
Abstract
MCP-1-induced protein (MCPIP, also known as ZC3H12A) has recently been uncovered to act as a negative regulator of inflammation. Expression of MCPIP was elevated in the ventricular myocardium of patients with ischemic heart failure. However, the role of MCPIP in the development of post-infarct cardiac inflammation and remodeling is unknown. The objective of the present study was to investigate whether MCPIP exerts an inhibitory effect on the cardiac inflammatory response and adverse remodeling after myocardial infarction (MI). Mice with cardiomyocyte-specific expression of MCPIP and their wild-type littermates (FVB/N) were subjected to permanent ligation of left coronary artery. The levels of MCPIP were significantly increased in the ischemic myocardium and sustained for 4 weeks after MI. Acute infarct size was comparable between groups. However, constitutive overexpression of MCPIP in the murine heart resulted in improved survival rate, decreased cardiac hypertrophy, less of fibrosis and scar formation, and better cardiac performance at 28 days after MI, along with a markedly reduced monocytic cell infiltration, less cytokine expression, decreased caspase-3/7 activities and apoptotic cell death compared to the wild-type hearts. Cardiomyocyte-specific expression of MCPIP also attenuated activation of cardiac NF-κB signaling and expression of inflammation-associated microRNAs (miR-126, -146a, -155, and -199a) when compared with the post-infarct wild-type hearts. In vitro, MCPIP expression suppressed hypoxia-induced NF-κB-luciferase activity in cardiomyocytes. In conclusion, MCPIP expression in the ischemic myocardium protects against adverse cardiac remodeling and dysfunction following MI by modulation of local myocardial inflammation, possibly through mitigating NF-κB signaling and suppressing inflammation-associated microRNA expression.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Adverse cardiac remodeling after myocardial infarction (MI) is a major factor contributing to the development of chronic heart failure. Extensive experimental evidence suggests persistent activation of the innate immune system is considered to be deleterious to the heart, although the initial immune response provides a short-term adaptive response to limit the cardiac injury [1, 14, 18, 28, 30]. In the infarcted heart, an excessive inflammatory activation is sufficient to cause maladaptive remodeling, by inducing death of surviving cardiomyocytes and enhancing ischemic myocardial injury [3, 9, 14, 22, 37]. All cells within the heart have the capacity to cause inflammatory response after being activated by activation of toll-like receptors (TLRs) and nuclear factor-kappaB (NF-κB) signaling pathways [14, 15, 28, 54]. Cardiomyocytes comprises about 20–30 % of all cells within the heart, and they also express TLRs and can be a significant source of innate immune responses [12, 15, 17, 29]. Extensive experimental evidence by gain- and loss-of-function studies suggests that persistent NF-κB activation in cardiomyocytes can cause cardiomyopathy and heart failure by mounting a robust inflammatory response that is common in the context of ischemic injury [17, 29, 58]. Various approaches that minimize inflammation to the heart have been shown to improve left ventricular remodeling and dysfunction following MI. For example, the TLR/IL-1 response inhibitor IRAK-M [7], and the NF-κB inhibitor A20 [25], were reported to attenuate post-infarction remodeling by negatively regulating the post-infarction inflammatory response. Such proteins, therefore, offer a unique ability to prevent the deleterious consequences of uncontrolled inflammation on post-MI tissue homeostasis and remodeling.
Monocyte chemoattractant protein-1 (MCP-1)-induced protein (MCPIP, also known as ZC3H12A or Regnase-1) was first identified as a ZCCCH-zinc finger protein which manifests a transcription factor-like activity [59], and regulates several biological function such as angiogenesis [39, 44, 47] and adipogenesis [57]. MCPIP is primarily expressed in monocytes and macrophages, and its transcription is rapidly induced in various types of cells upon NF-κB activation [49]. Recently, it has been revealed that MCPIP acts as a suppressor of cytokine signaling to modulate inflammation by negatively regulating NF-κB activation through its deubiquitinase activity [26] and/or by destabilizing a set of mRNAs encoding for inflammatory cytokine including IL-6 and IL-12p40 through its RNase activity [33, 49]. Intriguingly, MCPIP was also found to counteract Dicer by cleaving the terminal loops of pre-microRNAs (miRs), thus suppressing biosynthesis of mature miRs [52] that are recognized to regulate immune system homeostasis and cardiac remodeling [45, 48, 53]. Hence, MCPIP appears critically positioned at the interface to restrain inflammatory reaction. Given that the elevated expression of MCPIP in human myocardium with end-stage ischemic heart disease [59], we have investigated the potential role of MCPIP in regulating the post-infarction inflammatory response and cardiac remodeling. Here, we report that expression of MCPIP is up-regulated in murine ischemic hearts. Cardiomyocyte-targeted expression of MCPIP attenuates adverse post-MI remodeling and dysfunction, through modulation of local myocardial inflammatory reaction by inhibition of NF-κB activation and inflammation-associated miR production, indicating that up-regulation of MCPIP in ischemic myocardium may contribute to mitigate post-MI inflammatory response.
Methods
Animals
Transgenic mice (TG) with cardiac-specific expression of MCPIP were generated from the FVB/N strain under the control of α-myosin heavy chain promoter as described previously [42], and maintained in the Animal Facility at the University of Central Florida. FVB/N littermates were purchased from Harlan (Indianapolis, IN, USA) and served as wild-type (WT) controls. There was no obvious qualitative difference in the myocardial morphological and function between the WT and TG mice [42]. Male mice, 10–12 weeks old, were used for experiments. The experimental procedures in mice and protocol used in this study were approved by Animal Care and Use Committee of the University of Central Florida, in accordance with the Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources, 1996).
Surgical procedures of myocardial infarction
Mice were anesthetized with isoflurane (AErrane, Baxter, USA) and intubated with 20-gauge catheter, then placed in a supine position on a temperature-controlled heated pad to maintain body temperature at 37 °C. The tracheal tube was connected to a rodent ventilator (Harvard) at a rate of 120 per minute, tidal volume of 0.2 ml mixture of room air with 2.0 % isoflurane and 1.0 % oxygen. After left anterior thoracotomy and dissection of the pericardium, the left anterior descending (LAD) artery was ligated with a 7–0 polypropylene suture (Ethicon) between the left atrial appendage and the right ventricular outflow. Successful LAD occlusion was confirmed by local wall motion abnormality and myocardial color changes from bright red to white. The thoracic incision was closed using 6–0 Prolene sutures to adapt the ribs and 4–0 Prolene sutures were used to close the skin. Sham-operated animals were subjected to the same procedures without the LAD ligation and served as surgical controls. All operations were performed under an upright dissecting microscope. To assess the post-infarct mortality, mice were followed up and inspected daily from 24 h up to 28 days after surgery.
Assessment of cardiac function
The survival animals was followed up for 28 days to observe the heart failure syndromes such as respiratory stress, ascites, inactivity and hunched posture, and left ventricle (LV) function was assessed under anesthesia with inhalation of 1.5 % isoflurane via a nose cone before surgery and after 28 days of LAD ligation by echocardiography, using a 15 MHz probe (Sonos 4500, Philips) as we described previously [38].
