
Abstract
Macrophages play a key role in the initiation and progression of atherosclerotic plaques. Although a significant number of macrophages undergoes cell death during plaque development as a result of atherogenic stressors, advanced plaques are characterized by a large macrophage content. Macrophage accumulation is mediated by continuous recruitment of monocytes, reduced emigration of macrophages and poor phagocytosis of dead cells which may trigger secondary necrosis and amplification of plaque inflammation. Moreover, an increasing body of evidence indicates that macrophages have developed several strategies to survive and to proliferate in the adverse environment of an advanced atherosclerotic plaque. Macrophages contain organic molecules or enzymes that provide enhanced antioxidant protection. In addition, synthesis of anti-apoptotic proteins is upregulated and several cellular protection mechanisms such as the unfolded protein response and autophagy are activated in macrophages to promote cellular survival. In this review, we discuss these macrophage survival mechanisms that allow growth and destabilization of advanced atherosclerotic plaques.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
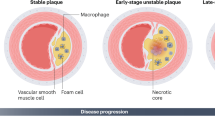
Atherosclerosis is a non-resolving inflammatory disease marked by atheromatous plaques in the intima of medium- and large-sized arteries [108]. Critical to plaque formation is the infiltration of circulating monocytes in the subendothelial space, where they differentiate into macrophages [50, 65]. Subsequently, macrophages internalize modified lipoproteins and turn into foam cells [50, 65]. Although a significant number of macrophage-derived foam cells undergoes apoptosis as a result of prolonged endoplasmic reticulum stress and other pro-apoptotic stimuli, an advanced plaque is characterized by a strong accumulation of macrophages. It should be noted that plaques with a large macrophage content are considered dangerous as they may easily rupture. Indeed, a large body of evidence indicates that macrophages contribute to plaque destabilization through cytokine secretion, the induction of smooth muscle cell (SMC) death and the release of matrix degrading enzymes [19, 24, 65]. A good understanding of the mechanisms leading to macrophage accumulation is therefore important to prevent plaque rupture and life-threatening clinical complications such as myocardial infarction and stroke. Several mechanisms contributing to macrophage accumulation have recently been identified. First, there is a persistent recruitment of monocytes into established atherosclerotic lesions [41, 75], particularly in the setting of hypercholesterolemia [94]. These monocytes can differentiate in two major types of macrophages, those that further promote inflammation, referred to as classically activated (or M1) macrophages, and those that promote resolution, referred to as alternatively activated (or M2) macrophages. In vitro experiments as well as limited in vivo data suggest that M1 and M2 macrophages coexist in human atheromas [7, 10], but an imbalance in this ratio may cause impaired resolution [25, 26, 55]. A third macrophage subset, namely Mox, has recently been identified in plaques of LDL receptor (LDLR) knockout mice [39], though little is known about their functionality in vivo [10]. Secondly, emigration of macrophages from atheromata is suppressed during hypercholesterolemia [54] and may lead to prolonged production of proteases, cytokines and procoagulant/thrombotic factors. Thirdly, apoptotic macrophages are not effectively cleared by viable phagocytes in advanced lesions [85], a condition known as defective efferocytosis, leading to secondary necrosis and amplification of inflammatory responses [95]. Finally, macrophages have developed several strategies to survive and to proliferate in the adverse environment of an advanced atherosclerotic plaque. In this review, we provide a comprehensive overview of these macrophage survival mechanisms that allow growth and destabilization of advanced atherosclerotic plaques.
Enhanced macrophage survival mediated by modified LDL (Table 1)
An initiating event in atherosclerosis is the accumulation of low density lipoprotein (LDL) in the subendothelial matrix where it undergoes minor modifications such as proteolysis, aggregation, hydrolysis of phosphatidylcholine and cholesteryl esters as well as oxidative changes [72]. Aggregated LDL (agLDL) stimulates foam cell formation in macrophages, but at the same time suppresses apoptosis by downregulating caspase-1 and -3 as well as by upregulating the anti-apoptotic cytokine interleukin-1β [27, 47]. Moreover, a human homologue of bovine ubiquitin-conjugating enzyme E2-25K, termed LIG (LDL-inducible gene), is upregulated in human macrophages after treatment with agLDL [27, 43]. Increased LIG mRNA expression is followed by polyubiquitination and increased ubiquitin-dependent degradation of intracellular pro-apoptotic proteins such as p53 [27, 43]. Because apoptosis caused by oxidized LDL (oxLDL) is p53 dependent [44], increased degradation of ubiquitinated p53 may represent an important anti-apoptotic event in agLDL-laden macrophages.
OxLDL exhibits a dramatic cytotoxic effect to vascular cells leading to apoptosis, as mentioned above, but may also stimulate necrosis and autophagic death [56, 60]. OxLDL is therefore involved in cell loss, formation of a lipid core, plaque rupture and subsequent thrombotic events. However, a large body of evidence indicates that low doses of oxLDL (<100 μg/ml) or short oxLDL treatment (4–6 h) leads to macrophage survival, DNA synthesis, and an enhanced proliferative response to macrophage-colony stimulating factor (M-CSF) and granulocyte macrophage-colony stimulating factor (GM-CSF) [32, 33]. Moreover, aggregation of lightly oxidized LDL potentiates dramatically its ability to stimulate macrophage DNA synthesis [31]. Importantly, exposure of macrophages to 100 μg/ml of minimally oxidized LDL induces proliferation and macrophage activation, whereas extensively oxidized LDL induces cell death at the same concentration [33]. Two hundred μg/ml of oxLDL causes cell death regardless of the oxidation degree [33]. These results suggest that oxLDL-induced cell death of macrophages is not self-evident, but indicate that the macrophage response to oxLDL (proliferation or cell death) largely depends on the oxidation degree, aggregation status, exposure time and concentration of oxLDL. Most interestingly, it is the lipid component of oxLDL that promotes macrophage survival and DNA synthesis [31]. As a consequence, also lipids isolated from human plaques promote macrophage survival [31].
Uptake of oxLDL occurs via a group of macrophage scavenger receptors (MSRs) such as class A MSR (MSR-A). Circulating monocytes elaborate MSR-A at undetectable levels, but when the cells differentiate into macrophages, they express high levels of MSR-A. It has been proposed that increased MSR-A expression promotes not only the uptake of lipid components, but also long-term protection of macrophages from apoptosis [52]. Because cholesterol-induced macrophage apoptosis requires engagement of MSR-A [20] and overexpression of MSR-A induces non-macrophage cell apoptosis [110], this hypothesis is unlikely. Indeed, recent evidence suggests that neither MSR-A nor other major pattern recognition receptors including CD36, Toll-like receptor 2 or 4, CD14 and RAGE are responsible for activating the oxLDL prosurvival pathway, and that the anti-apoptotic effect is not dependent on the uptake of oxLDL [79]. Therefore, several alternative mechanisms mediating oxLDL-induced macrophage survival have been proposed, even though not all of them are widely accepted. For example, oxLDL stimulates macrophages to release GM-CSF, which promotes autocrine macrophage growth. GM-CSF expression has been immunohistochemically confirmed in rabbit and human atherosclerotic plaques, and according to some groups, could suppress apoptotic death of macrophages [101]. However, others reported that the prosurvival effect of oxLDL is not inhibited by neutralizing GM-CSF antibodies and that macrophage survival induced by oxLDL does not depend on secretion of growth factors or cytokines [32, 37]. Eukaryotic elongation factor-2 (eEF2) kinase is activated in response to oxLDL, resulting in the inhibition of protein synthesis, and these effects have been considered to be part of a signaling pathway in which oxLDL can block macrophage apoptosis [14]. However, because macrophages in atherosclerotic plaques are stimulated, and not inhibited to undergo apoptosis after treatment with protein synthesis inhibitors [15, 16], also this mechanism seems unlikely. At present, the most plausible explanation for oxLDL-mediated macrophage survival, reported by several groups, is activation of the PI3K/Akt signaling pathway [8, 37]. Akt promotes cell survival directly by its ability to phosphorylate and inactivate several pro-apoptotic targets, including Bad and the forkhead transcription factors. Akt also exerts its anti-apoptotic effects indirectly by changing the expression level of genes that encode components of cell death, such as the Bcl-2 family members and through its effects on NF-κB and p53 [38]. According to recent evidence, activation of Akt by oxLDL requires engagement of the platelet-activating factor (PAF) receptor and a Gαi-coupled protein [81]. PAFR is involved in the uptake of oxLDL [80], but may require additional receptors (e.g. CD36) to induce oxLDL-mediated downstream events such as IL-8 and MCP-1 expression. Moreover, immune complexes of IgG with oxLDL engage Fcγ receptors and thereby activate Akt-dependent prosurvival mechanisms, also in monocytes [69]. Finally, it is noteworthy that Akt signaling in macrophages depends to a significant extent on constitutive Ca2+ influx, presumably through a mechanism that involves calmodulin and calmodulin-dependent kinase II [97].
