
Abstract
Objective
C1q/tumour necrosis factor-related protein 12 (CTRP12) is closely related to coronary artery disease and has an outstanding cardioprotective effect. However, whether CTRP12 participates in heart failure (HF) has not been well studied. This work aimed to explore the role and mechanism of CTRP12 in post-myocardial infarction (MI) HF.
Methods
Rats were subjected to left anterior descending artery ligation and then raised for six weeks to establish post-MI HF. Recombinant adeno-associated virus-mediated gene transfer was applied to overexpress or silence CTRP12 in rat hearts. RT-qPCR, Immunoblot, Echocardiography, Haematoxylin–eosin (HE) staining, Masson’s trichrome staining, TUNEL staining and ELISA were carried out.
Results
CTRP12 levels were decreased in the hearts of rats with post-MI HF. The overexpression of CTRP12 improved cardiac function and attenuated cardiac hypertrophy and fibrosis in rats with post-MI HF. CTRP12 silencing exacerbated cardiac dysfunction, hypertrophy and fibrosis in rats with post-MI HF. The cardiac apoptosis, oxidative stress and inflammatory response induced by post-MI HF were weakened by CTRP12 overexpression or aggravated by CTRP12 silencing. CTRP12 inhibited the activation of the transforming growth factor‐β activated kinase 1 (TAK1)-p38 mitogen‐activated protein kinase (MAPK)/c‐Jun N‐terminal kinase (JNK) pathway in the hearts of rats with post-MI HF. Treatment with the TAK1 inhibitor reversed the adverse effects of CTRP12 silencing on post-MI HF.
Conclusions
CTRP12 protects against post-MI HF by modulating the TAK1-p38 MAPK/JNK pathway. CTRP12 may be a therapeutic target for the treatment of post-MI HF.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Heart failure (HF) is an endpoint stage of numerous heart disorders, severely jeopardising the health of a large population worldwide [1]. Cardiac remodelling, including myocardial hypertrophy and fibrosis, is a vital element contributing to the occurrence and progression of HF [2]. Myocardial infarction (MI) has been acknowledged as the most important cause of HF [3]. Currently, the prevalence of post-MI HF continues to increase as people after MI survive for longer than in the past [4]. However, the reversal of cardiac remodelling remains a treatment challenge for preventing post-MI HF. Hence, discovering new genes that critically affect the progression of cardiac remodelling and determining the relevant mechanism may have great significance in developing an effective target gene therapy for post-MI HF.
C1q/tumour necrosis factor-related protein 12 (CTRP12), a newly discovered member of the CTRP family, is paralogue of adiponectin and a vital metabolic modulator linking glucose and lipid metabolism [5,6,7,8,9]. Thus, CTRP12 is related to metabolic disorders, including diabetes, obesity and liver steatosis [5,6,7]. Importantly, decreased serum levels of CTRP12 are associated with the severity of coronary artery disease [10]. CTRP12 inhibits the phenotypic switch of vascular smooth muscle cells and macrophages, which is conductive to retarding the development of atherosclerosis [11, 12]. The up-regulation of CTRP12 relieves lipopolysaccharide-evoked inflammatory injury of cardiomyocytes [13]. The overexpression of CTRP12 is able to protect cardiomyocytes from hypoxia/re-oxygenation-elicited damage by impeding apoptosis, oxidative stress and inflammatory response [14]. Therefore, CTRP12 is closely related to cardiovascular disease and has a remarkable cardioprotective function.
Transforming growth factor-β-activated kinase 1 (TAK1), a serine/threonine kinase, is a member of the mitogen-activated protein kinase (MAPK) kinase and mediates numerous aspects of cellular functions under pathological disorders [15]. TAK1 can be activated by various stimuli [16,17,18], which rapidly induces the phosphorylation of p38 MAPK and JNK [19] and affects the convergence of signals for cell survival, apoptosis, oxidative stress and inflammatory response [20,21,22]. TAK1-p38 MAPK/JNK pathway participates in numerous cardiovascular diseases, including HF [23,24,25]. The activation of TAK1-p38 MAPK/JNK pathway serves as a critical determinant for the development of cardiac hypertrophy and fibrosis [23, 24, 26, 27], thereby plays a vital role for cardiac remodelling in post-MI HF. The TAK1-p38 MAPK/JNK pathway is adjusted by diverse regulators and mechanisms. Elucidating the regulatory mechanism on the TAK1-p38 MAPK/JNK pathway is important for understanding the molecular mechanism underlying post-MI HF.
