
Abstract
Aberrant RAS/RAF signaling has been reported to be important for many tumor types including gliomas. Activation of the RAS/RAF pathway can result from oncogenic mutations of RAS/RAF itself. However, such mutations have only occasionally been reported in gliomas. In order to further elucidate the role of RAS/RAF pathway activation in a histopathological and genetic spectrum of glioma subtypes (n = 93), we evaluated different types of aberrations in this pathway. Hotspot mutation analysis of BRAF, NRAS, KRAS, and HRAS revealed only two mutations, V600M in BRAF and G10E in NRAS, both occurring in pure oligodendroglial tumors. However, CGH analysis of 87 tumors revealed copy number gains including the above mentioned oncogenes in 38 of the neoplasms (44%) and including the upstream growth factors EGF, PDGF, IGF, FGF, TGF and/or their receptors in 46 tumors (53%). Phosphorylated MAPK (i.e. the activated compound downstream the RAS/RAF pathway) was detected by immunohistochemistry using tissue micro-arrays in the majority of gliomas. Interestingly, a significant correlation was found for nuclear MAPK-P staining and the number of these copy number gains (≤ 2 and ≥ 3). These results indicate that RAS/RAF pathway activation in gliomas is achieved much more frequently by copy number gains including RAS/RAF and/or upstream growth factor (receptor) than by activating RAS/RAF mutations.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Gliomas form a heterogeneous group of tumors. In adult patients most of these can be classified based on their histopathological features as astrocytic, oligodendroglial, oligoastrocytic or ependymal tumors [17]. Additionally a malignancy grade is attributed to these gliomas based on histopathological features including nuclear atypia, mitotic activity, microvascular proliferation, and necrosis. Although histopathological classification is still the gold standard guiding therapy of glioma patients, it is increasingly clear that different genetic subtypes exist within these subgroups and that specific molecular genetic aberrations are of prognostic and therapeutic significance. Initially, molecular analysis revealed that genetic aberrations and pathways may differ between the different histopathological types: whereas a gain of chromosome 7(p) and a loss of chromosome 10(q) (+7/−10) are considered to be characteristic for glioblastoma multiforme (GBM; WHO grade IV astrocytoma), losses of 1p and 19q (−1p/−19q) are most frequently detected in oligodendrogliomas [11]. Subsequently, it was shown that different genetic subtypes exist even within the specific histopathological groups: secondary GBMs that develop via progression from a less malignant lesion frequently show p53 mutations, LOH 17p, and overexpression of PDGFR, while primary GBMs that occur without clinical evidence of a precursor lesion are often characterized by amplification or overexpression of EGFR (∼60%) ([18] and references therein). Similarly, different genetic subtypes have been described for oligodendroglial tumors, some of them showing loss of 1p and 19q (−1p/−19q), i.e. the aberrations considered to be characteristic for oligodendrogliomas, whereas others genetically show more resemblance to astrocytic tumors [12, 14, 16, 21, 31].
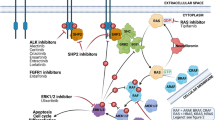
The RAS–RAF–MEK–ERK/MAPK pathway mediates cellular responses to growth signals, differentiation and programmed cell death (Fig. 1) [24]. Constitutive activation of this pathway has been described to occur through RAS and RAF mutations that affect only a few critical sites in these genes. Most BRAF mutations are located in exon 15 encoding the activation segment (which protects the substrate binding site) and to a lesser extent in exon 11 (which represents the glycine-rich P-loop mediated binding to ATP), whereas RAS mutations are detected in exons 2 and 3 affecting the GTP-binding domain. Mutation frequencies vary among the different tumor types [27, 29]. Moreover, it was reported for various tumor types that aberrations may only be detected in specific histopathological or genetic subtypes and that specific RAS and RAF mutations may be mutually exclusive [28, 29].

A schematic summary of the RAS/RAF pathway. Growth factor ligands (GF), including EGF, PDGF, TGFA, FGF, and IGF, bind to the extracellular portion of protein tyrosine kinase receptors (growth factor receptor, GF-R), inducing dimerization of two receptors and autophosphorylation of the catalytic domain tyrosine kinase residues. Receptor activation leads to activation of different downstream signaling pathways that promote proliferation, gene transcription and cell survival. For the RAS/RAF pathway, RAS is activated which in turn stimulates downstream effectors including RAF, MEK and ERK (=MAPK)
It has already been shown that the RAS pathway is important for glioma oncogenesis. In comparison with normal brain and low-grade astrocytomas, increased expression of RAS and high levels of RAS-GTP have been demonstrated in GBM cell-lines and tumor specimens [7, 10]. Furthermore, overexpression of HRAS in a transgenic mouse model resulted in multifocal astrocytomas [5]. The importance of increased RAS-activity in GBMs is further supported by studies demonstrating reduced growth of GBM cell lines when treated with dominant-negative RAS mutants or RAS pathway inhibitor drugs [10]. So far RAS/RAF mutations, however, have only occasionally been reported in gliomas [2–4, 19].