Besides aggregation and oxidation, partial enzymatic degradation of LDL by hydrolases secreted by vascular cells may take place in the subendothelium. Macrophages treated with enzymatically modified LDL (E-LDL) upregulate the anti-apoptotic gene Toso and show less apoptosis [90]. Toso directly binds the death adaptor FADD and this may disrupt the formation of the death-inducing signaling complex (DISC) [92]. Recent evidence showed that the anti-apoptotic function of Toso depends on RIP1 ubiquitination and involves the recruitment of FADD to a Toso/RIP1 protein complex [68]. In response to CD95L and TNFα, Toso also promotes the activation of MAPK and NF-κB signaling pathways [68].
Finally, it is noteworthy that hemoglobin (Hb)-modified LDL (HbLDL) can be found in atherosclerotic plaques. HbLDL is highly susceptible to oxidation and induces heme oxygenase (HO-1) in macrophages due to its lipid peroxide (LOOH) and heme content [3]. HO-1 induction in HbLDL-treated macrophages confers resistance against oxidant stress [3].
Enhanced antioxidant protection (Table 1)
Numerous studies suggest that oxidative stress and the production of intracellular reactive oxygen species (ROS) are key elements in the progression of atherosclerosis. Excessive synthesis of ROS and their byproducts are capable of causing oxidative damage to biomolecules including proteins, lipids and DNA. In accordance with this concept, elevated levels of base modifications and DNA strand breaks have been identified in both human and experimental atherosclerosis [57, 58]. Such damage is of potential pathobiologic significance, because many ROS-induced DNA modifications are promutagenic. DNA strand breaks, on the other hand, may account for a transient or permanent cell cycle arrest, chromosome rearrangements and, in case of extensive damage, induction of cell death or transformation events. Several enzymatic defense systems including base excision repair (Ref-1, PARP-1) or non-specific repair pathways (DNA-PK, p53) are upregulated particularly in plaque macrophages to prevent formation of oxidative DNA damage (Fig. 1) [57, 58]. Moreover, ascorbate, α-tocopherol and glutathione provide a considerable oxidant scavenging capacity to macrophages [30]. Yet, additional antioxidant mechanisms appear to be involved. For example, γ-interferon stimulation of macrophages by Th-1 cells causes the enzymatic breakdown of intracellular GTP to 7,8-dihydroneopterin which can either inhibit protein hydroperoxide formation by scavenging hydroxyl and peroxyl radicals, thereby generating 7,8-dihydroxanthopterin, or by rapidly reacting with the potent oxidant HOCl to form neopterin [30]. 7,8-dihydroneopterin also protects macrophages by scavenging oxidants generated in response to oxLDL and by decreasing CD36-mediated uptake of oxLDL [29]. Apart from 7,8-dihydroneopterin, γ-interferon catalyzes degradation of the amino acid tryptophan to a range of products including 3-hydroxyanthranilic acid (3HAA). This compound is a potent inhibitor of macrophage-mediated LDL oxidation, and thus may also reduce oxLDL concentrations within plaques [30].

Schematic overview of the cellular mechanisms that promote macrophage survival in early and advanced atherosclerotic plaques. In early plaques, activation of autophagy protects macrophages against adverse environmental effects (e.g. oxidative stress) by removing damaged proteins and organelles. Moreover, lipid droplets (LD) are a substrate for autophagy. Cholesteryl esters in LD are hydrolyzed in autolysosomes to generate free cholesterol for efflux via cholesterol transporter ABCA1. Apart from autophagy, the unfolded protein response (UPR) is activated in early plaques to protect macrophages against apoptosis by ER stress-inducing agents such as the accumulation of free cholesterol in the ER membrane. Macrophages esterify the cholesterol and try to prevent its toxic effects through efficient trafficking of the internalized cholesterol to acyl-CoA:cholesterol acyltransferase (ACAT) in the ER. If apoptosis would occur, apoptotic cells (AC) are rapidly cleared via phagocytosis. This event protects against cell death via several survival pathways involving PI3K/Akt and NF-κB. In advanced plaques, both autophagy and AC phagocytosis are defective. Defective autophagy results in the accumulation of damaged organelles and may sensitize macrophages to undergo cell death. Free, nonphagocytised AC will undergo secondary necrosis, contribute to the enlargement of the necrotic core and promote plaque progression. Furthermore, overstimulation of the UPR may occur in advanced plaques, which may lead to synthesis of the protein CHOP and induction of apoptosis. Because ROS-induced base modifications and DNA strand breaks frequently occur in advanced lesions, several enzymatic defense systems including base excision repair or non-specific repair pathways are upregulated, particularly in macrophages, to prevent formation of oxidative DNA damage or chromosome rearrangements
Mer tyrosine kinase plays several important roles in normal macrophage physiology such as regulation of cytokine secretion and clearance of apoptotic cells, but mediates also a prosurvival function in macrophages under conditions of oxidative stress. Indeed, Gas6-dependent phosphorylation of Mer has been reported in response to H2O2 treatment and leads to increased downstream anti-apoptotic signaling via Akt and Erk1/2 [1]. Protection against peroxynitrite in macrophages is mediated, at least in part, by cytosolic phospholipase A(2)-released arachidonic acid. This lipid messenger is metabolized by 5-lipoxygenase to 5-hydroxyeicosatetraenoic acid and causes the mitochondrial translocation of protein kinase Cα, an event associated with cytosolic accumulation of Bad and Bax [11, 12]. Under these conditions, the anti-mitochondrial permeability transition (MPT) activity of Bcl-2 is fully active and promotes macrophage survival. Finally, it is noteworthy that manganese superoxide dismutase (MnSOD) is upregulated in macrophages of atherosclerotic plaques [45]. This enzyme catalyzes the dismutation of superoxide anions into water and hydrogen peroxide. The latter compound is then broken down by catalase or glutathione peroxidase [36]. Potential triggers of MnSOD are TNF-α, IL-1 and oxLDL [30].
Probably the most important biological source of superoxide anions is the mitochondrial electron transport chain. Mitochondria possess a mechanism called ‘mild’ uncoupling, which reduces the production of ROS by the respiratory chain. This mechanism is in part regulated by the mitochondrial uncoupling protein 2 (UCP2) that uncouples ATP production from mitochondrial respiration and thereby converts the loss of potential energy in heat production [103, 106]. Differentiation of monocytes into macrophages increases UCP2 expression [102]. In line with this finding, it has been observed that UCP2 is abundantly expressed in subendothelial macrophages of atherosclerotic plaques and provides protection against oxidative damage [102]. Conversely, lack of UCP2 in macrophages accelerates atherosclerotic plaque development [5].