Reportedly, CTRP12 is closely related to cardiovascular diseases and has an outstanding cardioprotective effect [10, 11, 14, 28]. However, whether CTRP12 participates in HF has not been well studied. This work aimed to explore the role and mechanism of CTRP12 in post-MI HF. CTRP12 levels were decreased in the hearts of rats with post-MI HF. The overexpression of CTRP12 in rat myocardium by adeno-associated virus-mediated gene transfer improved cardiac function and attenuated cardiac hypertrophy and fibrosis in rats with post-MI HF. Moreover, the cardiac apoptosis, oxidative stress and inflammatory response induced by post-MI HF were weakened by cardiac CTRP12 overexpression. Molecular mechanism studies revealed that CTRP12 affected the activation of the TAK1-p38 MAPK/JNK pathway in the hearts of rats with post-MI HF. Reducing TAK1 could reverse the adverse effects of CTRP12 silencing on post-MI HF. Collectively, CTRP12 protects against post-MI HF by modulating the TAK1-p38 MAPK/JNK pathway. CTRP12 may be a therapeutic target for the treatment of post-MI HF.
Materials and methods
Animals and ethical statement
Male Sprague–Dawley rats were provided by the Laboratory Animal Centre of Air Force Medical University and raised as per routine procedures. Animal use and investigation was officially approved by the Institutional Animal Care and Use Committee. Experimental procedures complied with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health.
Preparation of rat models of post-MI HF
Rats were subjected to surgery of the left anterior descending coronary artery ligation according to previously described protocols [29]. MI was confirmed by ST-segment elevation. Sham rats underwent the identical protocols without ligation. After MI, rats were raised for a further six weeks to induce HF.
Recombinant adeno-associated virus (AAV)
Recombinant serotype 9 AAV encoding CTRP12 or sh-CTRP12 was constructed and produced by GenePharma (Shanghai, China). AAV-CTRP12 or AAV-sh-CTRP12 (1 × 1011 viral genome particles) was delivered into rats by intravenous injection via the tail vein two weeks before surgery. The increase or decrease of CTRP12 in rat hearts was validated by real-time quantitative PCR (RT-qPCR) or Immunoblotting.
Echocardiography
Echocardiography was carried out using the Vivid 7 ultrasound system (GE, Boston, MA, USA) to monitor the two-dimensional M-mode echocardiograms of rats. Left ventricular ejection fraction (LVEF) and left ventricular fractional shortening (LVFS) were calculated based on the measurements.
Sample collection
Rats were deeply anaesthetised and weighed. The blood samples were collected from rat abdominal aortas. The hearts and lungs were harvested and weighed.
Enzyme-linked immunosorbent assay (ELISA)
After blood coagulation, the serum was collected by centrifugation and stored at 4 °C for ELISA detection. The contents of N-terminal pro-brain natriuretic peptide (NT-proBNP), interleukin (IL)-1β, IL-6 and tumour necrosis factor (TNF)-α in serum was measured by corresponding ELISA kits purchased from Elabscience (Wuhan, China).
RT-qPCR
Heart tissues were homogenised in Beyozol and total RNA was isolated by the chloroform-isopropanol extraction protocol. First-strand cDNA was synthesised by reverse-transcription reaction using total RNA, reverse transcriptase, Oligo(dT) primers and dNTP Mix. The cDNA was amplified using SYBR Green qPCR Mix and forward/reverse primer mix on a real-time fluorescence quantitative PCR instrument. Relative mRNA levels were quantified by the 2−ΔΔCt methodology using GADPH for normalisation.
Immunoblot
Heart tissues were homogenised in RIPA buffer and the supernatants were collected by centrifugation. The protein content in the supernatants was determined by the BCA method. Protein samples were resolved by SDS-PAGE and then transferred onto a nitrocellulose membrane. For protein detection, membranes were blocked before immunoblotting with primary antibodies. After incubation at room temperature for 1 h, membranes were washed and blotted with HRP-conjugated secondary antibodies. Then, membranes were washed and exposed to ECL reagents. Proteins were visualised and images were captured in an Imaging System (Bio-Rad Laboratories, Inc). Primary antibodies included anti-CTRP12 (Biorbyt, Wuhan, China), anti-GAPDH (Biorbyt), anti-Collagen I (Biorbyt), anti-Collagen III (Biorbyt), anti-TAK1 (Biorbyt), anti-p-TAK1 (Biorbyt), anti-NF-κB p65 (CST, Shanghai, China), anti-p-NF-κB p65 (CST), anti-p38 MAPK (CST), anti-p-p38 MAPK (CST), anti-JNK (CST) and anti-p-JNK (CST).