The goal of the present study was to provide a more comprehensive overview of the genetic background of RAS/RAF pathway activation in gliomas. As previous studies usually evaluated a specific RAS gene or a specific glioma type, we first performed mutation analysis of BRAF and all three oncogenic RAS genes (NRAS, HRAS and KRAS) in a spectrum of histopathologically and genetically different gliomas (n = 93). We secondly evaluated the presence of gains of the regions containing the RAF/RAS genes in these tumors by CGH analysis (n = 87) as this can also result in activation of the corresponding pathways. For example, performing a parallel analysis of DNA copy number changes and mRNA levels using micro-arrays it was shown for breast cancer that indeed DNA copy number alterations (including low-level amplifications (gains) as well as high-level amplifications) can lead directly to deregulation of gene expression [25]. Thirdly, as increased copy number of upstream growth factors and their receptors may have a comparable (activating) effect as RAS/RAF mutations or gains, the presence of gains carrying the genes for such growth factor(s) or their receptors was evaluated in these tumors by CGH analysis (n = 87). Fourth, immunohistochemistry for phosphorylated (phospho-p42/44) MAPK (MAPK-P), the activated, downstream compound of the RAS/RAF pathway, was performed on tissue micro-arrays (containing at least three cores of 60 of these glial tumors) to evaluate the impact of the aberrations. As for the majority of tumors the histopathological as well as genetic subtype were known, the distribution of the detected aberrations among the different glioma types was evaluated. While RAS/RAF pathway activation is considered important for glioma oncogenesis [2–5, 7, 10, 19], our results corroborate that RAS/RAF mutations are only occasionally present in the spectrum of glioma types (2/93). However, as copy number gains including these RAS/RAF genes and of the genes of upstream growth factors and their receptors (especially EFG(R) and PDGF(R)) were detected in approximately half of the cases (38/87 and 46/87, respectively), and nuclear MAPK-P expression significantly correlated with the number of such gains, our study indicates that copy number gains may frequently be responsible for RAS/RAF pathway activation in gliomas.
Material and methods
Tumor samples
Ninety-three glioma samples were retrieved from our neuro-oncology archive at the Department of Pathology of the Radboud University Nijmegen Medical Centre (RUNMC), The Netherlands. The use of brain tumor tissue after completing histopathological diagnosis for research purposes has been approved by the ethics committee of the RUNMC and informed consent was given by the patients. Tumors were classified according to the WHO-2000 classification [17] and included 9 diffuse astrocytomas (WHO grade II; As), 1 anaplastic astrocytoma (WHO grade III; a-A), 19 glioblastomas multiforme (WHO grade IV; GBMs), 7 ependymomas (WHO grade II; Es), 2 anaplastic ependymomas (WHO grade III; a-Es), 12 oligodendrogliomas (WHO grade II; Os), 11 anaplastic oligodendrogliomas (WHO grade III; a-Os), 16 oligoastrocytomas (WHO grade II; OAs) and 16 anaplastic oligoastrocytomas (WHO grade III; a-OAs).
DNA isolation
Sections (50 μm) of formalin-fixed and paraffin-embedded tumor tissue were collected in a tube and incubated in 125 μl P-buffer (50 mM Tris–HCl pH 8.2, 100 mM NaCl, 1 mM EDTA, 0.5% Tween-20, 0.5% NP40, 20 mM DTT) for 15 min at 90°C. Protein digestion was performed by adding proteinase K (Roche Diagnostics GBMH, Mannheim, Germany) (final concentration of 0.5 mg/ml) and incubation at 55°C for 24 h followed by incubation at 37°C for 48 h with addition of 5 μl fresh proteinase K (20 mg/ml) every 24 h. Subsequently, DNA was purified using the DNeasy tissue kit (Qiagen, Venlo, The Netherlands) and the DNA concentration was measured using the NanoDrop spectrophotometer (NanoDrop Technologies, Wilmington, USA).
RAS/RAF mutation analysis
Hotspot mutations in N-RAS and H-RAS genes are often reported to be located on exons 1 and 2 as these are the first two translated exons. As, however, there is an exon before the first translated exon which is transcribed into RNA but not translated into a protein (5′ untranslated region), the hotspot mutations are actually located in exons 2 and 3. In this paper, we therefore refer to these exons as exon 2 and 3. For BRAF mutations, different reports refer to one and the same hotspot mutation codon in exon 15 as either codon 599 or 600. The initial mutations reported by Davies et al. were mapped to the DNA sequence NM 004333[gi;4757867] [4]. In July 2003, however, this sequence was updated to NM 004333[gi;33188458] with the insertion of 3 bp in the coding sequence resulting in a new codon number (600 instead of 599). In this paper, we will therefore refer to this codon as codon 600.
For mutation analysis, B-RAF exon 11 and 15 [4], N-RAS exon 2 and 3 [4], H-RAS exon 2 and 3 [1], and K-RAS exon 2 and 3 [9] primers and protocols were used as previously described [29]. Primer sequences used are shown in Table 1. Briefly, PCR amplification was performed in a total volume of 20 μl containing at least 40 ng template DNA, buffer IV (Integro, Dieren, The Netherlands), 200 μm of each deoxynucleotide triphospate, 3 mM MgCl2, 0.5 pmol of each primer, 30 μg bovine serum albumin (BSA, Sigma-Aldrich Chemie BV, Zwijndrecht, The Netherlands) and 0.25 units of thermostable DNA polymerase (Integro). An initial denaturation at 94°C for 5 min was followed by 30 cycles of denaturation at 94°C for 45 s, annealing at 60°C for 45 s, and extension at 72°C for 45 s with a final extension of 5 min at 72°C. DNA amplification was performed in a PTC 200 thermal cycler (MJ Research, Inc. Waltham, MA, USA). PCR products were purified using the PCR Product Pre-Sequencing kit (USB Corporation, Cleveland, OH, USA). A measure of 1–2 μl of the PCR product was used for the sequence reaction, using the Big Dye terminator kit and ABI PRISM 3700 DNA analyzer (Applied Biosystems, Foster City, CA, USA). Sequencing of both strands was performed.