Enhanced production of anti-apoptotic proteins (Table 1)
Apoptosis is a major event in advanced atherosclerosis targeting all cell types, including foam cells of macrophage origin [65]. Inhibition of macrophage apoptosis can occur by upregulating the multidomain protein Bcl-2 or one of its anti-apoptotic family members (Bcl-xL, Bcl-w, Mcl-1, A1/Bfl-1, Boo/Diva and NR-13). In particular, Bcl-2 is a key cell survival molecule and its almost all-round protective capacity has been interpreted as the result of an enormous variety of anti-apoptotic effects. Activities of Bcl-2 include a reduction in ROS levels [35], stabilization of lysosomal and mitochondrial membrane integrity [93, 112], enhancement of the proton efflux from mitochondria [89], regulation of intracellular calcium homeostasis [48], and prevention of cytochrome c release from mitochondria [111]. According to an immunohistochemical study using human coronary and carotid plaques, expression of Bcl-2 is similar in both control and atherosclerotic specimens, while the expression of Bcl-xL is higher in the plaque [83]. These findings suggest that Bcl-xL, but not Bcl-2, may act as an inducible protective factor in plaque macrophages. Indeed, recent evidence shows that the inactivation of the Bcl-x gene in macrophages stimulates their sensitivity to apoptosis, and results in more advanced atherosclerotic lesions in apoE-deficient mice [88]. However, even at basal levels, also Bcl-2 plays a protective role against macrophage apoptosis, specifically in advanced lesions, as macrophage-targeted deletion of Bcl-2 in apoE-deficient mice results in a 40–45 % increase in apoptotic cells [99].
Besides anti-apoptotic Bcl-2 proteins, macrophage apoptosis can be inhibited by the members of the inhibitor of apoptosis (IAP) protein family such as cIAP1, cIAP2, XIAP, NAIP, livin, survivin and apollon, which inhibit caspase activity. Survivin is clearly detectable by immunohistochemistry in macrophages infiltrating early lesions, but absent in advanced plaques [6]. In contrast, XIAP and particularly cIAP2 are both strongly upregulated in advanced plaques. Macrophages in culture upregulate survivin after stimulation with M-CSF [6], an inflammatory growth factor released by the atherosclerotic vessel wall. Conversely, prolonged treatment with oxLDL abolishes macrophage survivin expression and triggers apoptosis [6]. Raising XIAP or cIAP2 is not sufficient to block oxLDL-induced apoptosis, even though XIAP is important for macrophage survival. Survivin may thus have a biphasic role in atherosclerosis: it may promote macrophage survival and plaque progression in the early stages of atherosclerosis, but loss of survivin expression in advanced plaques, possibly due to prolonged oxLDL exposure, may contribute to apoptosis and plaque instability.
Next to the activity of IAPs, caspases can also be regulated by blocking their activation. For example, FLICE-inhibitory protein (FLIP), a caspase-8 homolog lacking proteolytic activity, is upregulated during macrophage differentiation and confers resistance to Fas-mediated apoptosis [73]. Furthermore, macrophages around the necrotic core of advanced human plaques show enhanced levels of the short isoform of caspase-2 (caspase-2S) that acts as an endogenous inhibitor of apoptotic cell death [59]. Indeed, overexpression of caspase-2S can inhibit nuclear changes associated with apoptosis [23]. Caspase-2S also prevents the maturation of apoptotic bodies, delays phosphatidylserine externalization on the plasma membrane of dying cells and prevents cleavage and activation of procaspase-2L [23]. Interestingly, the treatment of macrophages in culture with the DNA strand-breaking agents etoposide or camptothecin stimulates caspase-2S expression [59]. Given the high number of DNA strand breaks in advanced plaques [57, 58], these findings provide evidence for a survival factor in macrophage-derived foam cells of human atherosclerotic plaques that may be upregulated in response to DNA damage.
Induction of the unfolded protein response
Nascent proteins of the secretory pathway or proteins designed for trafficking to the cell membrane are post-translationally modified and folded in the endoplasmic reticulum (ER). Chaperones such as the glucose-regulated protein 78 (GRP78/BiP) are present in the ER lumen and associate with the newly synthesized peptides to improve their maturation. When folding is unsuccessful, the malfolded proteins are directed towards the cytoplasm where they are recognized by the proteasome and degraded. However, in stressful conditions, when there is an overload of malfolded proteins, a unique signaling cascade referred to as the unfolded protein response (UPR) is activated that confers cytoprotective advantage [34] (Fig. 1). Indeed, in vitro experiments showed that UPR activation occurs after monocyte to macrophage differentiation, particularly after a significant increase in de novo protein synthesis, and reduces macrophage cell death by ER stress-inducing agents [21]. Moreover, UPR markers are dramatically increased in very early intimal macrophages [113]. Because these cells rarely undergo apoptosis, it has been suggested that UPR activation is an important cellular mechanism for macrophage survival within early atherosclerotic lesions [21]. Interestingly, advanced lesions contain a number of molecules (e.g. oxidized lipids, saturated fatty acids) and processes (e.g. oxidative stress) that are known ER stressors [96]. Consistent with this finding, markers of UPR activation are not only markedly upregulated in macrophages from early lesions, but also in macrophage foam cells from fatty streaks and advanced atherosclerotic lesions [113]. However, unlike the UPR in early intimal macrophages, activation of the UPR in macrophages of advanced plaques leads to elevated levels of CHOP which triggers apoptosis execution pathways through a number of mechanisms including NADPH oxidase activation and subsequent ROS generation (Fig. 1) [96]. Using CHOP-deficient mice crossbred with ApoE or LDLR knockout mice, a causal link between CHOP expression, apoptosis and plaque necrosis has been demonstrated [98]. Thus at an early stage, the UPR may protect macrophages against death, but when ER stress is prolonged or severe, it causes cell death and this makes plaques more vulnerable and even prone to rupture.
Stimulation of autophagy (Fig. 1)
Autophagy is a subcellular mechanism for bulk destruction of cytoplasmic components such as protein aggregates and complete organelles via lysosomes. It occurs at basal levels in most tissues, but is activated by environmental stimuli (such as nutrient deprivation, low oxygen and hormones) or stress-related signals (e.g. accumulation of mutant proteins) [64]. The autophagic process thus maintains the balance between biogenesis and production of cell organelles, and destroys unwanted or damaged intracellular structures. In this way, cell survival is promoted in an unfavorable environment. LDLR knockout mice with a macrophage-specific deletion of the essential autophagy gene Atg5 develop plaques with increased apoptosis and oxidative stress as well as enhanced plaque necrosis, indicating that autophagy is anti-apoptotic and present in atherosclerotic plaques to protect macrophages against various atherogenic stressors including 7-ketocholesterol, ROS and inducers of ER stress (Fig. 1) [53]. Also of note, lipid droplets in macrophage foam cells are delivered to lysosomes via autophagy, where lysosomal acid lipase acts to hydrolyze lipid droplet-derived cholesteryl esters in order to generate free cholesterol mainly for ABCA1-dependent efflux (Fig. 1) [70]. This process is specifically induced upon macrophage cholesterol loading and may contribute to macrophage survival. Recent evidence suggests that basal levels of macrophage autophagy play an essential role in atheroprotection during early atherosclerosis [77]. However, autophagy becomes dysfunctional in the more advanced stages of atherosclerosis (Fig. 1) and its deficiency promotes atherosclerosis in part through activation of the inflammasome [77]. Autophagy can be intensified selectively in macrophages by specific drugs such as mammalian target of rapamycin (mTOR) inhibitors (e.g. everolimus) [62, 107] or Toll-like receptor 7 (TLR7) ligands (e.g. imiquimod) [19]. However, moderate drug-induced stimulation of macrophage autophagy via everolimus or imiquimod may trigger cytokine production, though in an autophagy-independent manner (via activation of p38 MAP kinase or NF-κB, which are off-target effects of everolimus and imiquimod, respectively) [19, 63]. As a consequence, imiquimod stimulates expression of vascular adhesion molecule-1, infiltration of T-lymphocytes, accumulation of macrophages and plaque enlargement [19]. These findings suggest that both inhibition and moderate drug-induced stimulation of macrophage autophagy is detrimental for the structure of the plaque. Nonetheless, extensive stimulation of macrophage autophagy, as demonstrated after stent-based delivery of everolimus, causes degradation of major portions of the cytosol which in turn leads to autophagy-mediated, non-apoptotic cell death and a marked reduction in macrophage content [62, 63, 107]. Given the numerous detrimental effects of macrophages in advanced plaques, this approach seems to be a promising strategy for the treatment of coronary artery disease [86].
