
Abstract
The constant ‘interchain pressure’ idea has been addressed, to evaluate if it is an adequate quantitative assumption to describe the fluid mechanics of oligomer diluted entangled NMMD polymer systems. The molecular stress function constitutive framework has been used with the constant interchain pressure assumption. Furthermore, the maximal extensibility based on the number of Kuhn steps in an entanglement has been used based on the relative Padé inverse Langevin function. The model predictions agree with the extensional measurements on all previously published poly(methyl methacrylate)s and almost all published oligomer diluted NMMD polystyrenes. The only deviation is on the most diluted and largest molecular weight case of an 18% 1880 kg/mol polystyrene in oligomer diluent. In this case, the maximal extensibility is not needed.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The idea of universality in flow dynamics of entangled monodisperse polymer systems is appealing and is the guideline in the earliest tube theories (De Gennes 1971; Doi and Edwards 1978). However, the lack of universality was recognized with the publication of extensional viscosity of narrow molar mass distributed (NMMD) polystyrene solutions and polystyrene (Bhattacharjee et al. 2002) and (Bach et al. 2003b) respectively. The recent publications (Huang et al. 2013a, 2013b) of extensional viscosity of oligomer diluted NMMD polystyrenes have cast new light on the flow dynamics of entangled polymers.
Concerning all the referred polystyrene measurements above, two recent ideas for constitutive concepts have been suggested to explain the fluid mechanics of entangled systems. One is the monomeric friction reduction (Yaoita et al. 2012; Ianniruberto et al. 2012), actually suggested before the appearance of the oligomer diluted polystyrene measurements by Huang et al. 2013a, ??b, and the other one is the modified interchain pressure (Wagner 2014), proposed more recently. Both seem capable of predicting the flow of the referred NMMD polystyrene melts and their respective dilutions. Particularly, the models concerning monomeric friction, the amount of changeable parameters (Ianniruberto 2015) and functions (Masubuchi et al. 2014) seem sufficient to model the flow of other NMMD polystyrene systems (Huang et al. 2015) and even systems other than polystyrenes (Sridhar et al. 2014) as well. The model by Wagner (2014) is based on an adaption of the originally suggested ‘interchain pressure’ model by Marrucci and Ianniruberto (2004) for monodisperse polymer melts. In the latter one, the tube deformation is controlled by the increasing thermal interchain or tube pressure, originating from the stretch of the surrounding chains. It explains the rheological measurements of NMMD polystyrene melts accurately (Wagner et al. 2005). In Wagner (2014), the flow behavior of entangled monodisperse polymer systems is explained as a unique consequence of the reduced glass transition temperature in solutions which increases the effect of a lateral spring force compared to monodisperse melts.
We have implied in Rasmussen and Huang (2014) that in an ideal dilution, when the diluent consists of Kuhn chains in random configurations, the thermal interchain tube pressure would be constant. This is a particular case for diluted systems as long as the length of all the diluent molecules are above two Kuhn steps and the molecules are in random configurations (Rasmussen 2015). This idea shows agreement with the extensional measurements on bi-disperse polystyrene and polyisoprene systems (Rasmussen 2015), and also the oligomer diluted NMMD polystyrenes measured by Huang et al. (2013a, ??b). Recently, extensional measurements on a wider concentration range of oligomer diluted NMMD polystyrenes (Huang et al. 2015), as well as oligomer diluted NMMD poly(methyl methacrylate)s (PMMA) (Wingstrand et al. 2015), have appeared. These measurements allow us to further evaluate if the idea of a constant ‘interchain pressure’ is an adequate assumption to describe the fluid mechanics of oligomer diluted entangled NMMD systems, which will be addressed in this paper.