Haematoxylin–eosin (HE) staining
Heart tissues were fixed by formalin and underwent dehydration by graded ethanol and vitrification by dimethylbenzene before being embedded in paraffin. Paraffin-embedded tissues were sliced into 4 μm sections. The sections were stained with haematoxylin for 3 min and then washed for 10 min. Then, the slices were stained with eosin for 2 min. After dehydration and vitrification, the slices were sealed with neutral resins and observed under a light microscope. Images were captured and the myocyte cross‐sectional area was measured with the Image-Pro Plus 6.0.
Masson’s trichrome staining
The slices of heart tissues were stained with Wiegert iron haematoxylin for 5 min and washed with distilled water. Then, the slices were stained with Masson ponceau-acid solution for 5 min. After washing with molybdenum phosphate for 5 min, the slices were stained with aniline blue solution for 5 min. After dehydration and vitrification, the slices were sealed with neutral resins and observed under a light microscope. Images were captured and collagen volume fraction (%) was calculated with the Image-Pro Plus 6.0.
TUNEL staining
Paraffin-embedded slices were immersed for dewaxing at 60 °C for 30 min. The slices were washed and incubated with Protease K solution for 20 min at 37 °C. The slices were washed and stained with TdT labelling solution (Elabscience, Wuhan, China) for 60 min at 37 °C in a humidifying box. The slices were washed and then stained with DAPI solution for 5 min at room temperature in a humidifying box. The slices were sealed with antifade mounting medium and observed under a luminescence microscope.
ROS detection
Heart tissues were cut into small pieces and digested at 37 °C for 30 min. The residual tissues were removed by filtration and cells were harvested by centrifugation. A total of 1 × 106 cells were collected and suspended in DCFH-DA solution (10 μM) and incubated for 30 min at 37 °C. Cells were centrifuged and the supernatants were removed. Cells were washed and suspended in washing buffer before being detected by a flow cytometer.
Statistical analysis
Data were analysed via GraphPad Prism 8 software. Results were expressed in the form of mean ± standard deviation. Student’s t test or one‐way analysis of variance was employed to compare the difference between two groups or between multiple groups, respectively. Values of < 0.05 indicate the differences are statistically significant.
Results
Myocardial CTRP12 expression under post-MI HF condition
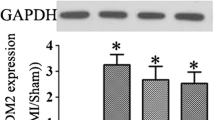
To assess the potential role of CTRP12 in post-MI HF, changes in CTRP12 in the hearts of rats with post-MI HF were analysed. Myocardial CTRP12 mRNA was down-regulated in rats with post-MI HF compared with sham rats (Fig. 1A). The protein level of CTRP12 was also decreased in the myocardium of rats with post-MI HF (Fig. 1B, C).

Myocardial CTRP12 expression in rats with post-MI HF. A RT-qPCR and B, C Immunoblot detection of CTRP12 expression in the myocardium of rats. **p < 0.01
Cardiac CTRP12 overexpression improves cardiac function in rats with post-MI HF
To detect the role of CTRP12 in post-MI HF, cardiac CTRP12 overexpression was achieved by AAV-CTRP12 infection in rats and its effects on cardiac function were determined. Infection of AAV-CTRP12 in rats led to marked increases in CTRP12 levels in rat myocardium (Fig. 2A, B). In rats with post-MI HF, the serum level of NT-proBNP was up-regulated (Fig. 2C) accompanied by decreased LVEF (Fig. 2D) and LVFS (Fig. 2E), indicating significant HF manifestations. Importantly, the serum level of NT-proBNP was down-regulated and LVEF and LVFS were increased in cardiac CTRP12-overexpressed rats with post-MI HF (Fig. 2C–E). To verify the cardioprotective role of CTRP12, the effects of CTRP12 loss on post-MI HF were further determined. The infection of AAV-sh-CTRP12 markedly depleted CTRP12 expression in the rat myocardium (Fig. 2F, G). On the contrary, cardiac dysfunction induced by post-MI HF including increased NT-proBNP and decreased LVEF and LVFS were aggravated in rats with cardiac CTRP12 silencing (Fig. 2H–J).