Other survival strategies
In addition to the strategies described above, many other cellular processes keep macrophages alive during atherogenesis. For example, phagocytic clearance of apoptotic cells by macrophages is very efficient in early plaques and protects against cell death (Fig. 1) [17, 78]. Phagocytosis of apoptotic cells also profoundly inhibits the proliferation of macrophages stimulated to proliferate by growth factors such as M-CSF [78]. The ability of apoptotic cells to promote macrophage survival and to inhibit proliferation appears to be the result of several survival pathways involving PI3K/Akt and NF-κB as well as inhibition of Erk1/2 [17, 78]. Conversely, phagocytosis of apoptotic cells in advanced plaques is defective and may result in secondary necrosis, expansion of the necrotic core and increased plaque vulnerability [85, 95]. Mechanisms that are responsible for defective clearance of apoptotic cells in advanced plaques include enhanced levels of oxidative stress [84, 85], competition with oxLDL for binding on macrophages [84], impaired autophagy [51, 76] and loss of UCP2 protein in deeper layers of the plaque [71, 102].
In contrast to phagocytosis of apoptotic cells, phagocytosis of modified LDL leads to large intracellular amounts of free cholesterol, which can be cytotoxic if not handled properly. Indeed, free cholesterol accumulates in the ER membrane, triggers ER dysfunction and activates an UPR-associated apoptotic response. Macrophages esterify the cholesterol and try to prevent its toxic effects through efficient trafficking of the internalized cholesterol to acyl-CoA:cholesterol acyltransferase (ACAT) in the ER [13]. Even when ACAT is rendered dysfunctional, as might occur in advanced atherosclerotic plaques, macrophages often remain viable through massive efflux of cholesterol via cholesterol efflux transporters such as ABCA1 and ABCG1.
Monocytes express the chemokine receptor CX3CR1 that binds to its ligand CX3CL1, also known as fractalkine. Absence of either CX3CR1 or CX3CL1 leads to reduced circulating monocyte numbers in mice, particularly the Gr1low subset that displays significantly higher CX3CR1 amounts [49]. Enforced expression of the human Bcl-2 transgene rescues the wild-type phenotype [49], suggesting that binding of CX3CL1 to its receptor is an important survival signal that promotes monocyte homeostasis and atherogenesis. Because there are significant differences in signaling downstream of mouse versus human CX3CR1, it remains to be determined whether the survival function of CX3CL1, as shown in several murine disease models, is also applicable in humans [109]. Moreover, the mechanism of monocyte survival via the CX3CR1–CX3CL1 axis is unclear and needs to be investigated [109].
Another important issue is that advanced atherosclerotic plaques reveal areas of low pH or low oxygen. A reduction in extracellular pH from 7.4 to 7.0 inhibits oxLDL-induced apoptosis of macrophages in vitro [28]. This inhibition of apoptosis is attributable at least in part to a decrease in the binding of oxLDL to cell surface receptors at low pH, thereby reducing endocytosis of oxLDL [28]. On the other hand, hypoxia prolongs macrophage survival by enhancing glycolysis [82]. Moreover, hypoxia mediates accumulation of hypoxia-inducible factors (Hifs) such as Hif-1α in macrophages [91]. Hifs are key proteins of the cellular adaption to low oxygen tensions. Recent evidence, however, showed that inhibition of Hif-signaling in combination with oxLDL treatment dramatically induces apoptosis independently of the oxygen tension [74]. This finding suggests that the Hif pathway is also involved in blocking the apoptotic effects of oxLDL.
Finally, it is noteworthy that prostaglandin E2, a major product of activated macrophages, regulates macrophage survival through EP4 receptor-dependent activation of PI3K/Akt and NF-κB signaling pathways. EP4 deficiency compromises macrophage survival and suppresses early atherosclerosis in LDLR knockout mice [4]. Furthermore, p38 MAPK is activated during atherogenesis [66]. Although p38 MAPK has been shown to play a crucial role in regulating apoptosis, p38 can have both pro- and anti-apoptotic functions depending on the apoptotic stimulus. Recent evidence showed that genetic ablation of p38 in macrophages suppresses activation of Akt and leads to enhanced macrophage apoptosis in advanced plaques [87]. These data demonstrate an important prosurvival role for p38 in macrophages during atherosclerosis.
Concluding remarks
Macrophages in atherosclerotic plaques possess a remarkable ability to survive different types of cellular stress. In this way, macrophages may accumulate in the vascular wall and contribute to plaque development. Because the mechanisms of macrophage survival are very diverse and not necessarily restricted to macrophages, inhibition of prosurvival pathways is probably not an ideal approach to prevent macrophage accumulation and plaque progression. Nonetheless, macrophages have become an excellent therapeutic target and interventions mediating macrophage loss can yield beneficial results [100]. Promising therapeutic strategies that provoke macrophage elimination in different stages of atherosclerosis include (1) lipid lowering, either in the circulation or from macrophages in the plaque, (2) direct and indirect targeting of adhesion molecules and chemokines involved in monocyte adhesion and transmigration, or (3) selective induction of macrophage death [18]. Even though these approaches are promising in animal studies (but not all, see for example [19, 104]), clinical trials are so far disappointing due to side effects or the lack of additional benefits [18]. Nonetheless, macrophages remain more than ever an important focus for ongoing research, not only for macrophage-specific delivery of drugs which may help to reduce adverse effects [2, 40], but also for modulation of the macrophage phenotype with growth factors or other compounds [9, 46], and for the detection or imaging of vulnerable plaques [22, 105]. Because the promotion of macrophage death (or the inhibition of macrophage survival) in atherosclerosis remains controversial [61], and hitherto, nobody really knows whether this strategy is beneficial for plaque regression, inhibition of monocyte infiltration or monocyte retention in the neointima may represent the best clinical targets, as demonstrated recently [42, 67, 75].