Constant interchain pressure
A diversity of constitutive models based on the tube idea (De Gennes 1971) have been published in the past decades, initiated with the constitutive model based on a purely orientational stress by Doi and Edwards (1978). Chain stretch was introduced by Marrucci and Grizzuti (1988) and the variety of the subsequent constitutive equations are all based on chain stretch. Particularly, considering the ‘interc- hain pressure’ idea, originally proposed by Marrucci and Ianniruberto (2004), three different approaches have currently been published (Wagner et al. 2005; Dhole et al. 2009; van Ruymbeke et al. 2010). Here, we use the molecular stress function (MSF) constitutive equation by Wagner et al. (2005, 2010), where the components of the stress tensor, σ ij, are defined as
E(x,t,t ′) is the displacement gradient tensor where its components are defined as \(E_{\text {ij}}(\boldsymbol {x},t,t^{\prime }) = \partial x_{\mathrm {i}}/{x_{\mathrm {j}^{\prime }}}\), i=1,2,3 and j=1,2,3. x=(x 1,x 2,x 3) are the Cartesian coordinates of a particle in the present time t and \((x^{\prime }_{1},x^{\prime }_{2}, x^{\prime }_{3})\) are the position of the same particle at the past time t ′. The angular brackets are the unit sphere integral given as \(\langle {\dots } \rangle = 1/(4 \pi ) {\int }_{|\mathbf {u}|=1} {\dots } d\mathbf {u}\). u is the unit vector. All relevant unit sphere integrals are analytically solved in Urakawa et al. (1995). The initial value of the molecular stress function is f(x,t ′,t ′)=1.
The memory function, M(t−t ′), used here is the one suggested by Baumgaertel, Schausberg, and Winter (BSW) (Baumgaertel et al. 1990; Baumgaertel and Winter 1992) given as
where h(x) is the Heaviside step function. τ m a x is the maximal relaxation time and τ c is the largest relaxation time of the glassy modes. \({G_{N}^{0}}\) is the plateau modulus. Particularly the fixed values of n e =0.2 and n g =0.7 (Rasmussen and Huang 2014) seem appropriate for all the involved samples discussed here. Some of the needed BSW parameters can be found in Rasmussen and Huang (2014). All the other linear viscoelastic parameters are obtained by fitting the mechanical spectroscopic measurements to the BSW model using the method from Rasmussen et al. (2000). The mechanical spectroscopic measurements from Huang et al. (2015) and Wingstrand et al. (2015) with the corresponding fittings are shown in Figs. 1 and 2 and the values of the parameters are listed in Table 1. 𝜃 represents the weight fraction of the long polymer chains in the dilution.

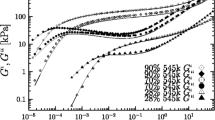
Loss, \(G^{\prime \prime }\) (open triangles and open circles) and storage moduli, \(G^{\prime }\) (filled triangles and bullets), both as a function of the angular frequency ω measured at 130 ∘C for the two polystyrene blends from (Huang et al. 2015). The bottom data are 18% 1880 kg/mole (triangles) and the top data are the 33% 910 kg/mole (circles/bullets), both diluted in the 4.29 kg/mole styrene oligomer. The solid lines are the least-square fittings to the BSW model in Eq. 3

Loss, \(G^{\prime \prime }\) (open boxes, open triangles, and open circles) and storage moduli, \(G^{\prime }\) (filled boxes, filled triangles, and bullets), both as a function of the angular frequency ω measured at 150 ∘C for the three PMMA blends from Wingstrand et al. (2015). The blends are (boxes) a 51% 86 kg/mole diluted in the 3.5 kg/mole oligomer, (circles/bullets) a 44% 270 kg/mole diluted in the 3.5 kg/mole oligomer and (triangles) a 43% 270 kg/mole diluted in the 2.1 kg/mole oligomer. The solid lines are the least-square fittings to the BSW model in Eq. 3
Rasmussen and Huang (2014) suggested that the thermal interchain or tube pressure stays constant if the tube contains chains in random configurations. Note that this is not a general constitutive idea for entangled systems, as it excludes dilutions with diluent molecules shorter than two Kuhn steps. Using the MSF constitutive approach (Wagner et al. 2005; Wagner and Rolón-Garrido 2010), the stretch evolution f(x,t,t ′) assuming a constant ‘interchain pressure’ can be calculated through