Effect of CTRP12 overexpression or knockdown on cardiac dysfunction induced post-MI HF. A, B Cardiac CTRP12 overexpression in rats infected with AAV-CTRP12 was validated via Immunoblot. Effects of CTRP12 overexpression on C serum levels of NT-proBNP, D LVEF and E LVFS. F, G Cardiac CTRP12 knockdown in rats infected with AAV-sh-CTRp12 was substantiated via Immunoblot. Effects of CTRP12 knockdown on H serum levels of NT-proBNP, I LVEF and J LVFS. **p < 0.01
Cardiac CTRP12 overexpression ameliorates cardiac hypertrophy and fibrosis in rats with post-MI HF
To further explore the role of CTRP12 in post-MI HF, the effects of CTRP12 overexpression on cardiac hypertrophy and fibrosis, two hallmarks of cardiac remodelling after HF, were further assessed. Cardiac hypertrophy manifestations, including increased heart weight/bodyweight (HW/BW) and lung weight (LW)/BW ratio (Fig. 3A, B) and larger heart size and cross-sectional area (Fig. 3C, D), occurred in rats with post-MI HF. Importantly, rats with cardiac CTRP12 overexpression exhibited lower HW/BW and LW/BW ratio (Fig. 3A, B) and smaller heart size and cross-sectional area (Fig. 3C, D). Rats with post-MI HF exhibited the disordered arrangement of myocardial fibre, massive collagen deposition and increased interstitial fibrosis (Fig. 3E, F). Moreover, post-MI HF also up-regulated the expression of collagen I and III in rat hearts (Fig. 3G, H. Post-MI HF-induced cardiac fibrosis and collagen expression were significantly attenuated in n cardiac CTRP12-overexpressed rats (Fig. 3E–H). On the contrary, cardiac hypertrophy (Fig. 4A–D) and fibrosis (Fig. 4E–H) induced by post-MI HF were aggravated in rats with cardiac CTRP12 silencing.

Effects of CTRP12 overexpression on cardiac hypertrophy induced by post-MI HF. Comparison of A HW/BW and B LW/BW ratio of rats among groups. C HE staining of heart sections and D quantification of the cross-sectional area. E Masson’s trichrome staining of heart sections and F quantification of fibrosis area. G, H Immunoblotting detection of collagen I and III in heat tissues. *p < 0.05 and **p < 0.01

Effects of CTRP12 knockdown on cardiac fibrosis induced by post-MI HF. Comparison of A HW/BW and B LW/BW ratio of rats among groups. C HE staining of heart sections and D quantification of cross-sectional area. E Masson’s trichrome staining of heart sections and F quantification of fibrosis area. G, H Immunoblotting detection of collagen I and III in heat tissues. *p < 0.05 and **p < 0.01
Cardiac CTRP12 overexpression relieves cardiac apoptosis and oxidative stress in rats with post-MI HF
The increased cardiac apoptosis and oxidative stress contribute to cardiac remodelling after HF. Indeed, increased cardiac apoptosis (Fig. 5A, B) and ROS generation (Fig. 5C, D) was observed in the hearts of rats with post-MI HF. Interestingly, cardiac overexpression of CTRP12 remarkably lowered cardiac apoptosis (Fig. 5A, B) and ROS generation (Fig. 5C, D) in rats with post-MI HF. On the contrary, cardiac silencing of CTRP12 exacerbated cardiac apoptosis (Fig. 5E, F) and ROS generation (Fig. 5G, H) in rats with post-MI HF.

Effects of CTRP12 overexpression or knockdown on cardiac apoptosis and oxidative stress induced by post-MI HF. A, B Effects of CTRP12 overexpression on cardiac apoptosis induced by post-MI HF were assessed by TUNEL assay. C, D Effect of CTRP12 overexpression on ROS generation induced by post-MI HF were evaluated by DCFH-DA staining. E, F Effects of CTRP12 knockdown on cardiac apoptosis induced by post-MI HF were determined by TUNEL assay. C, D Effect of CTRP12 knockdown on ROS generation induced by post-MI HF were examined by DCFH-DA staining. **p < 0.01
Cardiac CTRP12 overexpression weakened inflammatory response in rats with post-MI HF
The phosphorylation of NF-κB p65 protein in heart tissues (Fig. 6A, B) and serum levels of proinflammatory cytokines including IL-1β, IL-6 and TNF-α (Fig. 6C–E) were significantly elevated in rats with post-MI HF. Critically, cardiac overexpression of CTRP12 reduced the activation of NF-κB (Fig. 6A, B) and blocked the secretion of proinflammatory cytokines (Fig. 6C–E) in rats with post-MI HF. In contrast, cardiac silencing of CTRP12 enhanced the activation of NF-κB (Fig. 6F, G) and exacerbated the secretion of proinflammatory cytokines (Fig. 6H–J) in rats with post-MI HF.

Effects of CTRP12 overexpression or knockdown on inflammatory response induced by post-MI HF. A, B Effects of CTRP12 overexpression on the phosphorylation of NF-κB p65 protein were detected by Immunoblot. Effects of CTRP12 overexpression on the serum level of C IL-1β, D IL-6 and E TNF-α were monitored by ELISA. F, G Effects of CTRP12 knockdown on the phosphorylation of NF-κB p65 protein were examined by Immunoblot. Effects of CTRP12 knockdown on the serum level of H IL-1β, I IL-6 and J TNF-α were assessed by ELISA. **p < 0.01
Cardiac CTRP12 overexpression represses the activation of TAK1-p38 MAPK/JNK pathway induced by post-MI HF
The phosphorylation of TAK1, p38 MAPK and JNK in rat heart tissues was significantly increased by post-MI HF (Fig. 7A–D). Importantly, post-MI HF induced less up-regulation of phosphorylated TAK1, p38 MAPK and JNK in hearts from CTRP12-overexpressed rats (Fig. 7A–D). In contrast, the phosphorylation of TAK1, p38 MAPK and JNK induced by post-MI HF was remarkably enhanced in hearts from CTRP12-silenced rats (Fig. 7E–H).