Copy number analysis
In gliomas screened for RAS/RAF mutations, 87 of the 93 were genetically characterized by conventional comparative genomic hybridization (CGH) detecting copy number changes (>2 Mb) as described previously [14–16]. Briefly, control and tumor DNA were labeled by nick-translation with digoxigenin-dUTP and biotin-dUTP, respectively (Roche Molecular Biochemicals, The Netherlands), and precipitated in the presence of human COT-1 DNA (Invitrogen, Paisley, UK) and herring sperm DNA. The probe and the metaphase slides were denatured simultaneously. After hybridization and post-hybridization washes, biotin and digoxigenin were detected using streptavidin-FITC and sheep-anti-digoxigenin-TRITC (Roche Molecular Biochemicals). The chromosomes were counterstained with 4,6′-diamino-2-phenylindole-dihydrochloride (DAPI) (Merck, Darmstadt, Germany) and the slides were mounted in Fluoroguard (Biorad, Veenendaal, The Netherlands). For CGH analysis Quips CGH software (Applied Imaging, UK) was used. Detection thresholds for losses and gains were set at 0.8 and 1.2, respectively.
Immunohistochemical detection of MAPK-P
Tissue micro-arrays (TMAs) were constructed including 60 of the 87 CGH characterized gliomas. Representative regions were identified and 3 to 4 cores (2 mm diameter) were included in the TMAs which were subsequently stained using a polyclonal antibody against phosphorylated MAPK (phosphor-p44/42 MAPK, clone 9101, Cell Signaling, Danvers, USA) generously provided by Dr. Jeroen EM van Leeuwen (Department of Cell Biology, Nijmegen Centre for Molecular Life Sciences (NCMLS), RUNMC Nijmegen, The Netherlands), as described by the manufacturer. Testing different antibodies, this clone was identified as reliable for the detection of phosphorylated MAPK when using formalin fixed and paraffin embedded tissue. Analysis was performed when at least 3 evaluable cores were present (n = 60). All cores were individually scored for extent of staining (extensive = +++, moderate = ++, focal = +, absent = −) as well as for staining intensity (strong = s, moderate = m, weak = w, or absent = −). Based on the 3–4 individual cores, an average score was established for each tumor. As MAPK-P can be present in both the nucleus and cytoplasm, cytoplasmic and nuclear staining were scored separately.
For statistical analysis, extent and intensity of immunohistochemical stainings were converted into numerical data ranging from 0 to 3 (0 = − or −, 1 = + or w, 2 = ++ or m, 3 = +++ or s, addressing extent and intensity, respectively). Subsequently, both scores were multiplied and the product was defined as the immunohistochemical score which was used for statistical analysis using the Mann-Whitney U test. Nuclear and cytoplasmic staining were evaluated separately and P-values below 0.05 were considered to be statistically significant.
Results
Mutations in BRAF, NRAS, HRAS, or KRAS
Mutation analysis was performed for both exons of BRAF, NRAS, HRAS and KRAS containing the hotspot mutation site in a total of 93 different gliomas. Only 2 mutations were detected, these were found in histologically pure oligodendroglial tumors harboring -1p and -19q. As shown in Fig. 2a, tumor N78 (O) contains a G→A mutation in the hotspot codon 600 of BRAF exon 15, resulting in the substitution of valine by methionine (V600M). The second mutation was detected in N303 (a-O; Fig. 2b) and involved a G→A mutation in codon 10 of NRAS exon 2 resulting in an amino acid change of glycine by glutamate (G10E). Furthermore, some SNPs were detected during direct sequencing, the most frequent one being the C/T polymorphism in codon 27 of NRAS exon 2 (data not shown) showing an allele frequency of 45% (TT), 35% (CT), and 20% (CC) without resulting in a substitution of the histidine amino acid. Allele frequencies were not correlated with histopathological or genetic subtype.

BRAF and NRAS Mutations as detected in two pure oligodendroglial tumors. a A BRAF exon 15 mutation in a WHO grade II oligodendroglioma (N78). A guanine (G) to adenine (A) substitution at codon 600 is present resulting in an amino acid change of valine to methionine (V600M). b A NRAS exon 2 mutation in an anaplastic oligodendroglioma (WHO grade III) (N303). The guanine to adenine substitution at codon 10 results in an amino acid change of glycine to glutamate (G10E)
Gains including the genes for BRAF, NRAS, HRAS, and KRAS
Alternatively to oncogenic mutations, an increased copy number of the above mentioned oncogene(s) can result in activation of the RAS/RAF pathway. We performed CGH analysis (detecting chromosomal copy number aberrations >2 Mb) (87/93 cases) and studied the occurrence of gains including the BRAF, NRAS, HRAS, and KRAS genes located at chromosomal regions 7q35, 1p13, 11p15, and 12p12, respectively. Aberrations detected in the individual tumors are summarized in Table 2, whereas Table 3 shows an overview among the different histopathological subtypes represented by at least nine cases. Gains including the RAS/RAF genes occurred most frequently in WHO grade III and IV gliomas. In pure oligodendroglial tumors, 4/21 (19%) tumors were affected including gains carrying NRAS, HRAS, and BRAF (twice). These gains were present both in tumors with or without −1p/−19q. In mixed oligodendroglial tumors (i.e. oligoastrocytic tumors) 13/31 (42%) tumors showed gains including KRAS (2×), NRAS (1×), HRAS (1×), and BRAF (11×). In two cases, a gain including BRAF coincided with a gain including RAS (either HRAS or KRAS). A relatively high number of astrocytic tumors contained a gain including BRAF (17/27 cases; 63%), mainly in GBMs (13 of the 17 gains), which is not surprising as BRAF is located on chromosome 7(q35) and +7 is frequently detected in GBMs. Additionally, gains including HRAS and KRAS were detected in 2 As (2/9). In ependymal tumors, 4/9 (44%) tumors contained gain(s) involving KRAS (2×), BRAF (3×), or HRAS (1×). In two of these tumors a gain including BRAF coincided with either gains including KRAS or HRAS. In contrast to the other glioma types these aberrations occurred in the grade II (4/7) but not in the two a-Es.