as derived by Rasmussen and Huang (2014). τ R is the Rouse time. Notice that the dependence of x, t, and t ′ has been omitted in the notation. As the quantitative transition to a maximum extensibility the relative Padé (Cohen 1991) inverse Langevin function (Ye and Sridhar 2005)
is used. Here, λ m a x is the maximal relative stretch. In Rasmussen and Huang (2014), a maximal relative stretch of a polymer chain is the square root of the number of Kuhn steps between entanglements:
where N K is the number of Kuhn steps in a long polymer chain and Z is the number of entanglements in the chain. The number of Kuhn steps per chain, N K = M w /M K , is expected to be independent of dilution with a theoretically ideal solvent, most likely to be oligomers. M w is the weight average molecular weight of the long polymer chain, and M K is the Kuhn step molecular weight. A linear change with the concentration is expected for the number of entanglements Z = M w /M e,𝜃 = M w 𝜃/M e (Huang et al. 2013a), where M e,𝜃 and M e are the entanglement molecular weight of the diluted and undiluted long chain, respectively. M e,𝜃 = M e /𝜃 represents the change of the entanglement molecular weight with dilution. The plateau modulus for ideally diluted samples, as assumed here, changes as \(G_{N,\theta }^{0}=\theta ^{2} {G_{N}^{0}}\) (Huang et al. 2013a).
NMMD polystyrene
The actual values of the Rouse time depend on the used particular constitutive approach (Larson et al. 2003; Osaki et al. 2000; Osaki et al. 2001; Likhtman and McLeish 2002; Menezes and Graessley 1982). The vast majority of these different theoretical concepts as well as classical shear experimentations (Osaki et al. 1982; Menezes and Graessley 1982; Takahashi et al. 1993) suggest a dependence of the Rouse time as the square of the number of entanglements giving the ratio between the Rouse and maximal relaxation time of about τ R /τ m a x ∝Z −1.5.
In Rasmussen and Huang (2014), the dependency of the Rouse time was empirically found based on extensional experiments on NMMD polystyrene melts as
based on the ‘interchain pressure’ idea by Marrucci and Ianniruberto (2004) in the form by Wagner et al. (2005). The number of entanglements Z = M w /M e,𝜃 . The entanglement molecular weight for polystyrene has been reported within the range of 13.3 kg/mol to 18.1 kg/mol (Fetters et al. 2007). It creates an ambiguity in the estimation of the number of entanglements, Z. The calculation of the number of entanglements in Eq. 7 is based on the assumption of M e =13.3 kg/mol for polystyrene (Bach et al. 2003b). Note, though Eq. 7 is written as dependent of the M e , the fittings of the Rouse times were done based on the average molecular weight solely, whereas the maximum extensibility or relative stretch is dependent on this particular choice of M e .
For oligomer solutions of polystyrenes Rasmussen and Huang (2014) used a maximal relative stretch of \(\lambda _{max}=\sqrt {N_{K}/Z}= \sqrt {(M_{e}/M_{k})/\theta }\) where M e /M k =22.17. As mentioned before 𝜃 is the weight fraction of the long chain in the dilution. For the involved polystyrenes the molecular weight of one Kuhn step, M K , is 0.6 kg/mol (Fang et al. 2000).
In the study by Huang et al. (2015), emphasis was on keeping the number of entanglements constant while changing the fraction of the diluent and molecular weight of the polystyrene. The diluent was a M w = 4.33 kg/mol styrene oligomer with a polydispersity index (PDI) of P D I = M w /M n of 1.04. M n is the number average molecular weight. The diluent is considerable shorter than the lowest expected entanglement molecular weight of 13.3 kg/mol of polystyrene but contains 7 Kuhn steps. With increasing dilution it would be expected that the polymer could be stretched more. Actually one of the findings in Huang et al. (2015) is that the data clearly shows that the strain at the transition to a steady extensional stress increases with decreasing concentration of the long polymer.