Effects of CTRP12 overexpression or knockdown on TAK1-p38 MAPK/JNK pathway. Effects of CTRP12 A–D overexpression or E–H knockdown on the phosphorylation of TAK1, p38 MAPK and JNK in heart tissues of rats with post-MI HF were detected by Immunoblot. **p < 0.01
Restraining of TAK1 reverses CTRP12-knockdown-elicited deleterious effects on post-MI HF
To determine whether the TAK1-p38 MAPK/JNK pathway contributes to CTRP12-mediated cardioprotective effects on post-MI HF, we investigated the effect of TAK1 inhibition on CTRP12-knockdown-elicited deleterious effects on post-MI HF. TAK1 inhibitor NG25 was intraperitoneally injected into rats infected with AAV-sh-CTRP12. The increased phosphorylation of TAK1, p38 MAPK and JNK induced by CTRP12 knockdown in rats with post-MI HF was significantly restrained by NG25 (Fig. 8A–D). As expected, the deleterious effects of CTRP12 silencing on cardiac function (Fig. 8E, F), hypertrophy (Fig. 8G, H) and fibrosis (Fig. 8I) in rats with post-MI HF were obviously reversed by TAK1 inhibition. Moreover, the enhancing effects of CTRP12 silencing on cardiac apoptosis (Fig. 9A, B), ROS generation (Fig. 9C, D) and inflammatory response (Fig. 9E–G) in rats with post-MI HF were also decreased by TAK inhibition.

Effects of TAK1 inhibition on CTRP12-knockdown-elicited deleterious effects on cardiac function, hypertrophy and fibrosis of rats with post-MI HF. A–D Immunoblot detection of the phosphorylation of TAK1, p38 MAPK and JNK. Comparison of E LVEF and F LVFS of rats among groups. G HE staining and Masson’s trichrome of heart sections from different groups. Quantification of H cross-sectional area and I fibrosis area. *p < 0.05 and **p < 0.01

Effects of TAK1 inhibition on CTRP12-knockdown-elicited enhancing effects on cardiac apoptosis, ROS generation and inflammatory response of rats with post-MI HF. A, B TUNEL detection of cardiac apoptosis in rat heart tissues. C, D DCFH-DA staining of ROS level in rat heart tissues. ELISA detection of E IL-1β, F IL-6 and G TNF-α concentrations in rat serum. **p < 0.01
Discussion
This work has reported that cardiac CTRP12 overexpression is able to improve the cardiac function of rats with post-MI HF with the reduction of cardiac hypertrophy and fibrosis and the down-regulation of cardiac apoptosis, oxidative stress and inflammatory response. From a mechanistic perspective, our work showed that CTRP12 restrained the activation of TAK1-p38 MAPK/JNK pathway (Fig. 10), which may be the underlying basis for CTRP12-mediated cardioprotective effects on post-MI HF. This study indicates that CTRP12 may be a prospective candidate to prevent and treat post-MI HF.

A schematic diagram depicting the molecular mechanism of CTRP12 underlying pathological remodelling induced by post-MI HF
CTRP12 is closely related to numerous cardiovascular diseases. Serum levels of CTRP12 were found to be decreased in patients with coronary artery disease, which were inversely correlated with the severity of disease [10, 30]. The knockout of CTRP12 in mice enhanced neointimal thickening in the artery with wire-induced injury [11]. The overexpression of CTRP12 retarded the formation of atherosclerotic lesions in apolipoprotein E-deficient mice with a Western diet [12]. In vitro experiments demonstrated that CTRP12 could protect cardiomyocytes against destructive stimulus, such as lipopolysaccharide [13] and hypoxia/re-oxygenation [14]. However, whether CTRP12 participates in HF has not been well addressed. This work demonstrated decreased cardiac CTRP12 level in rats with post-MI HF, suggesting a possible link between CTRP12 and post-MI HF. Functional experiments elucidated that cardiac CTRP12 overexpression cardiac function and attenuated cardiac hypertrophy and fibrosis in rats with post-MI HF, whereas cardiac CTRP12 silencing demonstrated opposite effects. Therefore, these findings unveiled a cardioprotective effect for CTRP12 on post-MI HF. Interestingly, a recent study reported that CTRP12 repressed cardiac fibroblast transformation and cardiac fibrosis provoked by isoproterenol in mice [28], which was consistent with our results regarding the effect of CTRP12 on cardiac fibrosis. Collectively, CTRP12 protects against cardiac remodelling, which is beneficial to improve cardiac function after post-MI HF.