Histological and histomorphometric assessment
The extent of infarcted myocardium after 24 h of LAD ligation was determined by 1 % triphenyl tetrazolium chloride (TTC) (Sigma-Aldrich, St. Louis, USA) staining, after 1 % Evans blue dye was injected into the coronary circulation. After 28 days of LAD ligation, the hearts were collected, fixed in 10 % (V/V) buffered formalin and embedded in paraffin for histological analysis or snap frozen in liquid nitrogen and stored at −80 °C for RNA and protein extraction. For investigating infract size and myocardial fibrosis, equatorial regions of the heart (each heart was cut transversely into 3 pieces) were routinely processed and paraffin embedded. Sections were stained with hematoxylin and eosin, and Masson’s Trichrome using standard protocols for histomorphometric analysis. At least one section from each of three pieces of heart sample was examined. Infarct scar size was measured as a percentage of scar length to total LV circumference. Quantitative assessments for myocardial fibrotic area were performed on five sections in five randomly selected fields per section and expressed as interstitial collagen volume fraction using computerized imaging software (NIS-Elements AR 3.10) as described previously [38]. The expression of collagen type I in the ischemic myocardium was examined by immunohistochemistry using anti-collagen I antibody (L-19; Santa Cruz Biotechnology) and a peroxidase-conjugated secondary antibody (Santa Cruz Biotechnology). Peroxidase activity was visualized with DAB (diaminobenzidine, BD Biosciences), and scored semi-quantitatively by assigning a proportion score and an intensity score as we described previously [40]. The extent of cardiomyocyte hypertrophy was determined on Masson’s trichrome-stained transverse sections by measurement of the cardiomyocyte cross-sectional area using optical cursors with computerized imaging software. Cells with central nuclei as judged by eye were chosen for measurements. Cell death by apoptosis was assessed by terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) using the In Situ Cell Death Detection Kit, TMR red (Roche Diagnostics) as described previously [44]. Then, the slides were washed in PBS and stained with anti-sarcomeric α-actin antibody (Abcam Inc.) overnight at 4 °C followed by incubation with an FITC-conjugated secondary antibody (Chemicon International) in the dark at room temperature. The slides were washed in PBS and mounted with DAPI (Vector Laboratories). The total TUNEL-positive cells were calculated in five randomly selected fields for each animal, and six to eight animals were studied per group. The activity of caspase-3/7 was also determined using the Apo-ONE homogenous caspase-3/7 Assay kit (Promega) as described previously [44].
Immunohistochemistry and quantitative molecular pathology
Cell infiltration in the myocardium was examined by immunohistochemistry with FITC-conjugated anti-mouse CD11b/CD18 (Mac-1) and anti-mouse Mac-3 antibodies (Cedarlane Labs) overnight at 4 °C. Neovascularization in the ischemic myocardium was assessed by immunostaining with anti-platelet endothelial cell adhesion molecule-1 (PECAM-1/CD31) antibody (LifeSpan BioSciences) overnight at 4 °C and then with a Cy3-conjugated secondary antibody (Chemicon International). NF-κB activation in the ischemic myocardium was examined by immunostaining with anti-NF-κB p65 antibody (Santa Cruz Biotechnology) overnight at 4 °C and then with a Cy3-conjugated secondary antibody and nuclei were stained with DAPI (Vector Laboratories). All stained sections were subsequently washed in PBS, mounted and analyzed with a fluorescence microscopy equipped with a Nike digital camera and computerized imaging software. Five to ten different fields from three randomly selected sections per heart were used for quantitative analysis of cell infiltration, NF-κB p65 nuclear translocation and neovascularization in the infarcted myocardium, and five to six animals were examined per group.
RNA isolation and quantitative real-time RT-PCR (qRT-PCR)
Total RNA was extracted from myocardium using the miRCURYTM RNA isolation kit (Exiqon). The isolated RNA can be used for miR as well as mRNA analysis. cDNA synthesis for miR analysis was performed using the Universal MicroRNA cDNA Synthesis Kit II according to the manufacturer’s protocol (Exiqon). cDNA for mRNA analysis was performed using the High Capacity cDNA Reverse Transcription Kit according to the manufacturer’s protocol (Applied Biosystems). qRT-PCR was carried out in a 7500 Fast Real-time PCR system. All samples were run in duplicate. Relative expression was calculated using the comparative threshold cycle (Ct) method as we described previously [44].
qRT-PCR analyses for the mRNAs of MCPIP, TNF-α, IL-1β, IL-6, and β-actin were performed using Fast SYBR® Green Master Mix (Applied Biosystems) reagents with the following parameters: 95 °C for 30 s followed by 40 cycles of 95 °C, 5 s, 60 °C, 5 s, and 72 °C, 30 s. The mRNA level of β-actin was used as an internal control. The primers used were as follows: MCPIP: 5′-TGAGCCATGGGAAGAAGGAAGTCT-3′ and reverse, 5′-TGTGCTGGTCTGTGATAGGCACAT-3′; TNF-α: forward 5′-GTGGAACTGGCAGAAGAGGC-3′ and reverse, 5′-AGACAGAAGAGCGTGGTGGC-3′; IL-1β: forward, 5′-GGAAGATTCTGAAGAAGAGACGG-3′ and reverse, 5′-TGAGATTTTTAGAGTAACAGG-3′; IL-6: forward, 5′-ACAAGTCGGAGGCTT AATTACACAT-3′ and reverse, 5′-AATCAGAATTGCCATTGCACAA-3′; β-actin, forward, 5′-ATGTTTGAGACCTTCAACA-3′ and reverse, 5′-CACGTCAGACTTCATGATGG-3′.
qRT-PCR analyses for detection of miR-126, -146a, -155, -199a were performed using ExiLEMT SYBR® Green Master Mix (Exiqon) reagents. The primers for mature miR-126, -146a, -155, -199a, and U6 reference primers were obtained from Exiqon Inc. PCR reactions were performed using the following parameters: 95 °C for 2 min followed by 40 cycles of 95 °C, 15 s, and 60 °C, 30 s. U6 small nuclear RNA was used as endogenous control for data normalization.
Immunoblotting analysis
Myocardial cytosolic and nuclear proteins were extracted from mice myocardium with ice-cold tissue lysis buffer as we described previously [44]. Equal amounts of proteins were run on 12 % SDS-PAGE and transferred to nitrocellulose membranes. After blocking, membranes were incubated with the following primary antibodies: polyclonal rabbit anti-TNF-α, polyclonal rabbit anti-IL-1β, polyclonal rabbit anti-IL-6 (Santa Cruz Biotechnology), polyclonal rabbit anti-IкBα, anti-phospho-IкBα, anti-p65 (Cell Signaling Technology), and polyclonal goat anti-actin (Sigma) antibodies. The immune complexes were detected using appropriate peroxidase-labeled secondary antibodies (Santa Cruz Biotechnology) and chemiluminescence ECL kit (Amersham). Specific bands were quantified by densitometry using Alpha imager 2200 software.
Cell transfection and in vitro cardiomyocyte studies
H9c2 cardiomyocyte cells (CRL-1446; American Type Culture Collection, Manassas, VA, USA) were seeded at a concentration of 1.0 × 105 cells per well in six-well plates, and cotransfected with 1 μg of MCPIP plasmid DNA and 1 μg of NF-κB-luciferase reporter construct using the FuGENE 6 Transfection Reagent (Roche Applied Science, Indianapolis, IN, USA) in serum-free cell culture media as we described previously [43]. An expression plasmid of pSV40-β-galactosidase was also added as control for transfection efficiency. After 24 h exposure to the transfection mixture, the media on the transfected cells were removed and cells were subjected to hypoxia (1 % O2, 5 % CO2) in a HERAcell 150 CO2 incubator with O2 control (Thermo Fisher Scientific, Waltham, MA, USA) for 24 h under serum-free culture. Then, cells were harvested in passive lysis buffer (Promega) and luciferase activity was measured using the reporter luciferase assay system (Promega) and normalized to control β-galactosidase activity. Detailed description for the expression plasmids for MCPIP and its empty control vector was reported previously [59].
The neonatal murine cardiomyocytes were isolated from hearts removed from decapitated 1- to 3-day-old WT and TG mice as described previously [51]. Myocytes were enriched by the differential attachment to remove non-cardiomyocytes and seeded on a 96-well microtiter plates at a density of 1.0 × 105 cells/well. Cells were cultured in a 5 % CO2 atmosphere with DMEM containing 5 % fetal calf serum, 200 μM l-glutamine, and penicillin/streptomycin. To determine the activity of NF-κB, cells were cotransfected with 1 μl of NF-κB reporter and 1 μl of negative control reporter (NF-κB reporter kit, BPS Bioscience) using Lipofectamine 2000 (Life Technologies) in serum- and antibiotic-free Opti MEM I medium. After 24 h exposure to the transfection mixture, cells were subjected to hypoxia treatment for 24 h as described above. Luciferase activity was measured using Dual-Glo luciferase assay system (Promaga) by reading on an Envision Luminometer (Perkin Elmer) and expressed as the ratio of the firefly luminescence from the NF-κB reporter to Renilla luminescence from the control Renilla vector.