The start-up of extensional viscosity, \(\bar {\eta }^{+}\), from Huang et al. (2015) on the NMMD polystyrene dilutions are shown in Figs. 3, 4 and 5. Within the experimental accuracy the number of entanglements is similar for the different solutions. The values of Z are about 22 to 25. Notice that the experiments in Fig. 3 originates from Huang et al. (2013b) and the theoretical considerations in this figure are the same as in Rasmussen and Huang (2014). Further notice, that the formula from Rasmussen et al. (2010) has been applied to correct all the presented extensional viscosity from a potential shear contribution at small strain values.

The startup of extensional viscosity measurements, \(\bar {\eta }^{+}\), as a function of time at 130 ∘C. The symbols are the PS545k/o4k (52.5% 545 kg/mole diluted in the 4.29 kg/mole oligomer) blend with 𝜃=0.525 data from Huang et al. (2013a), measured at 129 ∘C, here time-temperature superposition shifted to 130 ∘C. The solid lines are the corresponding predictions to the data from Eq. 4. The dashed lines are the corresponding predictions to the data from Eq. 4 without a limit on the chain extensibility. The dotted line is the linear viscoelastic prediction based on the parameters listed in Table 1 using Eq. 3

The startup of extensional viscosity measurements, \(\bar {\eta }^{+}\), as a function of time at 130 ∘C. The symbols are the PS910k/o4k (33% 910 kg/mole diluted in the 4.29 kg/mole oligomer) blend with 𝜃=0.33 data from Huang et al. 2015, measured at 130 ∘C. The solid lines are the corresponding predictions to the data from Eq. 4. The dashed lines are the corresponding predictions to the data from Eq. 4 without a limit on the chain extensibility. The dotted line is the linear viscoelastic prediction based on the parameters listed in Table 1 using Eq. 3

The startup of extensional viscosity measurements, \(\bar {\eta }^{+}\), as a function of time at 130∘C. The symbols are the PS1880k/o4k (18% 1880 kg/mole diluted in the 4.29 kg/mole oligomer) blend with 𝜃=0.18 data from Huang et al. 2015, measured at 130 ∘C. The solid lines are the corresponding predictions to the data from Eq. 4. The dashed lines are the corresponding predictions to the data from Eq. 4 without a limit on the chain extensibility. The dotted line is the linear viscoelastic prediction based on the parameters listed in Table 1 using Eq. 3
The extensional viscosity for the samples at the same extensional conditions, of course relative to the relaxation time, should ideally be similar if the underlying physics only depends on the number of entanglements. This is actually the case in the constant interchain pressure model in Eq. 4 without a limit on the chain extensibility. This corresponds to c(f)=1 in Eq. 5. It basically is a state of universality with entanglement numbers. The dashed lines (- - -) in the figures are the calculations based on Eq. 4 without the limit on the chain extensibility. All the startup flow follows this assumption. Surprisingly, at the most diluted case (Fig. 5) of the 18% 1880 kg/mol, the maximum extensibility seems not needed. We have added the theoretical prediction based on Eq. 4, e.g., with the maximal extensibility as well. This is the solid lines (——) in Figs. 3, 4 and 5. In the most diluted case (Fig. 5), the maximal extensibility predicts steady extensional viscosity at values significantly below the measured ones. The transition to steady state in the cases of the dilutions with 52% (Fig. 3) and 33% (Fig. 4) long NMMD polystyrene polymers does match the idea of maximal extensibility based on the number of Kuhn steps between entanglements. The agreement in Figs. 3 and 4 was also observed for entangled bidisperse polystyrene systems, actually down to a concentration of 4% of the long polymers, in Rasmussen (2015). The most diluted sample, i.e. 13% 3140 kg/mol polystyrene, in Huang et al. (2015) shows a higher PDI of 1.38 than the commonly referred NMMD, and is therefore not compared with model predictions here. Notice that the model from Wagner (2014), which is also based on a ‘interchain pressure’ idea, does not include a limit based on the chain extensibility. The transition to the steady viscosity at high strain rates is controlled by the increase in the interchain pressure solely.