CTRP12 can influence apoptosis, oxidative stress and inflammation in response to different pathological stimuli. The treatment of vascular endothelial cells with CTRP12 protein decreased serum-deprivation-induced apoptosis [11]. The treatment of cultured macrophages with CTRP12 protein reduced lipopolysaccharide-stimulated secretion of inflammatory cytokines, including IL-6, TNF-α and monocyte chemotactic protein 1 [11]. The overexpression of CTRP12 restrained the inflammatory cytokine secretion, ROS production and apoptosis induced by lipopolysaccharide or hypoxia/re-oxygenation in cultured cardiomyocytes [13, 14]. The overexpression of CTRP12 alleviated the inflammatory response in lipid-laden macrophages [12]. The cardiac apoptosis, oxidative stress and inflammatory response contribute to cardiac remodelling in post-MI HF. Herein, we investigated whether CTRP12 affected these biological functions in post-MI HF. Crucially, cardiac CTRP12 overexpression was demonstrated to ameliorate cardiac apoptosis, ROS production and inflammation induced by post-MI HF in rats. Therefore, CTRP12 ameliorates cardiac remodelling in post-MI HF associated with blocking cardiac apoptosis, oxidative stress and inflammation.
CTRP12 exerts a vital role in signalling transduction. Reportedly, CTRP12 blocked the JNK activation induced by lipopolysaccharide in macrophages [11]. Moreover, CTRP12 restrained the p38 MAPK activation induced by isoproterenol in cardiomyocytes [28]. Considering that the JNK and p38 MAPK pathways play crucial roles in cardiac remodelling, we examined whether CTRP12 affected the activation of JNK and p38 MAPK pathways in post-MI HF. Importantly, the phosphorylation of JNK and p38 MAPK was increased in the myocardium of rats with post-MI HF, which was markedly attenuated by cardiac CTRP12 overexpression or aggravated by cardiac CTRP12 silencing. Moreover, we found that CTRP12 overexpression also blocked the phosphorylation of TAK1, a key upstream regulator of JNK and p38 MAPK. Additionally, the inhibition of TAK1 could reverse the enhancing effects of CTRP12 silencing on the activation of JNK and p38 MAPK. Our results showed that CTRP12 may affect the activation of JNK and p38 MAPK in post-MI HF by inhibiting TAK1. Furthermore, TAK1 inhibition also reversed the adverse effects of CTRP12 silencing on post-MI HF. In short, CTRP12 may ameliorate cardiac remodelling in post-MI HF by affecting the TAK1-p38 MAPK/JNK pathway.
Several concerns of this work should be noted. This work utilized the pharmacological inhibitor NG-25 to inactivate TAK1. However, NG-25 is a non-specific kinase inhibitor that also inhibits other kinases, such as MAP4K2, LYN, CSK, FER, p38α, ABL, ARG, and SRC [31]. Therefore, NG-25-mediated effect on post-MI HF may be not only related to TAK1. The PI3K/Akt signal cascade has been shown convincingly to be associated with post-MI HF [32]. It is worthy to investigate whether the PI3K/Akt signal cascade contributes to CTRP12-mediated effect on post-MI HF. Furthermore, CTRP12 is subject to cleavage and oligomerization leading to forms of the protein with different signalling preferences [33]. Proteolytic cleavage at the conserved Lys-91 by the proprotein convertase furin in the Golgi generates a shorter globular gCTRP12 isoform [33]. Full length CTRP12 isoform preferentially activates Akt signal cascade in H4IIE hepatocytes and 3T3-L1 adipocytes, while the gCTRP12 isoform preferentially activates MAPK signal cascade [33]. In this work, we have demonstrated a critical function of full length CTRP12 isoform in post-MI HF associated with regulation of the TAK1-p38 MAPK/JNK signal cascade. However, we did not specify the function of gCTRP12 isoform in post-MI HF. In this connection, further research is warranted to explore whether Akt signal cascade contributes to CTRP12-mediated protective effect on post-MI HF.
In summary, the current findings show that CTRP12 protects against post-MI HF by inhibiting activation of the TAK1-p38 MAPK/JNK pathway, providing novel insights into the underlying molecular mechanism for post-MI HF. Cardiac overexpression of CTRP12 can be regarded as a promising strategy for the treatment of post-MI HF.
Data availability
The datasets used during the present study are available from the corresponding author on reasonable request.