For visualization of NF-κB translocation, neonatal murine cardiomyocytes isolated from 1- to 3-day-old WT and TG mice were subjected to hypoxia treatment for 24 h as described above, fixed with 4 % paraformaldehyde, permeabilized with 0.1 % triton X-100, then immunostaining with an anti-NF-κB antibody directed against the p65 subunit (Cell Signaling Technology) overnight at 4 °C, followed by decoration with a FITC-conjugated secondary antibody (Chemicon International), and analyzed with a fluorescence microscopy equipped with a Nike digital camera and computerized imaging software.
Statistical analysis
All values are presented as mean ± standard deviation. Student’s t tests were used to compare means between two groups. Comparison of survival was performed by using a Kaplan–Meier method followed by the log-rank test. Significant differences between treatment groups were determined by two-way analysis of variance for multiple comparisons. A p value <0.05 was considered significant.
Results
Ischemic stress induces MCPIP expression in murine hearts
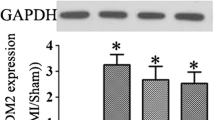
To investigate whether ischemia causes the expression of MCPIP in the hearts in vivo, we examined the levels of MCPIP in the hearts of WT mice after LAD ligation. The levels of MCPIP mRNA were significantly greater in early post-MI hearts (14 days) than in sham-operated controls, and the levels, although began to decline, were still high in 28 days post-MI hearts (Fig. 1a). Expression of MCPIP in the ischemic myocardium was confirmed by immunohistochemistry with MCPIP antibody, whereas no signal was detected with the IgG control probe (Fig. 1b). Cardiac myocytes and the infiltrated inflammatory cells in the heart were defined as the main cellular source of MCPIP expression. Immunoblots show that protein levels of MCPIP were markedly increased after MI compared to the sham controls (Fig. 1c). To further confirm the specific expression of MCPIP in the myocardium, the levels of MCPIP in cardiac tissues from WT, MCPIP knockout (KO), and transgenic (TG) mice were assessed by RT-PCR and Western blotting with the MCPIP-specific polyclonal antibody. Hearts from MCPIP TG mice markedly expressed MCPIP at mRNA and protein levels compared to WT mice, whereas no MCPIP expression was observed in the myocardium of MCPIP KO mice (Fig. 1d, e). The overall increase in MCPIP expression for the TG mice was about tenfold compared to WT sham control, and the levels of myocardial MCPIP mRNA in the TG mice showed no significant changes after MI (Fig. 1f).

MCPIP is up-regulated in the infarcted mouse heart. a WT mice were subjected to permanent ligation of LAD and expression of MCPIP in the infarcted hearts was examined by qRT-PCR at days 7, 14, and 28 after MI. Data were normalized to the sham-operated controls. *p < 0.05 versus the respective values of sham, n = 5. b Representative photomicrographs of immunohistochemical staining for myocardial MCPIP expression (brown) in the WT mice at day 14 after MI. c Proteins were extracted from pooled hearts of three animals, separated on 12 % SDS-PAGE, transferred onto a nitrocellulose membrane, and probed with anti-MCPIP polyclonal antibody. Densitometric quantification of immunoblot bands from pooled mice hearts normalized by β-actin protein levels. *p < 0.05 versus the respective values of sham controls. d, e Total RNA and proteins were isolated from pooled hearts of three animals and assayed by RT-PCR and immunoblots. *p < 0.05 versus the respective WT and MCPIP KO mice. f Transgenic mice hearts were subjected to permanent ligation of LAD for 7, 14 and 28 days and total RNA was isolated from pooled hearts of three animals, and changes in MCPIP expression were examined by qRT-PCR. Data were normalized to the sham-operated WT controls
Enhanced MCPIP expression improves post-MI survival and adverse cardiac remodeling
To examine the effect of MCPIP on ischemic heart, mice with cardiomyocyte-targeted expression of MCPIP and their littermates were subjected to permanent LAD ligation or a sham operation. Evans blue/TTC staining demonstrated that acute infarct size after 24 h of LAD ligation was comparable between WT (37.6 ± 1.8 %, n = 5) and TG mice (38.2 ± 1.6 %, n = 5). The survival of animals after MI was followed up for 28 days and the post-MI survival rates of WT and TG mice were compared. TG mice had significantly greater survival after MI compared with WT mice (76 versus 54 %, p < 0.05; Fig. 2a). 7 of 24 WT mice (29.2 %) and 4 of 23 TG mice (17.4 %, p < 0.05) died of LV rupture, which occurred frequently within 3–5 days post-MI. At 28 days after MI, the heart/body weight ratio (HW/BW) was lower in TG mice compared to WT mice (Fig. 2b). Histological analyses based on the trichrome-stained sections showed that the average scar size after 28 days MI was 37.8 ± 3.2 % in WT mice but only 27.9 ± 4.6 % in TG mice (Fig. 2c). Interstitial fibrosis was present in both WT and TG mice following MI, and the extent of fibrosis in the infarcted TG mice was much less than the infarcted WT mice (Fig. 2d). Quantitative assessments for fibrotic area showed a significant decrease in collagen volume fraction in the infarcted TG mice compared to the infracted WT mice (Fig. 2d). Analysis of the mRNA levels of fibrotic factors, such as transforming growth factor-β1 (TGF-β1), matrix metalloproteinase-9 (MMP-9) and tissue inhibitor of matrix metalloproteinase-1 (TIMP-1), showed a marked increase in the infarcted WT mice, whereas this increase in TGF-β1, MMP-9 and TIMP-1 was attenuated in the infarcted TG mice (Fig. 2e). Immunohistochemical staining also revealed lower immunoreactivity for collagen I in the myocardium of post-MI TG mice compared to the post-MI WT mice (Fig. 2f). Cardiac function was evaluated on day 28 after MI with echocardiography. Examples of M-mode echocardiogram obtained from WT and TG mice at 28 days after surgery are presented in Fig. 3a. In contrast to the sham-operated animals, permanent LAD ligation resulted in LV dilatation and functional impairment in both WT and TG mice. However, TG mice exhibited a better preservation of LV function and geometry as demonstrated by the greater fractional shortening with less ventricular dilatation compared to WT mice (Fig. 3b). Collectively, these results indicate that enhanced expression of MCPIP attenuated post-MI adverse remodeling and dysfunction.