NMMD poly(methyl methacrylate)
A variety of different stereoisomeric structures of PMMA have been published with different reported entanglement molecular weight. In Fuchs et al. (1996), M e values are reported in between of 6 and 8 kg/mol, whereas in Krishnamoorti et al. (2002), the value is reported as high as 12.5 kg/mol. The synthesis and the consequent tacticity of the two used NMMD PMMAs, a 86 kg/mol and a 270 kg/mol in Wingstrand et al. (2015), have not been reported. Wingstrand et al. (2015) calculated the PMMA entanglement molecular weight from the mechanical spectroscopic data, based on the assumption of identical linear viscoelasticity with the same number of entanglements Z, by comparing the PMMA samples with the NMMD polystyrene samples. The previously referred NMMD polystyrenes are all expected to be prepared by living anionic polymerization using the same initiator, resulting in similar tacticity. The M e value of the polystyrenes is assumed to be 13.3 kg/mol. The M e values of the two PMMAs are reported in between 5 and 6 kg/mol according to Wingstrand et al. (2015). Here, we use the value of 5.4 kg/mol for both involved PMMA melts.
The molecular weight of PMMA Kuhn step seems not to be very sensitive to tacticity. The value for an atactic PMMA is reported to be 0.6 kg/mol (Fetters et al. 2007), while for a 75% syndiotactic PMMA is about 0.66 kg/mol (Fetters et al. 1994). Therefore, the molecular weight of 0.6 kg/mol seems to be a reasonable average for the involved PMMA melts. The used oligomer diluents in Wingstrand et al. (2015) are a 2.1 and a 3.5 kg/mol PMMA with PDI of 1.1 and 1.04, respectively. The length of the oligomers are therefore expected to be considerably above two Kuhn steps and below the entanglement length. Furthermore, in similar structured, polymers such as polystyrene and polypropylene, it seems that the difference between atactic and isotactic structures (Fetters et al. 2007) only changes the Kuhn step length within a few percent.
The squared relation in Eq. 7 is a direct fitting of extensional experiments on NMMD polystyrene melt. They are based on the ‘interchain pressure’ idea by Marrucci and Ianniruberto (2004) in the form by Wagner et al. (2005). As the relevant Z values for the NMMD PMMA dilutions from Wingstrand et al. (2015) seem to be within the same interval as the NMMD polystyrene melts, we use the same proportionality for the Rouse time.
The Rouse time for the oligomer diluted PMMA samples is obtained based on a direct fitting to the measured extensional viscosity. This direct approach determines the initiation of strain hardening and is an adaption to the classical identification of the Rouse time in shear rheometry. Uniaxial extension flow is much more sensitive to changes in the Rouse time than shear flow. We empirically find the dependency of the Rouse time to the entanglements to be
for the PMMA systems. A linear change with the dilution is assumed as Z = M w 𝜃/M e . Note, though Eq. 8 is written as dependent of the M e , the fittings of the Rouse times were done based on the average molecular weight solely. Therefore, the values of the Rouse time are not dependent on the particular value of the entanglement length of M e =5.4 kg/mol. The start-up of extensional viscosity, \(\bar {\eta }^{+}\), from Wingstrand et al. (2015) is shown in Figs. 6 and 7 for the three different oligomer diluted PMMA samples. The dashed lines (- - -) in Figs. 6 and 7 are the calculations based on Eq. 4 without the limit given by the maximum extensibility, i.e. c(f)=1 in Eq. 5.