Abbreviations
- CTRP12:
-
C1q/tumour necrosis factor-related protein 12
- HF:
-
Heart failure
- MI:
-
Myocardial infarction
- TAK1:
-
Transforming growth factor‐β activated kinase 1
- MAPK:
-
Mitogen‐activated protein kinase
- JNK:
-
C‐Jun N‐terminal kinase
- AAV:
-
Adeno-associated virus
- LVEF:
-
Left ventricular ejection fraction
- LVFS:
-
Left ventricular fractional shortening
- ELISA:
-
Enzyme-linked immunosorbent assay
- NT-proBNP:
-
N-terminal pro-brain natriuretic peptide
- IL:
-
Interleukin
- HE:
-
Haematoxylin–eosin
- TNF:
-
Tumour necrosis factor
References
Disease GBD, Injury I, Prevalence C. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet. 2018;392:1789–858.
Bui AL, Horwich TB, Fonarow GC. Epidemiology and risk profile of heart failure. Nat Rev Cardiol. 2011;8:30–41.
Jenca D, Melenovsky V, Stehlik J, Stanek V, Kettner J, Kautzner J, et al. Heart failure after myocardial infarction: incidence and predictors. ESC Heart Fail. 2021;8:222–37.
Bhatt AS, Ambrosy AP, Velazquez EJ. Adverse remodeling and reverse remodeling after myocardial infarction. Curr Cardiol Rep. 2017;19:71.
Enomoto T, Ohashi K, Shibata R, Higuchi A, Maruyama S, Izumiya Y, et al. Adipolin/C1qdc2/CTRP12 protein functions as an adipokine that improves glucose metabolism. J Biol Chem. 2011;286:34552–628.
Wei Z, Peterson JM, Lei X, Cebotaru L, Wolfgang MJ, Baldeviano GC, et al. C1q/TNF-related protein-12 (CTRP12), a novel adipokine that improves insulin sensitivity and glycemic control in mouse models of obesity and diabetes. J Biol Chem. 2012;287:10301–15.
Tan SY, Little HC, Lei X, Li S, Rodriguez S, Wong GW. Partial deficiency of CTRP12 alters hepatic lipid metabolism. Physiol Genom. 2016;48:936–49.
Tan SY, Little HC, Sarver DC, Watkins PA, Wong GW. CTRP12 inhibits triglyceride synthesis and export in hepatocytes by suppressing HNF-4α and DGAT2 expression. FEBS Lett. 2020;594:3227–39.
Tan SY, Lei X, Little HC, Rodriguez S, Sarver DC, Cao X, et al. CTRP12 ablation differentially affects energy expenditure, body weight, and insulin sensitivity in male and female mice. Am J Physiol Endocrinol Metab. 2020;319:E146–62.
Nadimi Shahraki Z, Azimi H, Ilchi N, Rohani Borj M, Pourghadamyari H, Mosallanejad S, et al. Circulating C1q/TNF-related protein-12 levels are associated with the severity of coronary artery disease. Cytokine. 2021;144: 155545.
Ogawa H, Ohashi K, Ito M, Shibata R, Kanemura N, Yuasa D, et al. Adipolin/CTRP12 protects against pathological vascular remodelling through suppression of smooth muscle cell growth and macrophage inflammatory response. Cardiovasc Res. 2020;116:237–49.
Wang G, Chen JJ, Deng WY, Ren K, Yin SH, Yu XH. CTRP12 ameliorates atherosclerosis by promoting cholesterol efflux and inhibiting inflammatory response via the miR-155-5p/LXRα pathway. Cell Death Dis. 2021;12:254.
Zhou MQ, Jin E, Wu J, Ren F, Yang YZ, Duan DD. CTRP12 ameliorated lipopolysaccharide-induced cardiomyocyte injury. Chem Pharm Bull. 2020;68:133–9.
Jin AP, Zhang QR, Yang CL, Ye S, Cheng HJ, Zheng YY. Up-regulation of CTRP12 ameliorates hypoxia/re-oxygenation-induced cardiomyocyte injury by inhibiting apoptosis, oxidative stress, and inflammation via the enhancement of Nrf2 signaling. Hum Exp Toxicol. 2021;40:2087–98.
Wang W, Gao W, Zhu Q, Alasbahi A, Seki E, Yang L. TAK1: a molecular link between liver inflammation, fibrosis, steatosis, and carcinogenesis. Front Cell Dev Biol. 2021;9: 734749.
Zhu L, Lama S, Tu L, Dusting GJ, Wang JH, Liu GS. TAK1 signaling is a potential therapeutic target for pathological angiogenesis. Angiogenesis. 2021;24:453–70.