Gains including upstream growth factors and their receptors
Increased copy number of growth factors and their receptors upstream of RAF/RAS can have similar effects as activating mutations or increased copy number of the investigated oncogenes. Based on CGH analysis we evaluated the gains including the growth factor(s) and their receptors that are known to play a role upstream of RAS/RAF: EGF(R), PDGF(R), IGF(R), TGF(R), and FGF(R) (Tables 2 and 3). Overall, gains including growth factor (receptor) genes were more frequently detected in WHO grade III and IV than WHO grade II gliomas and occurred at an increasing frequency in pure oligodendroglial tumors (5/21; 24%), oligoastrocytic tumors (13/30; 43%) and astrocytic tumors (20/27; 74%). In ependymal tumors, however, most low-grade tumors were affected (6/7) and contained multiple gains whereas no gains were detected in both a-Es.
PDGF(R), EGF(R), and TGF(R) were most frequently affected (gains encompassing genes for these GF(R)s are detected in 47, 34 and 27 cases, respectively). Gains including EGF were only detected twice (in 2/9 Es) and only once this coincided with a gain including EGFR. In contrast, as described above, gains of chromosome 7 harboring the EGFR gene (7p12) were frequently detected, especially in GBMs. For PDGF, the majority of gains encompass PDGFA (30×) which is also localized on chromosome 7p(22) and therefore coincides with a gain of EGFR (29 cases). Other members of the PDGF(R) family are gained in tumors without gains on 7p (10 cases), in non-ependymal gliomas, PDGFD being the most frequent alternative (7×) whereas in ependymal tumors PDGFC and PDGFRA are the detected alternatives (3 and 3 cases, respectively). The highest frequency of gains encompassing TGF(R) was present in ependymal tumors (5/9), whereas in other histopathological glioma types this was up to approximately one third (3/21 pure oligodendroglial tumors, 4/30 oligoastrocytic tumors, and 8/27 astrocytic tumors). With the exception of gains including TGFBRII and TGFA (1×), other family members are included in gained regions at approximately the same frequency (TGFB1 and TGFBRI each six times, TGFBRIII five times, and TGFB2 and TGFB3 each four times). FGF(R) and IGF(R) are only occasionally affected by gains of different FGF(R) family members or IGFI (8× and 5×, respectively).
Immunohistochemical detection of phosphorylated MAPK
Activation (i.e. phosphorylation) of MAPK (MAPK-P) was evaluated using TMAs containing 60 cases analyzed for copy number changes (3–4 cores of each tumor). Results are summarized in Table 2. Overall, MAPK-P was detected in the majority of cases (53/60), usually showing cytoplasmic (site of phosphorylation) as well as nuclear (site of functional effect) staining (48/53 and 53/53, respectively). MAPK-P expression was absent in 7 cases either without (n = 3) or with (n = 4) copy number gains as described above. Statistical significant differences were detected for the MAPK-P nuclear staining patterns comparing cases with ≤ 2 and ≥ 3 gains encompassing the RAS/RAF or upstream growth factor (receptor) genes (P = 0.017), as well as for cases with or without a gain of chromosome 7 (P = 0.029). No significant correlation was detected for cytoplasmic staining (P = 0.061 and P = 0.076, respectively) nor for MAPK-P staining and histopathological classification.
Discussion
Neoplastic transformation is often a result of constitutive activation of genes in signaling pathways that regulate cell proliferation and differentiation. Indeed, the RAS–RAF–MEK–ERK/MAPK signaling pathway is constitutively activated in a large number of cancers.
There is ample evidence that such activation is also important for glioma oncogenesis [2–5, 7, 10, 19, 24, 28, 29]. This study provides an overview of aberrations detected in different types of gliomas that can result in RAS/RAF pathway activation. We not only evaluated mutations in BRAF and the oncogenic RAS family members (KRAS, HRAS, and NRAS) but also DNA copy number changes (gains) including these genes as well as the genes for the growth factors and their receptors upstream the RAS/RAF pathway, as the latter can have a comparable RAS/RAF pathway activating effect.
The rational for DNA copy number analysis is the fact that DNA copy number alterations, including low-level amplifications (gains) as well as high-level amplifications were reported to have a direct effect on gene expression, e.g. in a study of Pollack et al. who performed a parallel analysis of DNA copy number changes and mRNA levels using micro-arrays in breast cancer biopsies and cell-lines [25]. The importance of copy number changes is underscored by the recent discovery of an abundance of DNA copy number variations (CNVs) in the human genome ([26] and references therein). CNVs contribute to nucleotide diversity to an even larger extent than single nucleotide polymorphisms (SNPs). They influence gene expression, phenotypic variation and adaptation by disrupting genes and altering gene dosage, can cause disease as in microdeletion or microduplication disorders, and their contribution to human disease (susceptibility) may be greater than previously expected.
In our present study, copy number changes were evaluated using CGH analysis. Although such analysis does not provide gene specific information, it is clear that the gene will be lost or gained if its coding sequence is localized in the aberrant region detected by CGH. This was confirmed by comparing our conventional CGH data to our new array CGH data generated using a 32K array [30] as performed on approximately 25 gliomas for different research purposes. Furthermore, MLPA (multiplex ligation-dependent probe amplification) was used to detect gains/amplification of the EGFR gene (data not shown) and concordance between MLPA and CGH was found in 90% of the cases [13]. Using conventional CGH analysis, however, small gains of a low copy number will not be detected which might result in an underestimation of the gained regions (genes). Next to the widespread evaluation of DNA copy number gains and increase of corresponding mRNA levels as described above [25], it was clearly demonstrated in gliomas that an increased copy number of EGFR invariably results in increased expression of its coding mRNA and protein [6, 20, 32].
