido TC, Mann DM, Lee VM, Trojanowski JQ (1996) Full-length amyloid-beta (1–42(43)) and amino-terminally modified and truncated amyloid-beta 42(43) deposit in diffuse plaques. Am J Pathol 149:1823–1830
Jellinger KA (2002) Alzheimer disease and cerebrovascular pathology: an update. J Neural Transm 109:813–836
Love S (2004) Contribution of cerebral amyloid angiopathy to Alzheimer’s disease. J Neurol Neurosurg Psychiatry 75:1–4
Mackic JB, Weiss MH, Miao W, Kirkman E, Ghiso J, Calero M, Bading J, Frangione B, Zlokovic BV (1998) Cerebrovascular accumulation and increased blood-brain barrier permeability to circulating Alzheimer’s amyloid beta peptide in aged squirrel monkey with cerebral amyloid angiopathy. J Neurochem 70:210–215
Mak K, Yang F, Vinters HV, Frautschy SA, Cole GM (1994) Polyclonals to beta-amyloid(1–42) identify most plaque and vascular deposits in Alzheimer cortex, but not striatum. Brain Res 667:138–142
Mann DM, Iwatsubo T (1996) Diffuse plaques in the cerebellum and corpus striatum in Down’s syndrome contain amyloid beta protein (A beta) only in the form of A beta 42(43). Neurodegeneration 5:115–120
Mann DM, Iwatsubo T, Ihara Y, Cairns NJ, Lantos PL, Bogdanovic N, Lannfelt L, Winblad B, Maat-Schieman ML, Rossor MN (1996) Predominant deposition of amyloid-beta 42(43) in plaques in cases of Alzheimer’s disease and hereditary cerebral hemorrhage associated with mutations in the amyloid precursor protein gene. Am J Pathol 148:1257–1266
Maruyama K, Ikeda S, Ishihara T, Allsop D, Yanagisawa N (1990) Immunohistochemical characterization of cerebrovascular amyloid in 46 autopsied cases using antibodies to beta protein and cystatin C. Stroke 21:397–403
Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, Belle G van, Berg L (1991) The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 41:479–486
Natte R, Boer WI de, Maat-Schieman ML, Baelde HJ, Vinters HV, Roos RA, Duinen SG van (1999) Amyloid beta precursor protein-mRNA is expressed throughout cerebral vessel walls. Brain Res 828:179–183
Oide T, Takahashi H, Yutani C, Ishihara T, Ikeda S (2003) Relationship between lobar intracerebral hemorrhage and leukoencephalopathy associated with cerebral amyloid angiopathy: clinicopathological study of 64 Japanese patients. Amyloid 10:136–143
Olichney JM, Hansen LA, Hofstetter CR, Grundman M, Katzman R, Thal LJ (1995) Cerebral infarction in Alzheimer’s disease is associated with severe amyloid angiopathy and hypertension. Arch Neurol 52:702–708
Olichney JM, Ellis RJ, Katzman R, Sabbagh MN, Hansen L (1997) Types of cerebrovascular lesions associated with severe cerebral amyloid angiopathy in Alzheimer’s disease. Ann N Y Acad Sci 826:493–497
Preston SD, Steart PV, Wilkinson A, Nicoll JA, Weller RO (2003) Capillary and arterial cerebral amyloid angiopathy in Alzheimer’s disease: defining the perivascular route for the elimination of amyloid beta from the human brain. Neuropathol Appl Neurobiol 29:106–117
Revesz T, Holton JL, Lashley T, Plant G, Rostagno A, Ghiso J, Frangione B (2002) Sporadic and familial cerebral amyloid angiopathies. Brain Pathol 12:343–357
Revesz T, Ghiso J, Lashley T, Plant G, Rostagno A, Frangione B, Holton JL (2003) Cerebral amyloid angiopathies: a pathologic, biochemical, and genetic view. J Neuropathol Exp Neurol 62:885–898
Revesz T, Ghiso J, Plant G, Holton JL, Frangione B (2003) Inherited amyloidosis and neurodegenerations in Familial British and Danish Dementia. In: Dickson DW (ed) Neurodegeneration. The molecular pathology of dementia and movement disorders. ISN Neuropath Press, Basel, pp 380–385
Roher AE, Kuo YM, Esh C, Knebel C, Weiss N, Kalback W, Luehrs DC, Childress JL, Beach TG, Weller RO, Kokjohn TA (2003) Cortical and leptomeningeal cerebrovascular amyloid and white matter pathology in Alzheimer’s disease. Mol Med 9:112–122