References
Anwar A, Keating AK, Joung D, Sather S, Kim GK, Sawczyn KK, Brandao L, Henson PM, Graham DK (2009) Mer tyrosine kinase (MerTK) promotes macrophage survival following exposure to oxidative stress. J Leukoc Biol 86:73–79. doi:10.1189/jlb.0608334
Aouadi M, Tesz GJ, Nicoloro SM, Wang M, Chouinard M, Soto E, Ostroff GR, Czech MP (2009) Orally delivered siRNA targeting macrophage Map4k4 suppresses systemic inflammation. Nature 458:1180–1184. doi:10.1038/nature07774
Asatryan L, Ziouzenkova O, Duncan R, Sevanian A (2003) Heme and lipid peroxides in hemoglobin-modified low-density lipoprotein mediate cell survival and adaptation to oxidative stress. Blood 102:1732–1739. doi:10.1182/blood-2003-01-0293
Babaev VR, Chew JD, Ding L, Davis S, Breyer MD, Breyer RM, Oates JA, Fazio S, Linton MF (2008) Macrophage EP4 deficiency increases apoptosis and suppresses early atherosclerosis. Cell Metab 8:492–501. doi:10.1016/j.cmet.2008.09.005
Blanc J, Alves-Guerra MC, Esposito B, Rousset S, Gourdy P, Ricquier D, Tedgui A, Miroux B, Mallat Z (2003) Protective role of uncoupling protein 2 in atherosclerosis. Circulation 107:388–390. doi:10.1161/01.CIR.0000051722.66074.60
Blanc-Brude OP, Teissier E, Castier Y, Leseche G, Bijnens AP, Daemen M, Staels B, Mallat Z, Tedgui A (2007) IAP survivin regulates atherosclerotic macrophage survival. Arterioscler Thromb Vasc Biol 27:901–907. doi:10.1161/01.ATV.0000258794.57872.3f
Bouhlel MA, Derudas B, Rigamonti E, Dievart R, Brozek J, Haulon S, Zawadzki C, Jude B, Torpier G, Marx N, Staels B, Chinetti-Gbaguidi G (2007) PPARgamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab 6:137–143. doi:10.1016/j.cmet.2007.06.010
Boullier A, Li Y, Quehenberger O, Palinski W, Tabas I, Witztum JL, Miller YI (2006) Minimally oxidized LDL offsets the apoptotic effects of extensively oxidized LDL and free cholesterol in macrophages. Arterioscler Thromb Vasc Biol 26:1169–1176. doi:10.1161/01.ATV.0000210279.97308.9a
Brocheriou I, Maouche S, Durand H, Braunersreuther V, Le NG, Gratchev A, Koskas F, Mach F, Kzhyshkowska J, Ninio E (2011) Antagonistic regulation of macrophage phenotype by M-CSF and GM-CSF: implication in atherosclerosis. Atherosclerosis 214:316–324. doi:10.1016/j.atherosclerosis.2010.11.023
Butcher MJ, Galkina EV (2012) Phenotypic and functional heterogeneity of macrophages and dendritic cell subsets in the healthy and atherosclerosis-prone aorta. Front Physiol 3:44. doi:10.3389/fphys.2012.00044
Cantoni O, Tommasini I, Cerioni L, Palomba L, Carloni E, Guidarelli A (2005) Survival pathways triggered by peroxynitrite in cells belonging to the monocyte/macrophage lineage. Comp Biochem Physiol A Mol Integr Physiol 142:118–123. doi:10.1016/j.cbpb.2005.05.037
Cerioni L, Cantoni O (2007) ERK1/2 regulates two sequential steps promoting monocyte survival to peroxynitrite. J Cell Physiol 210:177–182. doi:10.1002/jcp.20837
Chang TY, Li BL, Chang CC, Urano Y (2009) Acyl-coenzyme A: cholesterol acyltransferases. Am J Physiol Endocrinol Metab 297:E1–E9. doi:10.1152/ajpendo.90926.2008
Chen JH, Riazy M, Smith EM, Proud CG, Steinbrecher UP, Duronio V (2009) Oxidized LDL-mediated macrophage survival involves elongation factor-2 kinase. Arterioscler Thromb Vasc Biol 29:92–98. doi:10.1161/ATVBAHA.108.174599
Croons V, Martinet W, Herman AG, Timmermans JP, De Meyer GRY (2007) Selective clearance of macrophages in atherosclerotic plaques by the protein synthesis inhibitor cycloheximide. J Pharmacol Exp Ther 320:986–993. doi:10.1124/jpet.106.113944
Croons V, Martinet W, Herman AG, Timmermans JP, De Meyer GRY (2009) The protein synthesis inhibitor anisomycin induces macrophage apoptosis in rabbit atherosclerotic plaques through p38 mitogen-activated protein kinase. J Pharmacol Exp Ther 329:856–864. doi:10.1124/jpet.108.149948
Cui D, Thorp E, Li Y, Wang N, Yvan-Charvet L, Tall AR, Tabas I (2007) Pivotal Advance: macrophages become resistant to cholesterol-induced death after phagocytosis of apoptotic cells. J Leukoc Biol 82:1040–1050. doi:10.1189/jlb.0307192
De Meyer I, Martinet W, De Meyer GRY (2012) Therapeutic strategies to deplete macrophages in atherosclerotic plaques. Br J Clin Pharmacol 74:246–263. doi:10.1111/j.1365-2125.2012.04211.x
De Meyer I, Martinet W, Schrijvers DM, Timmermans J-P, Bult H, De Meyer GRY (2012) Toll-like receptor 7 stimulation by imiquimod induces macrophage autophagy and inflammation in atherosclerotic plaques. Basic Res Cardiol. doi:10.1007/s00395-012-0269-1
DeVries-Seimon T, Li Y, Yao PM, Stone E, Wang Y, Davis RJ, Flavell R, Tabas I (2005) Cholesterol-induced macrophage apoptosis requires ER stress pathways and engagement of the type A scavenger receptor. J Cell Biol 171:61–73. doi:10.1083/jcb.200502078
Dickhout JG, Lhotak S, Hilditch BA, Basseri S, Colgan SM, Lynn EG, Carlisle RE, Zhou J, Sood SK, Ingram AJ, Austin RC (2011) Induction of the unfolded protein response after monocyte to macrophage differentiation augments cell survival in early atherosclerotic lesions. FASEB J 25:576–589. doi:10.1096/fj.10-159319
Dietrich T, Perlitz C, Licha K, Stawowy P, Atrott K, Tachezy M, Meyborg H, Stocker C, Grafe M, Fleck E, Schirner M, Graf K (2007) ED-B fibronectin (ED-B) can be targeted using a novel single chain antibody conjugate and is associated with macrophage accumulation in atherosclerotic lesions. Basic Res Cardiol 102:298–307. doi:10.1007/s00395-007-0652-5
Droin N, Rebe C, Bichat F, Hammann A, Bertrand R, Solary E (2001) Modulation of apoptosis by procaspase-2 short isoform: selective inhibition of chromatin condensation, apoptotic body formation and phosphatidylserine externalization. Oncogene 20:260–269. doi:10.1038/sj.onc.1204066
Erbel C, Dengler TJ, Wangler S, Lasitschka F, Bea F, Wambsganss N, Hakimi M, Bockler D, Katus HA, Gleissner CA (2011) Expression of IL-17A in human atherosclerotic lesions is associated with increased inflammation and plaque vulnerability. Basic Res Cardiol 106:125–134. doi:10.1007/s00395-010-0135-y
Feig JE, Parathath S, Rong JX, Mick SL, Vengrenyuk Y, Grauer L, Young SG, Fisher EA (2011) Reversal of hyperlipidemia with a genetic switch favorably affects the content and inflammatory state of macrophages in atherosclerotic plaques. Circulation 123:989–998. doi:10.1161/CIRCULATIONAHA.110.984146
Feig JE, Rong JX, Shamir R, Sanson M, Vengrenyuk Y, Liu J, Rayner K, Moore K, Garabedian M, Fisher EA (2011) HDL promotes rapid atherosclerosis regression in mice and alters inflammatory properties of plaque monocyte-derived cells. Proc Natl Acad Sci USA 108:7166–7171. doi:10.1073/pnas.1016086108