KRAS, HRAS, NRAS, and BRAF mutations
Mutations in B-RAF and RAS genes are frequently found in various forms of cancer. BRAF is often mutated in malignant melanomas and colorectal carcinomas and the most common oncogenic mutation occurs in the activation segment of its kinase domain [4]. It involves a valine to glutamate substitution at codon 600 and represents 90% of the BRAF mutations. Another relatively common B-RAF mutation affects the GXGXXG motif within the glycine-rich loop in exon 11 [4]. In contrast to the mutations in codon 600, exon 11 mutations are often associated with RAS mutations. RAS mutations are relative frequent in carcinomas of the pancreas, colon and thyroid [3]. The most common RAS mutations affect codons 12, 13 and 61, which all form part of the GTP-binding domain.
So far both BRAF and RAS mutations have been found in only a small fraction of human gliomas. A combined analysis of BRAF, NRAS, HRAS, and KRAS has been previously performed analyzing 94 GBMs [19]. Mutations were detected in either NRAS (n = 2) or BRAF (n = 3) in 5% of the cases. Furthermore, mutation analysis of BRAF was performed on a subset of 82 gliomas including 49 astrocytic (including 34 GBMs) and 33 oligodendroglial tumors and only 2 mutations were detected in GBMs (2/34; 6%) [2]. As RAS/RAF mutation analysis studies usually evaluated a specific subgroup of gliomas or a specific RAS/RAF oncogene, we conducted an extensive mutation analysis of all 3 oncogenic RAS genes and BRAF in different glioma types. In line with these previous reports, we analyzed that mutations were only sporadically (n = 2) detected in the 93 gliomas. Interestingly, in our series both mutations detected in either BRAF (V600M) or NRAS (G10E) occurred in two tumors with typical histopathological and genetic characteristics of pure oligodendroglial tumors. No mutations were detected in any of the other glioma types, not even in the GBMs (n = 28) for which a mutation rate of 5% has been reported [2, 19]. KRAS, HRAS, and NRAS mutations were investigated by us and Knobbe et al. [19] and we both only found evidence for involvement of NRAS (3/187 gliomas). This was rather surprising as of the different types of RAS proto-oncogenes (HRAS, KRAS, and NRAS) mutations are most frequently detected in KRAS and less often in NRAS. Comparing our results for BRAF mutation analysis on different glioma types with those of Basto et al. and Knobbe et al. [2, 19], we show that similar to NRAS, B-RAF mutations may occasionally (6/269) be involved in the development of not only GBMs but also of oligodendroglial tumors.
Both mutations detected (BRAF V600M and NRAS G10E) are different from the mutations most frequently reported in literature. An overview of RAS/RAF mutations can be found at the Catalogue of Somatic Mutations in Cancer, COSMIC, (http://www.sanger.ac.uk/cosmic) [8]. While the V600E substitution (also known as V599E) accounts for 90% of the BRAF mutations, over 30 other mutations in exon 15 are described. Usually, a negatively charged residue is inserted adjacent to a regulatory phosphorylation site, mimicking phosphorylation and thus leading to BRAF activation [4]. The V600M mutation detected by us has been previously identified in tumors of the stomach and large intestine (10/793 and 1/23 cases evaluated, respectively) and results in a non-polar amino acid. A substitution for a non-polar amino acid represents only 2% of the codon 600 mutations, most frequently involving V600M (6/2851 mutations in codon 600) and occasionally V600G (1/2851) or V600L (1/2851). The not yet reported N-RAS exon 2 mutation (G10E) may affect the intrinsic GTPase function, since codon 10 is part of the GTP-binding domain and results in constitutive activation of the RAS/RAF pathway. In line with previous experimental data, inhibition of the GTPase activity is the preferred mechanism of activation of oncogenic RAS proteins. The V600M mutation detected in our case did not coincide with other activating aberrations of the RAS/RAF pathway as evaluated in the current study. In contrast, the NRAS G10E mutation occurred in combination with an increased copy number of multiple upstream growth factors and/or their receptors (IGFI, FGF1, TGFB2, TGFB3, TGFBRIII, and PDGFRB).
Gains including the KRAS, HRAS, NRAS, and BRAF genes
Next to activating mutations, oncogene expression can be upregulated by an increased copy number. Using CGH we showed that indeed gains are detected in gliomas encompassing the RAS and BRAF genes. We show that especially WHO grade III and IV oligodendroglial and astrocytic tumors, and WHO grade II ependymal tumors (Es) contain such gains. The BRAF gene is located on chromosome 7q35 and a gain of the complete chromosome 7 is frequently detected, especially in GBMs and a-OAs. A gain including the BRAF gene was detected in approximately one third of the analyzed gliomas. Although mutation analysis suggests that NRAS is the most important RAS gene for gliomagenesis, an increased copy number of NRAS was only detected twice whereas KRAS or HRAS were gained in 5 and 4 cases, respectively, suggesting different modes of oncogene activation for the different RAS genes in gliomas. Multiple gains including the different RAS genes within a tumor were not detected and in 6 cases a gain encompassing BRAF coincided with a gain encompassing KRAS or HRAS (6/33 BRAF gains). Gains including the RAS genes can occur in tumors with or without −1p/−19q or +7/−10. In contrast, gains encompassing BRAF are present in the majority of gliomas of the +7/−10 genetic subtype (containing +7p and/or −10q). Overall 38/87 (44%) gliomas contained a gain including a RAS (11×) or BRAF (33×) gene suggesting that RAS/RAF pathway activation by an increased gene dosage through an increase in copy number is a rather common event in gliomas.