The startup of extensional viscosity measurements, \(\bar {\eta }^{+}\), as a function of time at 150 ∘C. The symbols are the PMMA270k/o2k (43% 270 kg/mole diluted in the 2.1 kg/mole oligomer) and PMMA270k/o4k (44% 270 kg/mole diluted in the 3.5 kg/mole oligomer) blend data with 𝜃=0.43 and 𝜃=0.44 respectively. The data are from Wingstrand et al. 2015 measured at 150 ∘C. The solid lines are the corresponding predictions to the data from Eq. 4. The dashed lines are the corresponding predictions to the data from Eq. 4 without a limit on the chain extensibility. The dotted lines are the linear viscoelastic prediction based on the parameters listed in Table 1 using Eq. 3

The startup of extensional viscosity measurements, \(\bar {\eta }^{+}\), as a function of time at 150 ∘C. The symbols are the PMMA86k/o4k (51% 86 kg/mole diluted in the 3.5 kg/mole oligomer) blend data with 𝜃=0.51 from Wingstrand et al. 2015, measured at 150 ∘C. The solid lines are the corresponding predictions to the data from Eq. 4. The dashed lines are the corresponding predictions to the data from Eq. 4 without a limit on the chain extensibility. The dotted line is the linear viscoelastic prediction based on the parameters listed in Table 1 using Eq. 3
The particular values of the entanglement molecular weight and Kuhn step length affect the value of the maximal extensibility. For the PMMA, we have used the Kuhn step length of the atactic PMMA of M K =0.6 kg/mol. It is expected to be independent of a dilution with an ideal solvent giving a maximal relative stretch of \(\lambda _{max}^{2}=M_{e,\theta }/M_{K}=9/\theta \). We have added the theoretical prediction with the maximal extensibility in Eq. 4 as well. This is the solid lines (——) in Figs. 6 and 7. The used limit on the chain extensibility predicts steady extensional viscosity at values only slightly above the measured values. Notice in Fig. 7 that the measured extensional viscosities from Wingstrand et al. (2015) at low extensional rates are somewhat above the expected value of three times the zero shear viscosity obtained from the mechanical spectroscopic measurements. Our model is of course bound to (three times) the zero shear viscosity from the mechanical spectroscopic measurements.
One particular issue are important to notice. In line with the majority of published ideas, the Rouse time relative to the relaxation time is expected to depend uniquely on the number of entanglements, Z. The difference between the Rouse time for the polystyrene and the PMMA in Eqs. 7 and 8 respectively is significant. This systematic difference is about a factor of 2.4. In the originally suggested ‘interchain pressure’ idea by Marrucci and Ianniruberto (2004), where the tube deformation is controlled by the increasing tube pressure originating from the stretch of the surrounding chains, the time scale is represented by the relaxation time of the squeezing pressure, τ p . τ p scales with the number of entanglements as the Rouse time, τ R , though they differ in the proportionality factor. Ideally, the difference between these two times is dependent on the number of Kuhn steps between entanglements solely. However, the exact relationship highly depends on the interpretation of the involved length scale of the friction factor in the model.
Summary and conclusion
The model predictions based on the constant interchain pressure assumption agree with the extensional measurements on oligomer diluted NMMD poly(methyl methacrylate)s from Wingstrand et al. (2015) and a wide range of oligomer diluted NMMD polystyrenes from Huang et al. (2015). With one exception, the transition to the steady-state viscosity can be explained by the finite chain extensibility based on the Kuhn steps in an entanglement. The exception is the most diluted polystyrene. In the case of an 18% 1880 kg/mol polystyrene in oligomer diluent, it seems not necessary to include the maximal extensibility within the limit of the measurements. When the maximal extensibility has an effect, it is responsible for the steady extensional viscosity. The time dependency is solely a consequence of the constant tube pressure assumption, which agrees with all the measured time dependent viscosities. Notice, when the constant tube pressure is responsible for the steady extensional viscosities as well agreement is also observed.
The assumption of universality in the Rouse time with number of entanglements seems to be challenged, as the proportionality factor for the Rouse time differs by a constant value of about 2.4 between oligomer diluted NMMD poly(methyl methacrylate)s and NMMD polystyrenes.