Cardiomyocyte-specific expression of MCPIP improves post-infarct survival and ischemic cardiomyopathy. a Kaplan–Meier graph of survival showed improved survival of MCPIP TG mice (p = 0.039; 76 % survival, n = 23) compared with WT mice (54 % survival, n = 24) after ligation of LAD for 28 days. b Heart weight/body weight ratio of WT and MCPIP TG mice at post-MI for 28 days. *p < 0.05 versus the respective sham controls. # p < 0.05 versus WT MI, n = 7 in each group. c Representative photomicrographs of the infarcted hearts at 28 days post-MI and stained with Masson’s trichrome. The infarct scar size was determined as a percentage of the scar length (blue) to total LV circumference (blue plus red) and showed a significant reduction in scar size in MCPIP TG mice. # p < 0.05 versus WT MI, n = 7 in each group. d Representative photomicrographs of Masson’s trichrome-stained cardiac tissue sections show the interstitial fibrosis at 28 days following MI, and the bar graphs represent the quantitative analysis of the interstitial fibrosis in both WT and MCPIP1 TG mice hearts. *p < 0.05 versus WT MI, n = 7 in each group. e Expression of fibrotic factors TGF-β1, MMP-9, and TIMP-1 in the myocardium following ligation of LAD for 28 days was determined by qRT-PCR. *p < 0.05 versus the respective sham controls; # p < 0.05 versus WT MI, n = 7 in each group. f Representative analysis of cardiac sections stained with collagen 1 (brown) and histogram showing a significant reduction of collagen 1 deposition in the myocardia of post-MI TG mice. *p < 0.05 versus WT MI, n = 4 in each group

Cardiomyocyte-specific expression of MCPIP improves post-infarct cardiac dysfunction. a Representative M-mode echocardiograms from WT and age-matched MCPIP TG mice show changes in LV dimensions after MI. b Quantitative analysis of the echocardiographic findings in WT and MCPIP TG mice after MI for 28 days. LVEDD, LV end-diastolic dimension; LVESD, LV end-systolic dimension; LVPWD, LV end-diastolic posterior wall dimension
Enhanced MCPIP expression reduces myocardial hypertrophy and apoptosis following MI
Cardiomyocyte cross-sectional area was measured to evaluate the extent of myocardial hypertrophy at 28 days following MI. The infarcted TG hearts displayed a much smaller cardiomyocyte cross-sectional area compared to the infarcted WT hearts (Fig. 4a). Consistent with this finding, qRT-PCR analysis using 28-day post-MI samples detected a markedly reduced expression of atrial natriuretic factor (ANF), a major hypertrophic marker, in the infarcted TG hearts compared to in the infarcted WT hearts (Fig. 4b). Vascular density was assessed to determine the effect of MCPIP on post-ischemic neovascularization using immunolabeling for PECAM-1. There was no significant difference in vascular density in the ischemic myocardium between the WT and TG mice at 28 days after MI (Fig. 4c). The extent of apoptotic cell death in the infarcted hearts was assessed by TUNEL and sarcomeric α-actin labeling, and counterstained with DAPI. The infarcted WT hearts showed a large number of nuclei that were TUNEL positive, whereas the infarcted TG hearts had much less TUNEL-positive nuclei (Fig. 4d). Quantitative assessments showed a significant decrease in apoptosis in both cardiomyocytes and non-cardiomyocytes in the infarcted TG mice compared to the infracted WT mice (Fig. 4d). This observation was also manifested by the reduced activities of caspase-3/7, a hallmark of the apoptotic process, in the infarcted TG hearts compared to the infarcted WT hearts (Fig. 4e).

Cardiomyocyte-specific expression of MCPIP attenuates adverse cardiac remodeling following MI. a Representative photomicrographs of H&E-stained tissue sections show cardiomyocyte cross-sectional area at 28 days following MI, and the bar graphs represent the quantitative analysis of the cardiomyocyte cross-sectional area in both WT and MCPIP TG mice after ligation of LAD for 28 days. *p < 0.05 versus WT MI, n = 7 in each group. b Expression of hypertrophy marker gene of ANF at 28 days following MI was determined by qRT-PCR. *p < 0.05 versus the respective sham controls; # p < 0.05 versus WT MI, n = 7 in each group. c Representative photomicrographs of immunohistochemical staining for CD31 (red fluorescence) at 28 days following MI, and the quantitative analysis of CD31 immunoreactivity showed no significant difference in the capillary density between WT and MCPIP TG hearts following MI for 28 days, n = 7 in each group. d Representative photomicrographs of TUNEL labeling for apoptotic cells at 28 days following MI sections, and MCPIP TG mice following MI had significant less TUNEL-positive cardiomyocytes and non-myocytes. *p < 0.05 versus WT MI, n = 7 in each group. SAA sarcomeric α-actin (green); TUNEL (pink); DAPI (blue). e Caspase-3/7 activity in heart homogenates derived from WT and MCPIP TG hearts following MI for 4 weeks was measured quantitatively using APO-One caspase-3/7 assay kit. *p < 0.05 versus the respective sham controls; # p < 0.05 versus WT MI, n = 7 in each group
Enhanced MCPIP levels suppress inflammatory activation following MI
MCP-1-induced protein is recognized as a negative regulator in inflammatory cytokine signaling [26, 33, 49], and cardiomyocytes have the capacity to cause cardiac inflammatory response upon ischemic stress [9, 12, 28, 29]. We examined whether cardiomyocyte-specific expression of MCPIP could affect post-MI inflammation. Sections from the infarcted WT and TG hearts (day 28 of MI) were labeled with antibody against Mac-1, a surface marker for both granulocytes and monocytes/macrophages, and antibody against Mac-3, a surface marker for activated macrophages, respectively (Fig. 5a, b). The number of positive cells for Mac-1 and Mac-3 was clearly less in the post-MI TG hearts compared to the post-MI WT hearts (Fig. 5a, b). To further evaluate inflammatory response in the ischemic myocardium, we measured the mRNA levels of TNF-α, IL-1β, and IL-6 in the post-MI hearts. Myocardial mRNA levels of TNF-α, IL-1β, and IL-6 were significantly increased in the post-MI hearts compared to the sham-operated controls (Fig. 5c–e). This increase in mRNA levels of all three cytokines was significantly attenuated in the post-MI TG hearts compared to that of the post-MI WT hearts, and mRNA levels of IL-6 in the post-MI TG hearts were as low as the levels found in the sham-operated hearts (Fig. 5e). This pattern of gene expression changes was also seen at the protein level (Fig. 5f–h), suggesting that cardiac-specific expression of MCPIP suppressed inflammatory response in the post-MI hearts.

Cardiomyocyte-specific expression of MCPIP attenuates cellular inflammatory response in the infarcted hearts. a, b Tissue sections from 28 days post-MI mice hearts were labeled with monocyte/macrophage markers Mac-1 (green fluorescence; A) and Mac-3 (green fluorescence; B). The bar graphs represent the quantification of Mac-1 and Mac-3 positive cells in the post-MI myocardium. *p < 0.05 versus WT MI, n = 7 in each group. c–e Expression of inflammatory cytokine genes for TNF-α, IL-1β, and IL-6 in the 28 days post-MI hearts was determined by qRT-PCR. *p < 0.05 versus the respective sham controls; # p < 0.05 versus WT MI, n = 5 in each group. f–h Proteins were extracted from pooled hearts of three animals of 28 days post-MI were assayed for levels of TNF-α, IL-1β, and IL-6 by immunoblots, and immunoreactivating bands were quantified by densitometric analysis. *p < 0.05 versus the respective sham controls; # p < 0.05 versus WT MI, n = 5 in each group
Enhanced MCPIP expression negatively regulates NF-κB activation in the post-MI hearts
MI promotes NF-κB-dependent inflammatory transcription and oxidative injury in the post-infarct myocardium [14, 17, 37]; we tested whether cardiac-specific expression of MCPIP could affect NF-κB activation in the post-infarct hearts. Sections from post-MI hearts (day 28) were labeled with antibody against NF-κB p65, and nuclei were stained with DAPI to assess the NF-κB p65 nuclear translocation. Immunofluorescence staining demonstrated that NF-κB p65 translocation into the nuclei in the infarcted TG hearts was much less compared to the infarcted WT hearts (Fig. 6a, b). This observation was confirmed by immunoblot analysis that showed a decrease in NF-κB p65 levels in nuclear extract of the infarcted TG hearts compared to the infarcted WT hearts (Fig. 6c). NF-κB p65 nuclear translocation requires the phosphorylation-dependent degradation of IκBα [6]. The levels of IκBα phosphorylation was determined by immunoblot analysis. The results showed an increased level of IκBα phosphorylation in the post-MI WT hearts and this increase was suppressed in the post-MI TG hearts (Fig. 6d), indicating that enhanced expression of MCPIP suppressed NF-κB activation in the infarcted heart.