Gains including the growth factor (receptor) genes upstream of RAS/RAF
As described above, activation of the RAS/RAF pathway through mutation in the RAS or BRAF genes occurs only occasionally in the different glioma types, whereas 44% of the gliomas contain gains including at least one of these genes. Alternatively, gains including growth factors and their receptors can result in RAS/RAF-pathway activation as well. Indeed such gains encompassing the genes for growth factors and their receptors that are known as upstream activators of the RAS/RAF pathway (EGF(R), PDGF(R), TGF(R), IGF(R) or FGF(R)) were detected in half of the gliomas (46/87). Interestingly, gains involving such growth factors and their receptors occurred in the majority of cases in which a gain encompassing RAS or BRAF was detected (90%; 34/38) and less frequently in cases without such gains (25%; 12/49). Gains including growth factor (receptor) genes are more frequently detected in WHO grade III and IV than WHO grade II gliomas and occur at an increasing frequency in pure oligodendroglial tumors (24%; 5/21), oligoastrocytic tumors (42%; 13/31) and astrocytic tumors (77%; 21/27). In ependymal tumors, however, WHO grade II tumors were frequently affected (85%, 6/7) and often contained multiple gains. In concordance with previous reports [22], in our group of gliomas EGF(R) and PDGF(R) are most frequently affected and additionally TGF(R) was affected at a slightly lower frequency than EGF(R).
Because of their chromosomal location, gains including the genes for EGFR (7p12), PDGFA (7p22), and BRAF (7q35) frequently coincide in +7/−10 tumors. A gain of chromosome 7 is, therefore, theoretically a rather effective way of RAS/RAF pathway activation which is detected in the majority of a-OAs (56%) and GBMs (76%) and less frequently in other glioma types. Gains encompassing EGF were only occasionally detected (2% of the cases) whereas gains including other members of the PDGF(R) family were somewhat more frequently detected (15% of the cases). These gains only occasionally coincided with those including EGFR or PDGFA (4/13 cases), suggesting that they may serve as alternatives for gains including EGFR or PDGFA occurring in tumors not containing a gain of chromosome 7. A gain of chromosome 4 would, therefore, also be an effective way of RAS/RAF pathway activation as this chromosome contains multiple growth factor (receptor) genes (FGFR3 (4p16), PDGFRA (4q12), EGF (4q25), FGF2 (4q27), and PDGFC (4q32)). However, chromosome 4 gains are only occasionally detected, and encompassing partial gains in 2 of the 3 tumors, involving only part of the growth factor (receptor) genes on this chromosome involved in the RAS/RAF pathway.
Immunohistochemical analysis of RAS/RAF pathway activation
Activation of the RAS/RAF pathway was evaluated using immunohistochemical detection of MAPK-P (i.e. activated MAPK downstream the RAS/RAF pathway) using TMAs, and was identified in the majority of cases in our present series. MAPK-P expression was absent in 7 cases either without (n = 3) or with (n = 4) copy number gains as described above. Although, the latter may suggest that these gains do not always result in MAPK activation, it is more likely that our approach results in an underestimation of MAPK-P due to either sampling artifacts when using TMAs or the detection threshold of immunohistochemical analysis. Furthermore, MAPK-P expression was detected in cases in which no copy number gains were detected which may reflect the underestimation of copy number gains due to the CGH resolution (as discussed above), may be the result of alternative changes (not detected in the present study) causing RAS/RAF pathway activation, or represent basic levels of MAPK-P. More refined techniques (e.g. immunoblotting, RNA-ISH, fluorescent immunohistochemistry) need to be applied to detect the actual impact of expression level changes for the genes described in the present study as well as for detection of activated downstream effector molecules.
Although more refined techniques will even further elucidate the exact role of the described copy number gains on RAS/RAF pathway activation, the significant correlation detected for nuclear MAPK-P (site of functional effect) and the number of gains (≤ 2 and ≥ 3) involving RAS/RAF or upstream growth factors and/or their receptors corroborates our hypothesis that these copy number gains, rather than activating RAS/RAF mutations, are involved in RAS/RAF pathway activation in gliomas. Furthermore, the significant correlation between MAPK-P expression and a gain of chromosome 7 confirms that this aberration, which is frequently detected in gliomas (especially in GBMs), can be rather effective in activating the RAS/RAF pathway.
In summary
Mutations in the exons containing the mutation hotspot sites of NRAS, KRAS, HRAS or BRAF were only sporadically detected in gliomas and can occur in different histopathological glioma types: GBMs (as reported by others) as well as pure oligodendroglial tumors (present study). Gains involving these genes, however, were detected in 44% (38 cases) of our series of gliomas. The BRAF gene is most frequently included in a gain (38% of the gliomas) and only occasionally coincides with gains including RAS (6 cases). Copy number gains including genes for upstream growth factors and their receptors were also frequently detected (53%; 46 cases) and usually coincide with gains encompassing RAS or BRAF (34 cases). Overall, 57% of the gliomas contained gains that can result in increased gene dosage and subsequent activation of the RAS/RAF pathway (50/87 cases), whereas only 2 (2/93) tumors contained a mutation. A significant correlation was indeed detected between nuclear MAPK-P (i.e. activated MAPK downstream the RAS/RAF pathway) staining and the number of genes involved in gains. Our results thus indicate that RAS/RAF pathway activation in gliomas is achieved by copy number gains rather than by activating mutations. Although it was shown for EGFR that copy number gains result in increased expression [6, 20, 32], and activation of MAPK was detected in the majority of cases using TMAs and immunohistochemistry, the actual consequences of the aberrations described in this study need to be further investigated using e.g. immunoblotting, RNA-ISH, or fluorescent immunohistochemistry, detecting expression level changes for the genes gained as well as for the detection of activated MAP and MAPK.