Furukawa Y, Kubo N, Kikuchi J, Tokura A, Fujita N, Sakurabayashi I (2000) Regulation of macrophage-specific gene expression by degenerated lipoproteins. Electrophoresis 21:338–346. doi:10.1002/(SICI)1522-2683(20000101)21:2<338:AID-ELPS338>3.0.CO;2-9
Gerry AB, Leake DS (2008) A moderate reduction in extracellular pH protects macrophages against apoptosis induced by oxidized low density lipoprotein. J Lipid Res 49:782–789. doi:10.1194/jlr.M700349-JLR200
Gieseg SP, Amit Z, Yang YT, Shchepetkina A, Katouah H (2010) Oxidant production, oxLDL uptake, and CD36 levels in human monocyte-derived macrophages are downregulated by the macrophage-generated antioxidant 7,8-dihydroneopterin. Antioxid Redox Signal 13:1525–1534. doi:10.1089/ars.2009.3065
Gieseg SP, Leake DS, Flavall EM, Amit Z, Reid L, Yang YT (2009) Macrophage antioxidant protection within atherosclerotic plaques. Front Biosci 14:1230–1246. doi:10.2741/3305
Hamilton JA, Jessup W, Brown AJ, Whitty G (2001) Enhancement of macrophage survival and DNA synthesis by oxidized-low-density-lipoprotein (LDL)-derived lipids and by aggregates of lightly oxidized LDL. Biochem J 355:207–214
Hamilton JA, Myers D, Jessup W, Cochrane F, Byrne R, Whitty G, Moss S (1999) Oxidized LDL can induce macrophage survival, DNA synthesis, and enhanced proliferative response to CSF-1 and GM-CSF. Arterioscler Thromb Vasc Biol 19:98–105. doi:10.1161/01.ATV.19.1.98
Han CY, Pak YK (1999) Oxidation-dependent effects of oxidized LDL: proliferation or cell death. Exp Mol Med 31:165–173
Hetz C (2012) The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol 13:89–102. doi:10.1038/nrm3270
Hockenbery DM, Oltvai ZN, Yin XM, Milliman CL, Korsmeyer SJ (1993) Bcl-2 functions in an antioxidant pathway to prevent apoptosis. Cell 75:241–251. doi:10.1016/0092-8674(93)80066-N
Hulten LM, Ullstrom C, Krettek A, van RD, Marklund SL, Dahlgren C, Wiklund O (2005) Human macrophages limit oxidation products in low density lipoprotein. Lipids Health Dis 4:6. doi:10.1186/1476-511X-4-6
Hundal RS, Salh BS, Schrader JW, Gomez-Munoz A, Duronio V, Steinbrecher UP (2001) Oxidized low density lipoprotein inhibits macrophage apoptosis through activation of the PI 3-kinase/PKB pathway. J Lipid Res 42:1483–1491
Jeong SJ, Pise-Masison CA, Radonovich MF, Park HU, Brady JN (2005) Activated AKT regulates NF-kappaB activation, p53 inhibition and cell survival in HTLV-1-transformed cells. Oncogene 24:6719–6728. doi:10.1038/sj.onc.1208825
Kadl A, Meher AK, Sharma PR, Lee MY, Doran AC, Johnstone SR, Elliott MR, Gruber F, Han J, Chen W, Kensler T, Ravichandran KS, Isakson BE, Wamhoff BR, Leitinger N (2010) Identification of a novel macrophage phenotype that develops in response to atherogenic phospholipids via Nrf2. Circ Res 107:737–746. doi:10.1161/CIRCRESAHA.109.215715
Kelly C, Jefferies C, Cryan SA (2011) Targeted liposomal drug delivery to monocytes and macrophages. J Drug Deliv 2011:727241. doi:10.1155/2011/727241
Keul P, Lucke S, von Wnuck LK, Bode C, Graler M, Heusch G, Levkau B (2011) Sphingosine-1-phosphate receptor 3 promotes recruitment of monocyte/macrophages in inflammation and atherosclerosis. Circ Res 108:314–323. doi:10.1161/CIRCRESAHA.110.235028
Keul P, Tolle M, Lucke S, von Wnuck LK, Heusch G, Schuchardt M, van der Giet M, Levkau B (2007) The sphingosine-1-phosphate analogue FTY720 reduces atherosclerosis in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol 27:607–613. doi:10.1161/01.ATV.0000254679.42583.88
Kikuchi J, Furukawa Y, Kubo N, Tokura A, Hayashi N, Nakamura M, Matsuda M, Sakurabayashi I (2000) Induction of ubiquitin-conjugating enzyme by aggregated low density lipoprotein in human macrophages and its implications for atherosclerosis. Arterioscler Thromb Vasc Biol 20:128–134. doi:10.1161/01.ATV.20.1.128
Kinscherf R, Claus R, Wagner M, Gehrke C, Kamencic H, Hou D, Nauen O, Schmiedt W, Kovacs G, Pill J, Metz J, Deigner HP (1998) Apoptosis caused by oxidized LDL is manganese superoxide dismutase and p53 dependent. FASEB J 12:461–467
Kinscherf R, Deigner HP, Usinger C, Pill J, Wagner M, Kamencic H, Hou D, Chen M, Schmiedt W, Schrader M, Kovacs G, Kato K, Metz J (1997) Induction of mitochondrial manganese superoxide dismutase in macrophages by oxidized LDL: its relevance in atherosclerosis of humans and heritable hyperlipidemic rabbits. FASEB J 11:1317–1328
Kohlstedt K, Trouvain C, Namgaladze D, Fleming I (2011) Adipocyte-derived lipids increase angiotensin-converting enzyme (ACE) expression and modulate macrophage phenotype. Basic Res Cardiol 106:205–215. doi:10.1007/s00395-010-0137-9
Kubo N, Kikuchi J, Furukawa Y, Sakai T, Ohta H, Iwase S, Yamada H, Sakurabayashi I (1997) Regulatory effects of aggregated LDL on apoptosis during foam cell formation of human peripheral blood monocytes. FEBS Lett 409:177–182. doi:10.1016/S0014-5793(97)00501-2
Lam M, Dubyak G, Chen L, Nunez G, Miesfeld RL, Distelhorst CW (1994) Evidence that BCL-2 represses apoptosis by regulating endoplasmic reticulum-associated Ca2 + fluxes. Proc Natl Acad Sci USA 91:6569–6573
Landsman L, Bar-On L, Zernecke A, Kim KW, Krauthgamer R, Shagdarsuren E, Lira SA, Weissman IL, Weber C, Jung S (2009) CX3CR1 is required for monocyte homeostasis and atherogenesis by promoting cell survival. Blood 113:963–972. doi:10.1182/blood-2008-07-170787
Ley K, Miller YI, Hedrick CC (2011) Monocyte and macrophage dynamics during atherogenesis. Arterioscler Thromb Vasc Biol 31:1506–1516. doi:10.1161/ATVBAHA.110.221127
Li W, Zou W, Yang Y, Chai Y, Chen B, Cheng S, Tian D, Wang X, Vale RD, Ou G (2012) Autophagy genes function sequentially to promote apoptotic cell corpse degradation in the engulfing cell. J Cell Biol 197:27–35. doi:10.1083/jcb.201111053
Liao HS, Kodama T, Geng YJ (2000) Expression of class A scavenger receptor inhibits apoptosis of macrophages triggered by oxidized low density lipoprotein and oxysterol. Arterioscler Thromb Vasc Biol 20:1968–1975. doi:10.1161/01.ATV.20.8.1968
Liao X, Sluimer JC, Wang Y, Subramanian M, Brown K, Pattison JS, Robbins J, Martinez J, Tabas I (2012) Macrophage autophagy plays a protective role in advanced atherosclerosis. Cell Metab 15:545–553. doi:10.1016/j.cmet.2012.01.022
Llodra J, Angeli V, Liu J, Trogan E, Fisher EA, Randolph GJ (2004) Emigration of monocyte-derived cells from atherosclerotic lesions characterizes regressive, but not progressive, plaques. Proc Natl Acad Sci USA 101:11779–11784. doi:10.1073/pnas.0403259101
Mantovani A, Garlanda C, Locati M (2009) Macrophage diversity and polarization in atherosclerosis: a question of balance. Arterioscler Thromb Vasc Biol 29:1419–1423. doi:10.1161/ATVBAHA.108.180497
Martinet W, De Meyer GRY (2009) Autophagy in atherosclerosis: a cell survival and death phenomenon with therapeutic potential. Circ Res 104:304–317. doi:10.1161/CIRCRESAHA.108.188318