References
Bach A, Almdal K, Rasmussen H K, Hassager O (2003b) Elongational viscosity of narrow molar mass distribution polystyrene. Macromolecules 36(14):5174–5179
Baumgaertel M, Winter H H (1992) Interrelation between continuous and discrete relaxation time spectra. J Non-Newtonian Fluid Mech 44(1):15–36
Baumgaertel M, Schausberger A, Winter H H (1990) The relaxation of polymers with linear flexible chains of uniform length. Rheol Acta 29(5):400–408
Bhattacharjee P K, Oberhauser J P, McKinley G H, Leal L G, Sridhar T (2002) Extensional rheometry of entangled solutions. Macromolecules 35(27):10131–10148
Cohen A (1991) A padé approximant to the inverse Langevin function. Rheol Acta 30(3):270–273
De Gennes PG (1971) Reptation of a polymer chain in the presence of fixed obstacles. J Chem Phys 55 (2):571–572
Dhole S, Leygue A, Bailly C, Keunings R (2009) A single segment differential tube model with interchain tube pressure effect. J Non-Newtonian Fluid Mech 161(1-3):10–18
Doi M, Edwards S F (1978) Dynamics of concentrated polymer systems. III. Constitutive equation. J Chem Soc Faraday Trans II 74(1):1818–1832
Fang J, Kröger M, Öttinger H C (2000) A thermodynamically admissible reptation model for fast flows of entangled polymers. II. Model predictions for shear and extensional flows. J Rheol 44(6):1293–1317
Fetters LJ, Lohse DJ, Colby RH (2007) Chain Dimensions and Entanglement Spacing. Physical Properties of Polymers Handbook 5(2):447–454
Fetters LJ, Lohse DJ, Richter D, Witten TA, Zirkel A (1994) Connection between polymer molecular weight, density, chain dimensions, and melt viscoelastic properties. Macromolecules 27(17):4639–4647
Fuchs K, Friedrich C, Weese J (1996) Viscoelastic Properties of Narrow-Distribution Poly(methyl methacrylates). Macromolecules 29(18):5893–5901
Huang Q, Mednova O, Rasmussen H K, Alvarez N J, Skov A L, Almdal K, Hassager O (2013a) Concentrated polymer solutions are different from melts: role of entanglement molecular weight. Macromolecules 46 (12):5026–5035
Huang Q, Alvarez N J, Matsumiya Y, Rasmussen H K, Watanabe H, Hassager O (2013b) Extensional rheology of entangled polystyrene solutions suggests importance of nematic interactions. ACS Macro Letters 2(8):741–744
Huang Q, Hengeller L, Alvarez N J, Hassager O (2015) Bridging the gap between polymer melts and solutions in extensional rheology. Macromolecules 48(12):4158–4163
Ianniruberto G (2015) Extensional flows of solutions of entangled polymers confirm reduction of friction coefficient. Macromolecules 48(17):6306–6312
Ianniruberto G, Brasiello A, Marrucci G (2012) Simulations of fast shear flows of PS oligomers confirm monomeric friction reduction in fast elongational flows of monodisperse PS melts as indicated by rheooptical data. Macromolecules 45(19):8058–8066
Krishnamoorti R, Graessley W W, Zirkel A, Richter D, Hadjichristidis N, Fetters L J, Lohse D J J (2002). Polym Sci, Part B Polym Phys 40:1768
Larson R G, Sridhar T, Leal L G, McKinley G H, Likhtman A E, McLeish T C B (2003) Definitions of entanglement spacing and time constants in the tube model. J Rheol 47(3):809–818
Likhtman A E, McLeish T C B (2002) Quantitative theory for linear dynamics of linear entangled polymers. Macromolecules 35(16):6332–6343
Marrucci G, Grizzuti N (1988) Fast flows of concentrated polymers—predictions of the tube model on chain stretching. Gazzetta Chimica Italiana 118(3):179–185
Marrucci G, Ianniruberto G (2004) Interchain pressure effect in extensional flows of entangled polymer melts. Macromolecules 37(10):3934–3942
Masubuchi Y, Matsumiya Y, Watanabe H (2014) Test of orientation/stretch-induced reduction of friction via primitive chain network simulations for polystyrene, polyisoprene, and Poly(n.butyl acrylate). Macromolecules 47(19):6768–6775