Regarding the distribution of the detected aberrations among the different glioma types we showed that copy number gains were most frequently detected in WHO grade III and IV gliomas with an increasing frequency in pure oligodendroglial tumors, oligoastrocytic tumors and astrocytic tumors. Interestingly, WHO grade II ependymal tumors were also relatively often affected. No correlation between histopathological subtype and MAPK-P staining were detected. Gains encompassing BRAF (7q35) were mainly detected in gliomas of the +7/−10 genetic subtype, whereas gains encompassing RAS were more evenly distributed among the different genetic glioma subtypes. As gains were studied using CGH detecting only copy number changes of at least 2 Mb and alternative mechanisms (next to an increased copy number) might also account for upregulation of gene expression and activation of corresponding pathway(s), our results are most likely an underestimation of the involvement of the above mentioned genes and pathway(s) in gliomas.
Abbreviations
- a-:
-
anaplastic tumor (WHO grade III)
- A:
-
diffuse astrocytoma (WHO grade II)
- BRAF:
-
v-raf murine sarcoma viral oncogene homologue B1
- E:
-
ependymoma (WHO grade II)
- EGF(R):
-
epidermal growth factor (receptor)
- ERK:
-
extracellular signal regulated kinase (= MAPK)
- FGF(R):
-
fibroblast growth factor (receptor)
- GBM:
-
glioblastoma multiforme (WHO grade IV astrocytoma)
- HRAS:
-
v-Ha-ras Harvey rat sarcoma viral oncogene homologue
- IGF(R):
-
insulin-like growth factor (receptor)
- KRAS:
-
v-Ki-ras2 Kirsten rat sarcoma viral oncogene homologue
- LOH:
-
loss of heterozygosity
- MAPK:
-
mitogen-activated protein kinase (= ERK)
- MEK:
-
MAPK/extracellular signal regulated kinase (= MAPKK)
- NRAS:
-
neuroblastoma RAS viral (v-ras) oncogene homologue
- O:
-
oligodendroglioma (WHO grade II)
- OA:
-
oligoastrocytoma (WHO grade II)
- PDGF(R):
-
platelet derived growth factor (receptor)
- RAS:
-
rat sarcoma viral oncogene homologue
- RAF:
-
v-raf murine sarcoma viral oncogene homologue
- TGF(R):
-
transforming growth factor (receptor)
- TMA:
-
tissue micro-array
- TP53:
-
tumor protein 53
References
Bastian BC, LeBoit PE, Pinkel D (2000) Mutations and copy number increase of HRAS in Spitz nevi with distinctive histopathological features. Am J Pathol 157:967–972
Basto D, Trovisco V, Lopes JM, Martins A, Pardal F, Soares P, Reis RM (2005) Mutation analysis of B-RAF gene in human gliomas. Acta Neuropathol (Berl) 109:207–210
Bos JL (1989) Ras oncogenes in human cancer: a review. Cancer Res 49:4682–4689
Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, Floyd Y, Gray K, Hall S, Hawes R, Hughes J, Kosmidou V, Menzies A, Mould C, Parker A, Stevens C, Watt S, Hooper S, Wilson R, Jayatilake H, Gusterson BA, Cooper C, Shipley J, Hargrave D, Pritchard-Jones K, Maitland N, Chenevix-Trench G, Riggins GJ, Bigner DD, Palmieri G, Cossu A, Flanagan A, Nicholson A, Ho JW, Leung SY, Yuen ST, Weber BL, Seigler HF, Darrow TL, Paterson H, Marais R, Marshall CJ, Wooster R, Stratton MR, Futreal PA (2002) Mutations of the BRAF gene in human cancer. Nature 417:949–954
Ding H, Roncari L, Shannon P, Wu X, Lau N, Karaskova J, Gutmann DH, Squire JA, Nagy A, Guha A (2001) Astrocyte-specific expression of activated p21-ras results in malignant astrocytoma formation in a transgenic mouse model of human gliomas. Cancer Res 61:3826–3836
Ekstrand AJ, James CD, Cavenee WK, Seliger B, Pettersson RF, Collins VP (1991) Genes for epidermal growth factor receptor, transforming growth factor alpha, and epidermal growth factor and their expression in human gliomas in vivo. Cancer Res 51:2164–2172
Feldkamp MM, Lala P, Lau N, Roncari L, Guha A (1999) Expression of activated epidermal growth factor receptors, Ras-guanosine triphosphate, and mitogen-activated protein kinase in human glioblastoma multiforme specimens. Neurosurgery 45:1442–1453
Forbes S, Clements J, Dawson E, Bamford S, Webb T, Dogan A, Flanagan A, Teague J, Wooster R, Futreal PA, Stratton MR (2006) COSMIC 2005. Br J Cancer 94:318–322
Fransen K, Klintenas M, Osterstrom A, Dimberg J, Monstein HJ, Soderkvist P (2004) Mutation analysis of the BRAF, ARAF and RAF-1 genes in human colorectal adenocarcinomas. Carcinogenesis 25:527–533
Guha A, Feldkamp MM, Lau N, Boss G, Pawson A (1997) Proliferation of human malignant astrocytomas is dependent on Ras activation. Oncogene 15:2755–2765
Jeuken JW, von Deimling A, Wesseling P (2004) Molecular pathogenesis of oligodendroglial tumors. J Neurooncol 70:161–181
Jeuken JWM, Boots-Sprenger SHE, Wesseling P (2004) Chromosomal imbalances in oligodendroglial tumors as detected by comparative genomic hybridization (CGH). In: Zhang W, Fuller GN (eds) Genomic and molecular neuro-oncology. Jones and Bartlett Publishers, Inc. Sudbury, pp 185–198
Jeuken JWM, Cornelissen S, Boots-Sprenger SHE, Gijssen S, Wesseling P (2006) Multiplex ligation-dependent probe amplification (MLPA): a diagnostic tool for simultaneous identification of different genetic markers in glial tumors. J Mol Diagnostics 8:433–443