Fechtner S, Fox DA, Ahmed S. Transforming growth factor beta activated kinase 1: a potential therapeutic target for rheumatic diseases. Rheumatology (Oxford). 2017;56:1060–8.
Sakurai H. Targeting of TAK1 in inflammatory disorders and cancer. Trends Pharmacol Sci. 2012;33:522–30.
Shim JH, Xiao C, Paschal AE, Bailey ST, Rao P, Hayden MS, et al. TAK1, but not TAB1 or TAB2, plays an essential role in multiple signaling pathways in vivo. Genes Dev. 2005;19:2668–81.
Aashaq S, Batool A, Andrabi KI. TAK1 mediates convergence of cellular signals for death and survival. Apoptosis. 2019;24:3–20.
Suzuki M, Asai Y, Kagi T, Noguchi T, Yamada M, Hirata Y, et al. TAK1 mediates ROS generation triggered by the specific cephalosporins through noncanonical mechanisms. Int J Mol Sci. 2020;21:9497.
Totzke J, Scarneo SA, Yang KW, Haystead TAJ. TAK1: a potent tumour necrosis factor inhibitor for the treatment of inflammatory diseases. Open Biol. 2020;10: 200099.
Zhao J, Jiang X, Liu J, Ye P, Jiang L, Chen M, et al. Dual-specificity phosphatase 26 protects against cardiac hypertrophy through TAK1. J Am Heart Assoc. 2021;10: e014311.
Guo S, Liu Y, Gao L, Xiao F, Shen J, Xing S, et al. TBC1D25 regulates cardiac remodeling through TAK1 signaling pathway. Int J Biol Sci. 2020;16:1335–48.
Rosenkranz S. TGF-beta1 and angiotensin networking in cardiac remodeling. Cardiovasc Res. 2004;63:423–32.
Xiao H, Zhang YY. Understanding the role of transforming growth factor-beta signalling in the heart: overview of studies using genetic mouse models. Clin Exp Pharmacol Physiol. 2008;35:335–41.
Li CY, Zhou Q, Yang LC, Chen YH, Hou JW, Guo K, et al. Dual-specificity phosphatase 14 protects the heart from aortic banding-induced cardiac hypertrophy and dysfunction through inactivation of TAK1-P38MAPK/-JNK1/2 signaling pathway. Basic Res Cardiol. 2016;111:19.
Wang X, Huang T, Xie H. CTRP12 alleviates isoproterenol induced cardiac fibrosis via inhibiting the activation of P38 pathway. Chem Pharm Bull. 2021;69:178–84.
Chen X, Wan W, Guo Y, Ye T, Fo Y, Sun Y, et al. Pinocembrin ameliorates post-infarct heart failure through activation of Nrf2/HO-1 signaling pathway. Mol Med. 2021;27:100.
Fadaei R, Moradi N, Kazemi T, Chamani E, Azdaki N, Moezibady SA, et al. Decreased serum levels of CTRP12/adipolin in patients with coronary artery disease in relation to inflammatory cytokines and insulin resistance. Cytokine. 2019;113:326–31.
Tan L, Nomanbhoy T, Gurbani D, Patricelli M, Hunter J, Geng J, et al. Discovery of type II inhibitors of TGFbeta-activated kinase 1 (TAK1) and mitogen-activated protein kinase kinase kinase kinase 2 (MAP4K2). J Med Chem. 2015;58:183–96.
Walkowski B, Kleibert M, Majka M, Wojciechowska M. Insight into the role of the PI3K/Akt pathway in ischemic injury and post-infarct left ventricular remodeling in normal and diabetic heart. Cells. 2022;11:1553.
Wei Z, Lei X, Seldin MM, Wong GW. Endopeptidase cleavage generates a functionally distinct isoform of C1q/tumour necrosis factor-related protein-12 (CTRP12) with an altered oligomeric state and signaling specificity. J Biol Chem. 2012;287:35804–14.
Funding
The work was supported by the Science and Technology Innovation and Development Fund of Tangdu Hospital (2019QYTS011).
Author information
Authors and Affiliations
Contributions
BB designed the work, performed the experiments and wrote the manuscript. ZJ, FW and CQ performed the experiments. HZ and DL performed data analysis and provided technical support. Yue Wu contributed to conceptualization and reviewed the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest.
Additional information
Responsible Editor: John Di Battista.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Bai, B., Ji, Z., Wang, F. et al. CTRP12 ameliorates post-myocardial infarction heart failure through down-regulation of cardiac apoptosis, oxidative stress and inflammation by influencing the TAK1-p38 MAPK/JNK pathway. Inflamm. Res. 72, 1375–1390 (2023). https://doi.org/10.1007/s00011-023-01758-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00011-023-01758-4