Cardiomyocyte-specific expression of MCPIP suppresses NF-κB activation in the infarcted hearts and neonatal cardiomyocytes. a Tissue sections from 28 days post-MI hearts from WT and MCPIP TG mice were labeled with NF-κB p65 subunit (red fluorescence) and nuclei were counterstained with DAPI (blue fluorescence) and the images are merged (pink fluorescence). b The bar graphs represented the quantification of NF-κB p65 positive cells in the 28 days post-MI hearts. MCPIP TG hearts had significantly less NF-κB p65 positive cells. *p < 0.05 versus WT, n = 7 in each group. c, d Levels of NF-κB p65 nuclear translocation and IκBα phosphorylation in the 28 days post-MI hearts were assayed by immunoblots in the nuclear and the cytosolic extracts from WT and MCPIP TG hearts, and the immunoreactivating bands were quantified by densitometric analysis normalized by internal controls. *p < 0.05 versus the respective sham controls; # p < 0.05 versus WT MI, n = 5 in each group. e Cultured H9c2 cardiomyocytes were transfected with empty vector or with MCPIP expression construct for 24 h, and then cells were transfected with an NF-κB-luciferase reporter plasmid for 24 h and subjected to hypoxic condition for 24 h. Luciferase activity from cell lysates was determined. Cells expressing MCPIP had significantly reduced NF-κB-luciferase activity. *p < 0.05 versus untreated cells; # p < 0.05 versus cells transfected with empty vector. f Neonatal murine cardiomyocytes isolated from WT and TG mice were transferred with NF-κB promoter and negative control promoter for 24 h, and then subjected to hypoxia condition for 24 h. Luciferase activity was measured as described in the “Methods”. Cardiomyocytes from TG mice had significantly reduced NF-κB-luciferase activity. g Hypoxia induced nuclear translocation of NF-κB in cardiomyocytes from WT mice, which markedly reduced cardiomyocytes derived from TG mice. NF-κB nuclear translocation was detected by immunofluorescence using an anti-NF-κB antibody directed against the p65 subunit (green)
NF-κB activation is required for inflammatory gene expression in response to hypoxia [13]. To test whether MCPIP regulates hypoxia-induced NF-κB activity in cardiomyocytes, cultured H9c2 cardiomyocytes were transfected with MCPIP plasmid and NF-κB-luciferase reporter construct, then subjected to hypoxia for 24 h. Cells transfected with NF-κB reporter construct showed a significant increase of luciferase activity when stimulated with hypoxia; however, this effect was drastically decreased when cells were preinfected with MCPIP plasmid for 24 h (Fig. 6e). Similar results were obtained in neonatal murine cardiomyocytes isolated from WT and TG mice. Hypoxia induced a significant increase of luciferase activity in cardiomyocytes derived from WT mice, but not TG mice (Fig. 6f). Moreover, hypoxia induced a significant increase in nuclear translocation of NF-κB in cardiomyocytes derived from WT mice, whereas NF-κB is retained in the cytoplasm of cardiomyocytes derived from TG mice (Fig. 6g), thus demonstrating the functional consequences of cardiomyocyte-specific expression of MCPIP on NF-κB activation in cardiomyocytes.
Enhanced MCPIP expression influences miRNA expression in the post-MI hearts
Several miRs, like miR-126, -146a, -155, and -199a, are known to be involved in inflammation [5, 8, 48] and post-MI remodeling [19, 45, 53]. Since MCPIP was reported to inhibit miR biogenesis [52], we determined whether expression of these miRs can also be suppressed by cardiomyocyte-specific expression of MCPIP. Expression of these miRs in the 28 days post-MI hearts were analyzed by qRT-PCR, and normalized by their respective sham-operated controls. Results show that the infracted myocardium of WT mice had a significant up-regulation of miR-126, -146a, -155, and -199a, whereas expression of these miRs was not significantly altered in the infarcted myocardium of TG mice (Fig. 7), suggesting that cardiomyocyte-specific expression of MCPIP suppresses the production of inflammation-associated miRs.

Cardiomyocyte-specific expression of MCPIP suppresses the expression of inflammation-associated miRs in the infarcted heart. The expression of inflammation-associated miRs in the 28 days post-MI hearts was examined by qRT-PCR. Expression levels for miR-126 (a), miR-146a (b), miR-155 (c), and miR-199a (d) were induced significantly in the post-MI WT hearts compared to that of the respective sham controls, whereas the increased expression of these miRs was suppressed in MCPIP TG hearts following MI. *p < 0.05 versus the respective sham controls; # p < 0.05 versus WT MI, n = 5 in each group
Discussion
Emerging evidence suggests that uncontrolled chronic immune activation is a major pathogenetic factor for this deleterious remodeling process [1, 14, 18, 20, 28, 30, 37]. Therefore, anti-inflammatory strategies to control chronic immune activation in the heart should be therapeutically relevant in preventing the progression of post-MI heart failure [7, 11, 22, 25, 32]. We show here for the first time that ischemia induces induction of MCPIP in the murine heart and overexpression of MCPIP in cardiomyocytes improves post-infarct remodeling and function in vivo, and this outcome is associated with suppression of cardiac inflammation through inhibiting NF-κB activation and reducing inflammation-associated miRs expression in the infarcted heart. These findings suggest that MCPIP functions as a suppressor for the post-infarct cardiac inflammation and could be a potential target for ischemic heart disease.
MCPIP mRNA has previously been identified in whole heart homogenates and shown to be up-regulated in the end-stage ischemic heart of human [59]. However, whether intrinsic cardiomyocytes or circulating and/or non-cardiomyocytes account for cardiac MCPIP expression was not known. We show here that, MCPIP is expressed in murine cardiomyocytes after MI. Even though cardiomyocytes in the infarct zone undergo a dramatic loss, MCPIP was still robustly expressed in the ischemic myocardium, indicating that other type of cells, such as inflammatory cells and non-cardiomyocytes could be the source of MCPIP expression. In fact, we found MCPIP mRNA in isolated, cultured cardiomyocytes, smooth muscle cells and endothelial cells upon oxidative stress (e.g., H2O2), and inflammatory cytokine (e.g., TNF-α) stimulation (data not shown). In response to MI, cardiomyocytes are able to initiate an adaptation response by activation of NF-κB that exerts not only proinflammatory actions but also triggers expression of anti-inflammatory agents that inhibit the inflammatory reaction [22, 25, 32]. MCPIP expression has been shown to be dependent on activation of NF-κB [49] and to negatively regulate inflammatory response in many types of cells [26, 49]. It is possible that MCPIP may play an important role in the resolution of post-infarct inflammation.
To address the cardiac effects of MCPIP, we used transgenic mice with cardiomyocyte-specific expression of MCPIP. After LAD ligation, hearts of MCPIP expressing mice showed significantly better cardiac functional and histomorphological parameters as compared to WT mice together with a significantly reduced cardiac inflammation. These results suggest that myocardial MCPIP expression suppresses post-infarct inflammatory reaction and protects heart from adverse remodeling. Several distinct mechanisms have been reported to explain the inhibitory effects of MCPIP on the inflammatory response. First, MCPIP is thought to inhibit NF-κB signaling by removing polyubiquitin chain from TNF receptor-associated factor (TRAF)-2 and -6 [26]. Second, MCPIP may act as an RNase, thus destabilizing a set of cytokine-encoding mRNAs such as IL-6, IL-1β and IL-12p40 [33, 49]. Results of our study showed that cardiomyocyte-specific expression of MCPIP inhibited expression of proinflammatory cytokines (TNF-α, IL-β and IL-6), leading to reduced infiltration of Mac-1-, and Mac-3-positive inflammatory leukocytes in the infarcted heart and attenuated expression of pro-fibrotic factors (TGF-β1, MMP-9 and TIMP-1) and apoptotic cell death. The decreased proinflammatory cytokine expression by cardiomyocyte-specific expression of MCPIP likely reflects its RNase activity to degrade cytokine-encoding mRNAs caused by ischemic damage. The decreased number of inflammatory leukocytes in the infarcted myocardium of MCPIP expressing mice may reflect a global attenuation of inflammatory signaling pathways responsible for recruitment of inflammatory cells [37]. Previous studies have shown that inflammatory cytokines and infiltrating leukocytes may induce death of surviving cardiomyocytes in the infracted myocardium, extending ischemic myocardial injury and contribute to sustained inflammation and result in the development of heart failure [3, 12, 14, 18, 20, 37]. A strong up-regulation of TNF-α in microinfarction after microembolization is associated with ventricular dysfunction [9]. MMP-9 is highly induced in response to inflammatory cytokines, and enhanced expression of MMP-9 has been linked to post-MI remodeling [4, 10, 21], and MMP-9 gene deletion or treatment with an MMP-9 active site inhibitor has been shown to modulate the cellular inflammatory response and improve post-MI remodeling [10, 21]. Thus, cardiomyocyte-specific expression of MCPIP may modulate the cardiomyocyte function via timely resolving the post-infarct inflammatory process, leading to improve survival and post-MI cardiac remodeling. Although MCPIP was reported to promote inflammatory angiogenesis [39, 44, 47], no significant difference in the immunohistochemically detectable capillary density in the post-MI hearts was observed between the two groups, probably due to targeted MCPIP expression in cardiomyocytes, but not in the endothelial cells or monocytic cells that are known to be involved in post-ischemic neovascularization [2].