Jeuken JWM, Sprenger SHE, Boerman RH, von Deimling A, Teepen HLJM, van Overbeeke JJ, Wesseling P (2001) Subtyping of oligo-astrocytic tumours by comparative genomic hybridisation. J Pathol 194:81–87
Jeuken JWM, Sprenger SHE, Vermeer H, Kappelle AC, Boerman RH, Wesseling P (2002) Chromosomal imbalances in primary oligodendroglial tumors and their recurrences; clues for malignant progression as detected by CGH. J Neurosurg 96:559–564
Jeuken JWM, Sprenger SHE, Wesseling P, Macville MVE, von Deimling A, Teepen HLJM, van Overbeeke JJ, Boerman RH (1999) Identification of subgroups of high-grade oligodendroglial tumors by comparative genomic hybridization. J Neuropathol Exp Neurol 58:606–612
Kleihues P, Cavenee WK (2000) World Health Organization classification of tumours. Pathology and genetics. Tumours of the nervous system. International Agency for Research on Cancer (IARC) Press, Lyon
Kleihues P, Ohgaki H (2000) Primary and secondary glioblastomas: from concept to clinical diagnosis. Neuro-Oncology 1:44–51
Knobbe CB, Reifenberger J, Reifenberger G (2004) Mutation analysis of the Ras pathway genes NRAS, HRAS, KRAS and BRAF in glioblastomas. Acta Neuropathol 108:467–470
Libermann TA, Nusbaum HR, Razon N, Kris R, Lax I, Soreq H, Whittle N, Waterfield MD, Ullrich A, Schlessinger J (1985) Amplification, enhanced expression and possible rearrangement of EGF receptor gene in primary human brain tumours of glial origin. Nature 313:144–147
Maintz D, Fiedler K, Koopmann J, Rollbrocker B, Nechev S, Lenartz D, Stangl AP, Louis DN, Schramm J, Wiestler OD, von Deimling A (1997) Molecular genetic evidence for subtypes of oligoastrocytomas. J Neuropathol Exp Neurol 56:1098–1104
Newton HB (2003) Molecular neuro-oncology and development of targeted therapeutic strategies for brain tumors. Part 1: growth factor and Ras signaling pathways. Expert Rev Anticancer Ther 3:595–614
Osterstrom A, Dimberg J, Fransen K, Soderkvist P (2002) Expression of cytosolic and group X secretory phospholipase A(2) genes in human colorectal adenocarcinomas. Cancer Lett 182:175–182
Peyssonnaux C, Eychene A (2001) The Raf/MEK/ERK pathway: new concepts of activation. Biol Cell 93:53–62
Pollack JR, Sorlie T, Perou CM, Rees CA, Jeffrey SS, Lonning PE, Tibshirani R, Botstein D, Borresen-Dale AL, Brown PO (2002) Microarray analysis reveals a major direct role of DNA copy number alteration in the transcriptional program of human breast tumors. Proc Natl Acad Sci USA 99:12963–12968
Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, Fiegler H, Shapero MH, Carson AR, Chen W, Cho EK, Dallaire S, Freeman JL, Gonzalez JR, Gratacos M, Huang J, Kalaitzopoulos D, Komura D, MacDonald JR, Marshall CR, Mei R, Montgomery L, Nishimura K, Okamura K, Shen F, Somerville MJ, Tchinda J, Valsesia A, Woodwark C, Yang F, Zhang J, Zerjal T, Zhang J, Armengol L, Conrad DF, Estivill X, Tyler-Smith C, Carter NP, Aburatani H, Lee C, Jones KW, Scherer SW, Hurles ME (2006) Global variation in copy number in the human genome. Nature 444:444–454
Tannapfel A, Sommerer F, Benicke M, Katalinic A, Uhlmann D, Witzigmann H, Hauss J, Wittekind C (2003) Mutations of the BRAF gene in cholangiocarcinoma but not in hepatocellular carcinoma. Gut 52:706–712
Trovisco V, Vieira dC I, Soares P, Maximo V, Silva P, Magalhaes J, Abrosimov A, Guiu XM, Sobrinho-Simoes M (2004) BRAF mutations are associated with some histological types of papillary thyroid carcinoma. J Pathol 202:247–251
van Dijk MC, Bernsen MR, Ruiter DJ (2005) Analysis of mutations in B-RAF, N-RAS, and H-RAS genes in the differential diagnosis of Spitz nevus and spitzoid melanoma. Am J Surg Pathol 29:1145–1151
Vissers LE, Veltman JA, van Kessel AG, Brunner HG (2005) Identification of disease genes by whole genome CGH arrays. Hum Mol Genet 14(Spec No. 2):R215–R223
von Deimling A (1997) Neoplasia. Molecular genetic classification of astrocytic and oligodendroglial tumors. Brain Pathol 7:1311–1313
Wong AJ, Bigner SH, Bigner DD, Kinzler KW, Hamilton SR, Vogelstein B (1987) Increased expression of the epidermal growth factor receptor gene in malignant gliomas is invariably associated with gene amplification. Proc Natl Acad Sci USA 84:6899–6903
Acknowledgements
We thank Dr. Jeroen EM van Leeuwen for generously providing the phospho-p44/42 MAPK antibody, Jos Rijntjes for the immunohistochemical stainings, and Susan van de Kieboom-Hageman for construction of the tissue micro-arrays.
Author information
Authors and Affiliations
Corresponding author
Additional information
This research was funded by the KWF Dutch Cancer Society grant KUN2004-3143. PW was sponsored by Dutch Cancer Society grant KUN2003-2975.
Rights and permissions
About this article
Cite this article
Jeuken, J., van den Broecke, C., Gijsen, S. et al. RAS/RAF pathway activation in gliomas: the result of copy number gains rather than activating mutations. Acta Neuropathol 114, 121–133 (2007). https://doi.org/10.1007/s00401-007-0239-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-007-0239-0