Martinet W, Knaapen MW, De Meyer GRY, Herman AG, Kockx MM (2001) Oxidative DNA damage and repair in experimental atherosclerosis are reversed by dietary lipid lowering. Circ Res 88:733–739. doi:10.1161/hh0701.088684
Martinet W, Knaapen MW, De Meyer GRY, Herman AG, Kockx MM (2002) Elevated levels of oxidative DNA damage and DNA repair enzymes in human atherosclerotic plaques. Circulation 106:927–932. doi:10.1161/01.CIR.0000026393.47805.21
Martinet W, Knaapen MW, De Meyer GRY, Herman AG, Kockx MM (2003) Overexpression of the anti-apoptotic caspase-2 short isoform in macrophage-derived foam cells of human atherosclerotic plaques. Am J Pathol 162:731–736. doi:10.1016/S0002-9440(10)63869-2
Martinet W, Schrijvers DM, De Meyer GRY (2011) Necrotic cell death in atherosclerosis. Basic Res Cardiol 106:749–760. doi:10.1007/s00395-011-0192-x
Martinet W, Schrijvers DM, De Meyer GRY (2011) Pharmacological modulation of cell death in atherosclerosis: a promising approach towards plaque stabilization? Br J Pharmacol 164:1–13. doi:10.1111/j.1476-5381.2011.01342.x
Martinet W, Verheye S, De Meyer GRY (2007) Everolimus-induced mTOR inhibition selectively depletes macrophages in atherosclerotic plaques by autophagy. Autophagy 3:241–244
Martinet W, Verheye S, De Meyer I, Timmermans JP, Schrijvers DM, Van Brussel I, Bult H, De Meyer GRY (2012) Everolimus triggers cytokine release by macrophages: rationale for stents eluting everolimus and a glucocorticoid. Arterioscler Thromb Vasc Biol 32:1228–1235. doi:10.1161/ATVBAHA.112.245381
Mizushima N, Komatsu M (2011) Autophagy: renovation of cells and tissues. Cell 147:728–741. doi:10.1016/j.cell.2011.10.026
Moore KJ, Tabas I (2011) Macrophages in the pathogenesis of atherosclerosis. Cell 145:341–355. doi:10.1016/j.cell.2011.04.005
Muslin AJ (2008) MAPK signalling in cardiovascular health and disease: molecular mechanisms and therapeutic targets. Clin Sci (Lond) 115:203–218. doi:10.1042/CS20070430
Nagy N, Melchior-Becker A, Fischer JW (2010) Long-term treatment with the AT1-receptor antagonist telmisartan inhibits biglycan accumulation in murine atherosclerosis. Basic Res Cardiol 105:29–38. doi:10.1007/s00395-009-0051-1
Nguyen XH, Lang PA, Lang KS, Adam D, Fattakhova G, Foger N, Kamal MA, Prilla P, Mathieu S, Wagner C, Mak T, Chan AC, Lee KH (2011) Toso regulates the balance between apoptotic and nonapoptotic death receptor signaling by facilitating RIP1 ubiquitination. Blood 118:598–608. doi:10.1182/blood-2010-10-313643
Oksjoki R, Kovanen PT, Lindstedt KA, Jansson B, Pentikainen MO (2006) OxLDL-IgG immune complexes induce survival of human monocytes. Arterioscler Thromb Vasc Biol 26:576–583. doi:10.1161/01.ATV.0000201041.14438.8d
Ouimet M, Franklin V, Mak E, Liao X, Tabas I, Marcel YL (2011) Autophagy regulates cholesterol efflux from macrophage foam cells via lysosomal acid lipase. Cell Metab 13:655–667. doi:10.1016/j.cmet.2011.03.023
Park D, Han CZ, Elliott MR, Kinchen JM, Trampont PC, Das S, Collins S, Lysiak JJ, Hoehn KL, Ravichandran KS (2011) Continued clearance of apoptotic cells critically depends on the phagocyte Ucp2 protein. Nature 477:220–224. doi:10.1038/nature10340
Pentikainen MO, Oorni K, Ala-Korpela M, Kovanen PT (2000) Modified LDL: trigger of atherosclerosis and inflammation in the arterial intima. J Intern Med 247:359–370. doi:10.1046/j.1365-2796.2000.00655.x
Perlman H, Pagliari LJ, Georganas C, Mano T, Walsh K, Pope RM (1999) FLICE-inhibitory protein expression during macrophage differentiation confers resistance to fas-mediated apoptosis. J Exp Med 190:1679–1688. doi:10.1084/jem.190.11.1679
Poitz DM, Augstein A, Weinert S, Braun-Dullaeus RC, Strasser RH, Schmeisser A (2011) OxLDL and macrophage survival: essential and oxygen-independent involvement of the Hif-pathway. Basic Res Cardiol 106:761–772. doi:10.1007/s00395-011-0186-8
Potteaux S, Gautier EL, Hutchison SB, van RN, Rader DJ, Thomas MJ, Sorci-Thomas MG, Randolph GJ (2011) Suppressed monocyte recruitment drives macrophage removal from atherosclerotic plaques of Apoe-/- mice during disease regression. J Clin Invest 121:2025–2036. doi:10.1172/JCI43802
Qu X, Zou Z, Sun Q, Luby-Phelps K, Cheng P, Hogan RN, Gilpin C, Levine B (2007) Autophagy gene-dependent clearance of apoptotic cells during embryonic development. Cell 128:931–946. doi:10.1016/j.cell.2006.12.044
Razani B, Feng C, Coleman T, Emanuel R, Wen H, Hwang S, Ting JP, Virgin HW, Kastan MB, Semenkovich CF (2012) Autophagy links inflammasomes to atherosclerotic progression. Cell Metab 15:534–544. doi:10.1016/j.cmet.2012.02.011
Reddy SM, Hsiao KH, Abernethy VE, Fan H, Longacre A, Lieberthal W, Rauch J, Koh JS, Levine JS (2002) Phagocytosis of apoptotic cells by macrophages induces novel signaling events leading to cytokine-independent survival and inhibition of proliferation: activation of Akt and inhibition of extracellular signal-regulated kinases 1 and 2. J Immunol 169:702–713
Riazy M, Chen JH, Yamamato Y, Yamamato H, Duronio V, Steinbrecher UP (2011) OxLDL-mediated survival of macrophages does not require LDL internalization or signalling by major pattern recognition receptors. Biochem Cell Biol 89:387–395. doi:10.1139/o11-035
Rios FJ, Gidlund M, Jancar S (2011) Pivotal role for platelet-activating factor receptor in CD36 expression and oxLDL uptake by human monocytes/macrophages. Cell Physiol Biochem 27:363–372. doi:10.1159/000327962
Rios FJ, Koga MM, Ferracini M, Jancar S (2012) Co-stimulation of PAFR and CD36 is required for oxLDL-induced human macrophages activation. PLoS ONE 7:e36632. doi:10.1371/journal.pone.0036632
Roiniotis J, Dinh H, Masendycz P, Turner A, Elsegood CL, Scholz GM, Hamilton JA (2009) Hypoxia prolongs monocyte/macrophage survival and enhanced glycolysis is associated with their maturation under aerobic conditions. J Immunol 182:7974–7981. doi:10.4049/jimmunol.0804216
Saxena A, McMeekin JD, Thomson DJ (2002) Expression of Bcl-x, Bcl-2, Bax, and Bak in endarterectomy and atherectomy specimens. J Pathol 196:335–342. doi:10.1002/path.1040
Schrijvers DM, De Meyer GRY, Herman AG, Martinet W (2007) Phagocytosis in atherosclerosis: molecular mechanisms and implications for plaque progression and stability. Cardiovasc Res 73:470–480. doi:10.1016/j.cardiores.2006.09.005
Schrijvers DM, De Meyer GRY, Kockx MM, Herman AG, Martinet W (2005) Phagocytosis of apoptotic cells by macrophages is impaired in atherosclerosis. Arterioscler Thromb Vasc Biol 25:1256–1261. doi:10.1161/01.ATV.0000166517.18801.a7
Schrijvers DM, De Meyer GRY, Martinet W (2011) Autophagy in atherosclerosis: a potential drug target for plaque stabilization. Arterioscler Thromb Vasc Biol 31:2787–2791. doi:10.1161/ATVBAHA.111.224899