Failing hearts exhibit chronic activation of NF-κB and sustained inflammation [16, 29], and blockage of NF-κB has been shown to protect heart from advanced remodeling after MI [17, 25, 58]. MCPIP was reported to inhibit NF-κB activation in other cell types [26, 49]. We analyzed the effect of cardiomyocyte-specific expression of MCPIP on the NF-κB activation in the heart following MI, by examining IкBα phosphorylation and p65 subunit translocation in the infarcted myocardium. Our results show clearly that cardiomyocyte-specific expression of MCPIP inhibits the activation of NF-κB in the ischemic myocardium. This inhibitory effect was also demonstrated by using the NF-κB-luciferase assay in the H9c2 myofibroblasts and neonatal murine cardiomyocytes. We found that overexpression of MCPIP was able to reduce the NF-κB-luciferase activity and p65 subunit nuclear translocation induced by hypoxia in neonatal murine cardiomyocytes in vitro. These data are therefore, in general, consistent with our previous data showing suppression of LPS-induced cardiac NF-κB activity by MCPIP [42]. Although not directly studied, the suppression of NF-κB activation in the infarcted hearts by MCPIP might be attributed to its deubiquitinase activity, because our recent findings indicated that overexpression of MCPIP substantially inhibited TGF-β-activated kinase (TAK-1) (C. Menaa, and P. E. Kolattukudy, unpublished), which is activated by ubiquitinated proteins [6]. This hypothesis is further supported by the recent mutantgenesis studies showing MCPIP-dependent inhibition of NEMO liner ubiquitylation diminishes sequential activation of TAK-1, resulting in the decreased NF-κB activation [41]. Thus, further studies are needed to identify the ubiquitinated protein(s) that are substrates for MCPIP in the ischemic myocardium.
Recent studies have indicated that mature miR levels are altered in ischemic hearts from animal models and patients, and may represent potential targets to the therapeutic modulation of post-infarct remodeling [45, 53]. For example, miRs associated with regulation of the inflammatory pathway (i.e., miR-146a, -155, and -126,) and oxidative stress (i.e., -199a) have been consistently found to be strongly up-regulated in ischemic hearts and failing heart [8, 50, 53, 56]. MCPIP was previously reported to be able to cleave the terminal loops of precursor miRs, thus suppressing the biosynthesis of mature miRs [52]. Our current study confirmed that these miRs (i.e., miR-146a, -155, -126, and -199a) were up-regulated in the post-MI WT hearts, whereas the expression of these miRs was suppressed by the presence of MCPIP, suggesting that down-regulation of these miRs may be beneficial to the post-MI remodeling. Taken together, our observations indicate a broader role for MCPIP in protecting against post-infarct cardiac inflammation and adverse remodeling.
A growing body of evidence suggests that events underlying myocardial adaptation to ischemia injury involve the signaling of the innate immune response [1, 4, 11, 24, 55]. It has been shown that a low-dose of TNF-α reduces infarct size when given before sustained ischemia–reperfusion [24]. Administration of a sublethal dose of LPS reduces subsequent myocardial infarction and improves cardiac functions [3, 55]. This preconditioning-like protection of the heart against subsequent ischemic injury has been linked with induction of cellular protective proteins (i.e., HSPs) [23] and reduction of cellular inflammatory response [34, 46]. MCPIP is up-regulated in cardiomyocytes by inflammatory agents (i.e., LPS) and has been shown to protect against LPS-induced myocardial dysfunction [42]. Mice with cardiac-targeted expression of MCP-1 manifest myocardial inflammation, MCPIP expression and preconditioning-like cardioprotection against ischemia–reperfusion injury and post-infarction remodeling [31, 35]. MCPIP was also reported to participate in LPS preconditioning-induced neuroprotection against brain ischemic injury by preventing cellular inflammatory response [27]. The cardioprotective role of MCPIP uncovered in the present study is consistent with these observations and raises the possibility that targeted MCPIP expression in cardiomyocytes may be involved in “preconditioning” of the heart against subsequent uncontrolled activation of innate immune response by the ischemic injury. This could be one of the reasons leading to increased survival of MCPIP TG mice within a week after MI and restricted inflammatory response in adverse remodeling. Further studies using different models (e.g., myocarditis) will need to fully understand the roles of MCPIP in the modulation of inflammatory response, granulation tissue formation and long-term adverse cardiac remodeling.
In conclusion, our findings show a complex picture in which MCPIP affects inflammatory response involved in the processes of post-infarction cardiac remodeling. Our data demonstrate that cardiomyocytes express MCPIP upon ischemic stress and provide a functional relevance to this expression. The underlying mechanisms of MCPIP on cardiomyocytes involve suppression of inflammatory response and timely resolution of inflammation, and thereby, improvement of myocardial remodeling. The recognition of MCPIP as an innate anti-inflammatory protein in the heart may offer a potential therapeutic strategy that target MCPIP and could limit the severity of ischemic cardiac injury. However, there are many cellular components that contribute to the production of inflammatory cytokines in the heart [15, 18, 36]. Cytokines are released by immune cells that infiltrate the heart, with significant consequences on the pathogenesis of post-infarct remodeling [14, 20, 24, 37], further studies to establish the roles of MCPIP in myeloid cells, in the context of post-infarct cardiac remodeling are also critical to design therapeutic strategies.
References
Arslan F, de Kleijn DP, Pasterkamp G (2011) Innate immune signaling in cardiac ischemia. Nat Rev Cardiol 8:292–300. doi:10.1038/nrcardio.2011.38
Baer C, Squadrito ML, Iruela-Arispe ML, De Palma M (2013) Reciprocal interactions between endothelial cells and macrophages in angiogenic vascular niches. Exp Cell Res 319:1626–1634. doi:10.1016/j.yexcr.2013.03.026
Baumgarten G, Kim SC, Stapel H, Vervölgyi V, Bittig A, Hoeft A, Meyer R, Grohé C, Knuefermann P (2006) Myocardial injury modulates the innate immune system and changes myocardial sensitivity. Basic Res Cardiol 101:427–435. doi:10.1007/s00395-006-0597-0
Blömer N, Pachel C, Hofmann U, Nordbeck P, Bauer W, Mathes D, Frey A, Bayer B, Vogel B, Ertl G, Bauersachs J, Frantz S (2013) 5-Lipoxygenase facilitates healing after myocardial infarction. Basic Res Cardiol 108:367. doi:10.1007/s00395-013-0367-8
Boldin MP, Baltimore D (2012) MicroRNAs, new effectors and regulators of NF-κB. Immunol Rev 246:205–220. doi:10.1038/ni.f.209