Menezes E V, Graessley W W (1982) Nonlinear rheological behavior of polymer systems for several shear-flow histories. J Polym Sci B Polym Phys 20(10):1817–1833
Osaki K, Nishizawa K, Kurata M (1982) Material time constant characterizing the nonlinear viscoelasticity of entangled polymeric systems. Macromolecules 15(4):1068–1071
Osaki K, Inoue T, Isomura T (2000) Stress overshoot of polymer solutions at high rates of shear. J Polym Sci B Polym Phys 38(14):1917–1925
Osaki K, Inoue T, Uematsu T, Yamashita Y (2001) Evaluation methods of the longest Rouse relaxation time of an entangled polymer in a semidilute solution. J Polym Sci B Polym Phys 39(14):1704–1712
Rasmussen H K (2015) Interchain tube pressure effect in the flow dynamics of bi-disperse polymer melts. Rheol Acta 54(1):9–18
Rasmussen H K, Christensen J H, Gttsche S J (2000) Inflation of polymer melts into elliptic and circular cylinders. J Non-Newtonian Fluid Mech 93(2-3):245–263
Rasmussen H K, Bejenariu A G, Hassager O, Auhl D (2010) Experimental evaluation of the pure configurational stress assumption in the flow dynamics of entangled polymer melts. J Rheol 54(6):1325–1336
Rasmussen HK, Huang Q (2014) Interchain tube pressure effect in extensional flows of oligomer diluted nearly monodisperse polystyrene melts. Rheologica Acta 53(3):199–208
Sridhar T, Acharya M, Nguyen D, Bhattacharjee P K (2014) On the extensional rheology of polymer melts and concentrated solutions. Macromolecules 47(1):379–386
Takahashi M, Isaki T, Takigawa T, Masuda T (1993) Measurement of biaxial and uniaxial extensional flow behavior of polymer melts at constant strain rates. J Rheol 37(5):827–846
Urakawa O, Takahashi M, Masuda T, Golshan Ebrahimi N (1995) Damping functions and chain relaxation in uniaxial and biaxial extensions: comparison with the Doi-Edwards theory. Macromolecules 28(21):7196–7201
van Ruymbeke E, Nielsen J, Hassager O (2010) Linear and nonlinear viscoelastic properties of bidisperse linear polymers: mixing law and tube pressure effect. J Rheol 54(5):1155–1172
Wagner M H (2014) Scaling relations for elongational flow of polystyrene melts and concentrated solutions of polystyrene in oligomeric styrene. Rheol Acta 53(10-11):765–777
Wagner M H, Rolón-Garrido V H (2010) The interchain pressure effect in shear rheology. Rheol Acta 49 (5):459–471
Wagner M H, Kheirandish S, Hassager O (2005) Quantitative prediction of transient and steady-state elongational viscosity of nearly monodisperse polystyrene melts. J Rheol 49(6):1317–1327
Wingstrand S L, Alvarez N J, Huang Q, Hassager O (2015) Linear and nonlinear universality in the rheology of polymer melts and solutions. Phys Rev Lett 115(7):078302
Yaoita T, Isaki T, Masubuchi Y, Watanabe H, Ianniruberto G, Marrucci G (2012) Primitive chain network simulation of elongational flows of entangled linear chains: stretch/orientation-induced reduction of monomeric friction. Macromolecules 45 (6):2773–2782
Ye X, Sridhar T (2005) Effects of the polydispersity on rheological properties of entangled polystyrene solutions. Macromolecules 38(8):3442–3449
Acknowledgments
We thank Sara L. Wingstrand for sending the data of the oligomer diluted PMMA samples used in this paper. QH would like to acknowledge financial support from the Aage og Johanne Louis −Hansen Foundation. HKR would like to acknowledge financial support from the Otto Mønsted Foundation.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Rasmussen, H.K., Huang, Q. Constant interchain pressure effect in extensional flows of oligomer diluted polystyrene and poly(methyl methacrylate) melts. Rheol Acta 56, 27–34 (2017). https://doi.org/10.1007/s00397-016-0982-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00397-016-0982-z