Saido TC, Iwatsubo T, Mann DM, Shimada H, Ihara Y, Kawashima S (1995) Dominant and differential deposition of distinct beta-amyloid peptide species, A beta N3(pE), in senile plaques. Neuron 14:457–466
Thal DR, Ghebremedhin E, Rub U, Yamaguchi H, Tredici KD, Braak H (2002) Two types of sporadic cerebral amyloid angiopathy. J Neuropathol Exp Neurol 61:282–293
Thal DR, Ghebremedhin E, Orantes M, Wiestler OD (2003) Vacular pathology in Alzheimer disease: correlation of cerebral amyloid angiopathy and arteriosclerosis/lipohyalinosis with cognitive decline. J Neuropathol Exp Neurol 62:1287–1301
Tian J, Shi J, Bailey K, Mann DM (2003) Negative association between amyloid plaques and cerebral amyloid angiopathy in Alzheimer’s disease. Neurosci Lett 352:137–140
Tian J, Shi J, Bailey K, Mann DM (2004) Relationships between arteriosclerosis, cerebral amyloid angiopathy and myelin loss from cerebral cortical white matter in Alzheimer’s disease. Neuropathol Appl Neurobiol 30:46–56
Vinters HV (1987) Cerebral amyloid angiopathy. A critical review. Stroke 18:311–324
Vinters HV, Secor DL, Pardridge WM, Gray F (1990) Immunohistochemical study of cerebral amyloid angiopathy. III. Widespread Alzheimer A4 peptide in cerebral microvessel walls colocalizes with gamma trace in patients with leukoencephalopathy. Ann Neurol 28:34–42
Vonsattel JP, Myers RH, Hedley-Whyte ET, Ropper AH, Bird ED, Richardson EP Jr (1991) Cerebral amyloid angiopathy without and with cerebral hemorrhages: a comparative histological study. Ann Neurol 30:637–649
Weller RO, Nicoll JA (2003) Cerebral amyloid angiopathy: pathogenesis and effects on the ageing and Alzheimer brain. Neurol Res 25:611–616
Weller RO, Massey A, Newman TA, Hutchings M, Kuo YM, Roher AE (1998) Cerebral amyloid angiopathy: amyloid beta accumulates in putative interstitial fluid drainage pathways in Alzheimer’s disease. Am J Pathol 153:725–733
Weller RO, Massey A, Kuo YM, Roher AE (2000) Cerebral amyloid angiopathy: accumulation of A beta in interstitial fluid drainage pathways in Alzheimer’s disease. Ann N Y Acad Sci 903:110–117
Xu D, Yang C, Wang L (2003) Cerebral amyloid angiopathy in aged Chinese: a clinico-neuropathological study. Acta Neuropathol 106:89–91
Yamada M, Tsukagoshi H, Otomo E, Hayakawa M (1987) Cerebral amyloid angiopathy in the aged. J Neurol 234:371–376
Zekry D, Duyckaerts C, Belmin J, Geoffre C, Moulias R, Hauw JJ (2003) Cerebral amyloid angiopathy in the elderly: vessel walls changes and relationship with dementia. Acta Neuropathol 106:367–373
Zlokovic BV, Ghiso J, Mackic JB, McComb JG, Weiss MH, Frangione B (1993) Blood-brain barrier transport of circulating Alzheimer’s amyloid beta. Biochem Biophys Res Commun 197:1034–1040
Acknowledgements
The authors thank Mrs. Veronika Rappelsberger and Mrs. Barbara Weidinger for excellent laboratory work, Erich Mitter-Ferstl, PhD, for secretarial and computer work, and Prof. Roy Weller, Southampton, for critical remarks. Part of the study was supported by the Society for the Support of Research in the Field of Experimental Neurology and by Consultatio GmbH, both Vienna, Austria.
Author information
Authors and Affiliations
Corresponding author
Additional information
An erratum to this article can be found at http://dx.doi.org/10.1007/s00401-004-0866-7
Rights and permissions
About this article
Cite this article
Attems, J., Lintner, F. & Jellinger, K.A. Amyloid β peptide 1–42 highly correlates with capillary cerebral amyloid angiopathy and Alzheimer disease pathology. Acta Neuropathol 107, 283–291 (2004). https://doi.org/10.1007/s00401-004-0822-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-004-0822-6