Chen J, Chen ZJ (2013) Regulation of NF-κB by ubiquitination. Curr Opin Immunol 25:4–12. doi:10.1016/j.coi.2012.12.005
Chen W, Saxena A, Li N, Sun J, Gupta A, Lee DW, Tian Q, Dobaczewski M, Frangogiannis NG (2012) Endogenous IRAK-M attenuates postinfarction remodeling through effects on macrophages and fibroblasts. Arterioscler Thromb Vasc Biol 32:2598–2608. doi:10.1161/ATVBAHA.112.300310
Corsten MF, Papageorgiou A, Verhesen W, Carai P, Lindow M, Obad S, Summer G, Coort SL, Hazebroek M, van Leeuwen R, Gijbels MJ, Wijnands E, Biessen EA, De Winther MP, Stassen FR, Carmeliet P, Kauppinen S, Schroen B, Heymans S (2012) MicroRNA profiling identifies microRNA-155 as an adverse mediator of cardiac injury and dysfunction during acute viral myocarditis. Circ Res 111:415–425. doi:10.1161/CIRCRESAHA.112.267443
Dörge H, Schulz R, Belosjorow S, Post H, van de Sand A, Konietzka I, Frede S, Hartung T, Vinten-Johansen J, Youker KA, Entman ML, Erbel R, Heusch G (2002) Coronary microembolization: the role of TNF-alpha in contractile dysfunction. J Mol Cell Cardiol 34:51–62. doi:10.1006/jmcc.2001.1489
Ducharme A, Frantz S, Aikawa M, Rabkin E, Lindsey M, Rohde LE, Schoen FJ, Kelly RA, Werb Z, Libby P, Lee RT (2000) Targeted deletion of matrix metalloproteinase-9 attenuates left ventricular enlargement and collagen accumulation after experimental myocardial infarction. J Clin Invest 106:55–62. doi:10.1172/JCI8768
Duerr GD, Heinemann JC, Suchan G, Kolobara E, Wenzel D, Geisen C, Matthey M, Passe-Tietjen K, Mahmud W, Ghanem A, Tiemann K, Alferink J, Burgdorf S, Buchalla R, Zimmer A, Lutz B, Welz A, Fleischmann BK, Dewald O (2014) The endocannabinoid-CB2 receptor axis protects the ischemic heart at the early stage of cardiomyopathy. Basic Res Cardiol 109:425. doi:10.1007/s00395-014-0425-x
Fallach R, Shainberg A, Avlas O, Fainblut M, Chepurko Y, Porat E, Hochhauser E (2010) Cardiomyocyte Toll-like receptor 4 is involved in heart dysfunction following septic shock or myocardial ischemia. J Mol Cell Cardiol 48:1236–1244. doi:10.1016/j.yjmcc.2010.02.020
Fitzpatrick SF, Tambuwala MM, Bruning U, Schaible B, Scholz CC, Byrne A, O’Connor A, Gallagher WM, Lenihan CR, Garvey JF, Howell K, Fallon PG, Cummins EP, Taylor CT (2011) An intact canonical NF-κB pathway is required for inflammatory gene expression in response to hypoxia. J Immunol 186:1091–1096. doi:10.4049/jimmunol.1002256
Frangogiannis NG (2014) The immune system and the remodeling infarcted heart: cell biological insights and therapeutic opportunities. J Cardiovasc Pharmacol 63:185–195. doi:10.1038/nrcardio.2014.28
Fujiu K, Nagai R (2013) Contributions of cardiomyocyte-cardiac fibroblast-immune cell interactions in heart failure development. Basic Res Cardiol 108:357. doi:10.1007/s00395-013-0357-x
Grabellus F, Levkau B, Sokoll A, Welp H, Schmid C, Deng MC, Takeda A, Breithardt G, Baba HA (2002) Reversible activation of nuclear factor-κB in human end-stage heart failure after left ventricular mechanical support. Cardiovasc Res 53:124–130. doi:10.1016/S0008-6363(01)00433-3
Hamid T, Guo SZ, Kingery JR, Xiang X, Dawn B, Prabhu SD (2011) Cardiomyocyte NF-κB p65 promotes adverse remodelling, apoptosis, and endoplasmic reticulum stress in heart failure. Cardiovasc Res 89:129–138. doi:10.1093/cvr/cvq274
Heusch G, Libby P, Gersh B, Yellon D, Böhm M, Lopaschuk G, Opie L (2014) Cardiovascular remodelling in coronary artery disease and heart failure. Lancet 383:1933–1943. doi:10.1016/S0140-6736(14)60107-0
Heymans S, Corsten MF, Verhesen W, Carai P, van Leeuwen RE, Custers K, Peters T, Hazebroek M, Stöger L, Wijnands E, Janssen BJ, Creemers EE, Pinto YM, Grimm D, Schürmann N, Vigorito E, Thum T, Stassen F, Yin X, Mayr M, de Windt LJ, Lutgens E, Wouters K, de Winther MP, Zacchigna S, Giacca M, van Bilsen M, Papageorgiou AP, Schroen B (2013) Macrophage microRNA-155 promotes cardiac hypertrophy and failure. Circulation 128:1420–1432. doi:10.1161/CIRCULATIONAHA.112.001357
Hofmann U, Frantz S (2013) How can we cure a heart “in flame”? A translational view on inflammation in heart failure. Basic Res Cardiol 108:356. doi:10.1007/s00395-013-0356-y
Hughes BG, Schulz R (2014) Targeting MMP-2 to treat ischemic heart injury. Basic Res Cardiol 109:424. doi:10.1007/s00395-014-0424-y
Kain V, Prabhu SD, Halade GV (2014) Inflammation revisited: inflammation versus resolution of inflammation following myocardial infarction. Basic Res Cardiol 109:444. doi:10.1007/s00395-014-0444-7
Kaucsár T, Bodor C, Godó M, Szalay C, Révész C, Németh Z, Mózes M, Szénási G, Rosivall L, Sőti C, Hamar P (2014) LPS-induced delayed preconditioning is mediated by Hsp90 and involves the heat shock response in mouse kidney. PLoS ONE 9:e92004. doi:10.1371/journal.pone.0092004
Kleinbongard P, Schulz R, Heusch G (2011) TNFα in myocardial ischemia/reperfusion, remodeling and heart failure. Heart Fail Rev 16:49–69. doi:10.1007/s10741-010-9180-8
Li HL, Zhuo ML, Wang D, Wang AB, Cai H, Sun LH, Yang Q, Huang Y, Wei YS, Liu PP, Liu DP, Liang CC (2007) Targeted cardiac overexpression of A20 improves left ventricular performance and reduces compensatory hypertrophy after myocardial infarction. Circulation 115:1885–1894. doi:10.1161/CIRCULATIONAHA.106.656835
Liang J, Saad Y, Lei T, Wang J, Qi D, Yang Q, Kolattukudy PE, Fu M (2010) MCP-induced protein 1 deubiquitinates TRAF proteins and negatively regulates JNK and NF-kappaB signaling. J Exp Med 207:2959–2973. doi:10.1084/jem.20092641
Liang J, Wang J, Saad Y, Warble L, Becerra E, Kolattukudy PE (2011) Participation of MCP-induced protein 1 in lipopolysaccharide preconditioning-induced ischemic stroke tolerance by regulating the expression of proinflammatory cytokines. J Neuroinflammation 8:182–192. doi:10.1186/1742-2094-8-182
Lin L, Knowlton AA (2014) Innate immunity and cardiomyocytes in ischemic heart disease. Life Sci 100:1–8. doi:10.1016/j.lfs.2014.01.062
Maier HJ, Schips TG, Wietelmann A, Krüger M, Brunner C, Sauter M, Klingel K, Böttger T, Braun T, Wirth T (2012) Cardiomyocyte-specific IκB kinase (IKK)/NF-κB activation induces reversible inflammatory cardiomyopathy and heart failure. Proc Natl Acad Sci USA 109:11794–11799. doi:10.1073/pnas.1116584109
Mann DL (2011) The emerging role of innate immunity in the heart and vascular system: for whom the cell tolls. Circ Res 108:1133–1145. doi:10.1161/CIRCRESAHA.110.226936
Martire A, Fernandez B, Buehler A, Strohm C, Schaper J, Zimmermann R, Kolattukudy PE, Schaper W (2003) Cardiac overexpression of monocyte chemoattractant protein-1 in transgenic mice mimics ischemic preconditioning through SAPK/JNK1/2 activation. Cardiovasc Res 57:523–534. doi:10.1016/S0008-6363(02)00697-1
Maskrey BH, Megson IL, Whitfield PD, Rossi AG (2011) Mechanisms of resolution of inflammation: a focus on cardiovascular disease. Arterioscler Thromb Vasc Biol 31:1001–1006. doi:10.1161/ATVBAHA.110.213850