Seimon TA, Wang Y, Han S, Senokuchi T, Schrijvers DM, Kuriakose G, Tall AR, Tabas IA (2009) Macrophage deficiency of p38alpha MAPK promotes apoptosis and plaque necrosis in advanced atherosclerotic lesions in mice. J Clin Invest 119:886–898. doi:10.1172/JCI37262
Shearn AI, Deswaerte V, Gautier EL, Saint-Charles F, Pirault J, Bouchareychas L, Rucker EB III, Beliard S, Chapman J, Jessup W, Huby T, Lesnik P (2012) Bcl-x inactivation in macrophages accelerates progression of advanced atherosclerotic lesions in Apoe(−/−) mice. Arterioscler Thromb Vasc Biol 32:1142–1149. doi:10.1161/ATVBAHA.111.239111
Shimizu S, Eguchi Y, Kamiike W, Funahashi Y, Mignon A, Lacronique V, Matsuda H, Tsujimoto Y (1998) Bcl-2 prevents apoptotic mitochondrial dysfunction by regulating proton flux. Proc Natl Acad Sci USA 95:1455–1459
Sigruener A, Buechler C, Bared SM, Grandl M, Aslanidis C, Ugocsai P, Gehrmann M, Schmitz G (2007) E-LDL upregulates TOSO expression and enhances the survival of human macrophages. Biochem Biophys Res Commun 359:723–728. doi:10.1016/j.bbrc.2007.05.169
Sluimer JC, Gasc JM, van Wanroij JL, Kisters N, Groeneweg M, Sollewijn G, Cleutjens JP, van den Akker LH, Corvol P, Wouters BG, Daemen MJ, Bijnens AP (2008) Hypoxia, hypoxia-inducible transcription factor, and macrophages in human atherosclerotic plaques are correlated with intraplaque angiogenesis. J Am Coll Cardiol 51:1258–1265. doi:10.1016/j.jacc.2007.12.025
Song Y, Jacob CO (2005) The mouse cell surface protein TOSO regulates Fas/Fas ligand-induced apoptosis through its binding to Fas-associated death domain. J Biol Chem 280:9618–9626. doi:10.1074/jbc.M413609200
Susin SA, Zamzami N, Castedo M, Hirsch T, Marchetti P, Macho A, Daugas E, Geuskens M, Kroemer G (1996) Bcl-2 inhibits the mitochondrial release of an apoptogenic protease. J Exp Med 184:1331–1341
Swirski FK, Pittet MJ, Kircher MF, Aikawa E, Jaffer FA, Libby P, Weissleder R (2006) Monocyte accumulation in mouse atherogenesis is progressive and proportional to extent of disease. Proc Natl Acad Sci USA 103:10340–10345. doi:10.1073/pnas.0604260103
Tabas I (2005) Consequences and therapeutic implications of macrophage apoptosis in atherosclerosis: the importance of lesion stage and phagocytic efficiency. Arterioscler Thromb Vasc Biol 25:2255–2264. doi:10.1161/01.ATV.0000184783.04864.9f
Tabas I (2010) The role of endoplasmic reticulum stress in the progression of atherosclerosis. Circ Res 107:839–850. doi:10.1161/CIRCRESAHA.110.224766
Tano JY, Vazquez G (2011) Requirement for non-regulated, constitutive calcium influx in macrophage survival signaling. Biochem Biophys Res Commun 407:432–437. doi:10.1016/j.bbrc.2011.03.048
Thorp E, Li G, Seimon TA, Kuriakose G, Ron D, Tabas I (2009) Reduced apoptosis and plaque necrosis in advanced atherosclerotic lesions of Apoe-/- and Ldlr-/- mice lacking CHOP. Cell Metab 9:474–481. doi:10.1016/j.cmet.2009.03.003
Thorp E, Li Y, Bao L, Yao PM, Kuriakose G, Rong J, Fisher EA, Tabas I (2009) Brief report: increased apoptosis in advanced atherosclerotic lesions of Apoe−/− mice lacking macrophage Bcl-2. Arterioscler Thromb Vasc Biol 29:169–172. doi:10.1161/ATVBAHA.108.176495
Tiwari RL, Singh V, Barthwal MK (2008) Macrophages: an elusive yet emerging therapeutic target of atherosclerosis. Med Res Rev 28:483–544. doi:10.1002/med.20118
Ujihara M, Nomura K, Yamada O, Shibata N, Kobayashi M, Takano K (2001) Granulocyte-macrophage colony-stimulating factor ensures macrophage survival and generation of the superoxide anion: a study using a monocytic-differentiated HL60 subline. Free Radic Biol Med 31:1396–1404. doi:10.1016/S0891-5849(01)00711-0
Van De Parre TJ, Martinet W, Verheye S, Kockx MM, Van Langenhove G, Herman AG, De Meyer GRY (2008) Mitochondrial uncoupling protein 2 mediates temperature heterogeneity in atherosclerotic plaques. Cardiovasc Res 77:425–431. doi:10.1093/cvr/cvm003
Van De Parre T, Martinet W, Verheye S, De Meyer GRY (2007) Uncoupling protein 2-mediated thermogenesis in vulnerable atherosclerotic plaques. Eurointervention 3:275–279
Van Herck JL, De Meyer GRY, Martinet W, Bult H, Vrints CJ, Herman AG (2010) Proteasome inhibitor bortezomib promotes a rupture-prone plaque phenotype in ApoE-deficient mice. Basic Res Cardiol 105:39–50. doi:10.1007/s00395-009-0054-y
Van Herck JL, De Meyer GRY, Martinet W, Salgado RA, Shivalkar B, De Mondt R, Van De Ven H, Ludwig A, Van Der Veken P, Van Vaeck L, Bult H, Herman AG, Vrints CJ (2010) Multi-slice computed tomography with N1177 identifies ruptured atherosclerotic plaques in rabbits. Basic Res Cardiol 105:51–59. doi:10.1007/s00395-009-0052-0
Verheye S, De Meyer GRY, Van Langenhove G, Knaapen MW, Kockx MM (2002) In vivo temperature heterogeneity of atherosclerotic plaques is determined by plaque composition. Circulation 105:1596–1601. doi:10.1161/01.CIR.0000012527.94843.BF
Verheye S, Martinet W, Kockx MM, Knaapen MW, Salu K, Timmermans JP, Ellis JT, Kilpatrick DL, De Meyer GRY (2007) Selective clearance of macrophages in atherosclerotic plaques by autophagy. J Am Coll Cardiol 49:706–715. doi:10.1016/j.jacc.2006.09.047
Weber C, Noels H (2011) Atherosclerosis: current pathogenesis and therapeutic options. Nat Med 17:1410–1422. doi:10.1038/nm.2538
White GE, Greaves DR (2012) Fractalkine: a survivor’s guide: chemokines as antiapoptotic mediators. Arterioscler Thromb Vasc Biol 32:589–594. doi:10.1161/ATVBAHA.111.237412
Yang H, Chen SC, Tang YQ, Dai YL (2011) Effect of overexpression of human SR-AI on oxLDL uptake and apoptosis in 293T cells. Int Immunopharmacol 11:1752–1757. doi:10.1016/j.intimp.2011.07.001
Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Peng TI, Jones DP, Wang X (1997) Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science 275:1129–1132. doi:10.1126/science.275.5303.1129
Zhao M, Eaton JW, Brunk UT (2000) Protection against oxidant-mediated lysosomal rupture: a new anti-apoptotic activity of Bcl-2? FEBS Lett 485:104–108. doi:10.1016/S0014-5793(00)02195-5
Zhou J, Lhotak S, Hilditch BA, Austin RC (2005) Activation of the unfolded protein response occurs at all stages of atherosclerotic lesion development in apolipoprotein E-deficient mice. Circulation 111:1814–1821. doi:10.1161/01.CIR.0000160864.31351.C1
Acknowledgments
This work was supported by the Fund for Scientific Research-Flanders and the University of Antwerp.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Martinet, W., Schrijvers, D.M. & De Meyer, G.R.Y. Molecular and cellular mechanisms of macrophage survival in atherosclerosis. Basic Res Cardiol 107, 297 (2012). https://doi.org/10.1007/s00395-012-0297-x
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00395-012-0297-x