Matsushita K, Takeuchi O, Standley DM, Kumagai Y, Kawagoe T, Miyake T, Satoh T, Kato H, Tsujimura T, Nakamura H, Akira S (2009) Zc3h12a is an RNase essential for controlling immune responses by regulating mRNA decay. Nature 458:1185–1190. doi:10.1038/nature07924
Merry HE, Wolf PS, Fitzsullivan E, Keech JC, Mulligan MS (2010) Lipopolysaccharide pre-conditioning is protective in lung ischemia-reperfusion injury. J Heart Lung Transplant 29:471–478. doi:10.1016/j.healun.2009.11.005
Morimoto H, Takahashi M, Izawa A, Ise H, Hongo M, Kolattukudy PE, Ikeda U (2006) Cardiac overexpression of monocyte chemoattractant protein-1 in transgenic mice prevents cardiac dysfunction and remodeling after myocardial infarction. Circ Res 99:891–899. doi:10.1161/01.RES.0000246113.82111.2d
Müller J, Gorressen S, Grandoch M, Feldmann K, Kretschmer I, Lehr S, Ding Z, Schmitt JP, Schrader J, Garbers C, Heusch G, Kelm M, Scheller J, Fischer JW (2014) Interleukin-6-dependent phenotypic modulation of cardiac fibroblasts after acute myocardial infarction. Basic Res Cardiol 109:440. doi:10.1007/s00395-014-0440-y
Nian M, Lee P, Khaper N, Liu P (2004) Inflammatory cytokines and post-myocardial infarction remodeling. Circ Res 94:1543–1553. doi:10.1161/01.RES.0000130526.20854.fa
Niu J, Azfer A, Deucher MF, Goldschmidt-Clermont PJ, Kolattukudy PE (2006) Targeted cardiac expression of soluble Fas prevents the development of heart failure in mice with cardiac-specific expression of MCP-1. J Mol Cell Cardiol 40:810–820. doi:10.1016/j.yjmcc.2006.03.010
Niu J, Azfer A, Zhelyabovska O, Fatma S, Kolattukudy PE (2008) Monocyte chemotactic protein (MCP)-1 promotes angiogenesis via a novel transcription factor, MCP-1-induced protein (MCPIP). J Biol Chem 283:14542–14551. doi:10.1074/jbc.M802139200
Niu J, Gilliland MG, Jin Z, Kolattukudy PE, Hoffman WH (2014) MCP-1and IL-1β expression in the myocardia of two young patients with Type 1 diabetes mellitus and fatal diabetic ketoacidosis. Exp Mol Pathol 96:71–79. doi:10.1016/j.yexmp.2013.11.001
Niu J, Shi Y, Xue J, Miao R, Huang S, Wang T, Wu J, Fu M, Wu ZH (2013) USP10 inhibits genotoxic NF-κB activation by MCPIP1-facilitated deubiquitination of NEMO. EMBO J 32:3206–3219. doi:10.1038/emboj.2013.247
Niu J, Wang K, Graham S, Azfer A, Kolattukudy PE (2011) MCP-1-induced protein attenuates endotoxin-induced myocardial dysfunction by suppressing cardiac NF-кB activation via inhibition of IкB kinase activation. J Mol Cell Cardiol 51:177–186. doi:10.1016/j.yjmcc.2011.04.018
Niu J, Wang K, Kolattukudy PE (2011) Cerium oxide nanoparticles inhibit oxidative stress and nuclear factor-κB activation in H9c2 cardiomyocytes exposed to cigarette smoke extract. J Pharmacol Exp Ther 338:53–61. doi:10.1124/jpet.111.179978
Niu J, Wang K, Zhelyabovska O, Saad Y, Kolattukudy PE (2013) MCP-1-induced protein promotes endothelial-like and angiogenic properties in human bone marrow monocytic cells. J Pharmacol Exp Ther 347:288–297. doi:10.1124/jpet.113.207316
Papait R, Greco C, Kunderfranco P, Latronico MV, Condorelli G (2013) Epigenetics: a new mechanism of regulation of heart failure? Basic Res Cardiol 108:361. doi:10.1007/s00395-013-0361-1
Rosenzweig HL, Lessov NS, Henshall DC, Minami M, Simon RP, Stenzel-Poore MP (2004) Endotoxin preconditioning prevents cellular inflammatory response during ischemic neuroprotection in mice. Stroke 35:2576–2581. doi:10.1161/01.STR.0000143450.04438.ae
Roy A, Zhang M, Saad Y, Kolattukudy PE (2013) Antidicer RNAse activity of monocyte chemotactic protein-induced protein-1 is critical for inducing angiogenesis. Am J Physiol Cell Physiol 305:C1021–C1032. doi:10.1152/ajpcell.00203.2013
Schroen B, Heymans S (2012) Small but smart–microRNAs in the centre of inflammatory processes during cardiovascular diseases, the metabolic syndrome, and ageing. Cardiovasc Res 93:605–613. doi:10.1093/cvr/cvr268
Skalniak L, Mizgalska D, Zarebski A, Wyrzykowska P, Koj A, Jura J (2009) Regulatory feedback loop between NF-κB and MCP-1-induced protein 1 RNase. FEBS J 276:5892–5905. doi:10.1111/j.1742-4658.2009.07273.x
Song XW, Li Q, Lin L, Wang XC, Li DF, Wang GK, Ren AJ, Wang YR, Qin YW, Yuan WJ, Jing Q (2010) MicroRNAs are dynamically regulated in hypertrophic hearts, and miR-199a is essential for the maintenance of cell size in cardiomyocytes. J Cell Physiol 225:437–443. doi:10.1002/jcp.22217
Sreejit P, Kumar S, Verma RS (2008) An improved protocol for primary culture of cardiomyocyte from neonatal mice. In Vitro Cell Dev Biol Anim 44:45–50. doi:10.1007/s11626-007-9079-4
Suzuki HI, Arase M, Matsuyama H, Choi YL, Ueno T, Mano H, Sugimoto K, Miyazono K (2011) MCPIP1 ribonuclease antagonizes Dicer and terminates microRNA biogenesis through precursor microRNA degradation. Mol Cell 44:424–436. doi:10.1016/j.molcel.2011.09.012
Topkara VK, Mann DL (2011) Role of microRNAs in cardiac remodeling and heart failure. Cardiovasc Drugs Ther 25:171–182. doi:10.1007/s10557-011-6289-5
Valen G, Yan ZQ, Hansson GK (2001) Nuclear factor kappa-B and the heart. J Am Coll Cardiol 38:307–314. doi:10.1016/S0735-1097(01)01377-8
Valeur HS, Valen G (2009) Innate immunity and myocardial adaptation to ischemia. Basic Res Cardiol 104:22–32. doi:10.1007/s00395-008-0756-6
van de Vrie M, Heymans S, Schroen B (2011) MicroRNA involvement in immune activation during heart failure. Cardiovasc Drugs Ther 25:161–170. doi:10.1007/s10557-011-6291-y
Younce CW, Azfer A, Kolattukudy PE (2009) MCP-1 (monocyte chemotactic protein-1)-induced protein, a recently identified zinc finger protein, induces adipogenesis in 3T3-L1 pre-adipocytes without peroxisome proliferator-activated receptor gamma. J Biol Chem 284:27620–27628. doi:10.1074/jbc.M109.025320
Zhang XQ, Tang R, Li L, Szucsik A, Javan H, Saegusa N, Spitzer KW, Selzman CH (2013) Cardiomyocyte-specific p65 NF-κB deletion protects the injured heart by preservation of calcium handling. Am J Physiol Heart Circ Physiol 305:H1089–H1097. doi:10.1152/ajpheart.00067.2013
Zhou L, Azfer A, Niu J, Graham S, Choudhury M, Adamski FM, Younce C, Binkley PF, Kolattukudy PE (2006) Monocyte chemoattractant protein-1 induces a novel transcription factor that causes cardiac myocyte apoptosis and ventricular dysfunction. Circ Res 98:1177–1185. doi:10.1161/01.RES.0000220106.64661.71
Acknowledgments
This work was supported by Grant HL-69458 from National Institutes of Health. The authors thank Edilu Becerra for the care of the animals used in this study.
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Niu, J., Jin, Z., Kim, H. et al. MCP-1-induced protein attenuates post-infarct cardiac remodeling and dysfunction through mitigating NF-κB activation and suppressing inflammation-associated microRNA expression. Basic Res Cardiol 110, 26 (2015). https://doi.org/10.1007/s00395-015-0483-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00395-015-0483-8