
Abstract
Accumulation of biglycan, a small leucine-rich proteoglycan, in the neointima precedes the retention of lipids and accumulation of macrophages during early atherosclerosis. Biglycan is therefore considered a pro-atherogenic proteoglycan that might play a key role in atherogenesis. On the other hand biglycan ensures in part establishment of stable collagen networks. Aim of the present study was to determine whether telmisartan affects biglycan accumulation in a murine model of accelerated atherosclerosis and whether collagen matrix is affected. ApoE−/−-mice on Western diet were chronically (12 weeks) treated either with telmisartan (10 mg/kg) or hydralazine (500 mg/l drinking water) and systolic arterial blood pressure was determined by tail cuff plethysmography. Animals were killed and aortic plaque score, plaque morphology and extracellular matrix as well as cellular plaque composition were analyzed at the aortic root. Furthermore, expression of biglycan and enzymes involved in collagen cross-linking were analyzed in the aorta. Telmisartan and hydralazine lowered systolic arterial blood pressure to the same extent. Biglycan accumulation in the aorta and the aortic root was significantly reduced by telmisartan but not by hydralazine. The amount of collagen and collagen fibril density, macrophages and SMCs was not affected by either treatment as determined by analysis of picrosirius red staining, mac2 and α-SM-actin. Furthermore, telmisartan inhibited aortic plaque score and aortic root plaque size compared to mice receiving hydralazine and untreated controls. The current study shows that telmisartan reduces biglycan accumulation and inhibits atherosclerosis independently of blood pressure lowering and without affecting the collagenous plaque matrix. Thus, biglycan is a pleiotropic target of telmisartan that might contribute to the anti-atherogenic effects of this AT1-antagonist.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Atherosclerosis progresses over decades from the initial intimal thickenings to complex expanded lesions of the arterial vessel wall [16]. This process is not only characterized by accumulation of macrophage-derived foam cells, lipid deposition and cytokine release [37, 38], but also by phase-specific accumulation and extensive remodeling of extracellular matrix (ECM) [14, 23]. Proteoglycans, hyaluronan and collagen comprise both functionally and structurally important group of ECM molecules that are deposited in the neointimal areas of atherosclerotic plaques [32].
Proteoglycans are key components of the ECM due to their multifaceted functions such as control of growth factor activity, control of collagen fibrillogenesis, signaling and lipid retention [33, 35]. The latter three functions have among other proteoglycans been attributed to the small leucine-rich repeat proteoglycan (SLRP) biglycan [18, 19]. Biglycan consists of a protein core containing leucine-rich repeats and two glycosaminoglycan (GAG) chains consisting of either chondroitin sulphate or dermatan sulphate [8].
Biglycan emerged to be important for a variety of processes that are relevant for vascular functions and pathophysiology. First, soluble biglycan has recently been shown to signal through toll-like receptors 2 and 4, leading to macrophage activation, which in turn contributes to systemic inflammation as shown in a murine model of sepsis [18]. Second, biglycan confers LDL-binding properties mediated by the glycosaminoglycan chains. The binding of LDL has been shown in vitro in biglycan overexpressing smooth muscle cells [15]. Furthermore, in human pathological intimal thickening biglycan accumulation precedes and co-localizes to lipid deposits and subsequent macrophage invasion [13]. This property places biglycan at the center of the “response-to-retention” hypothesis of atherogenesis assuming that increased biglycan expression in the arterial wall causes LDL retention in the ECM, facilitates LDL-modification and therefore initiates and accelerates the early events in the pathophysiology of atherogenesis [22, 34]. Third, as a collagen binding proteoglycan, biglycan regulates the collagen fibril diameter in the adventitia. This was demonstrated by disruptive aortic aneurysm occurring in biglycan deficient mice [6] and may explain the association of low biglycan expression with abdominal aneurysms in humans [25]. Biglycan deficient mice showed also impaired scar formation after myocardial infarction leading to left ventricular rupture and hemodynamic insufficiency [31]. Biglycan expression is regulated by growth factors such as platelet derived growth factor BB, transforming growth factor beta 1 (TGFβ1), interleukin 1 and angiotensin II (Ang II) [20, 21, 28]. Furthermore, in case of Ang II biglycan expression responds to AT1-receptor stimulation and subsequent release of TGFβ1 as shown in cardiac fibroblasts [1, 26].
Considering the diverse and phase-specific functions proposed for biglycan during atherosclerosis, one might propose that biglycan contributes to lipid retention and inflammation at the earliest stages of atherosclerosis. On the other hand biglycan is likely also involved in the stability of the collagenous matrix of atherosclerotic plaques and might confer plaque stability. The aim of the present study was to elucidate whether chronic treatment of apoE−/−-mice with the AT1 antagonist telmisartan (1) reduces biglycan accumulation in pre-existing atherosclerotic vascular lesions and (2) how this affects plaque collagen matrix and lipid retention in the matrix.
Materials and methods
Animals
Male apoE−/−-mice were obtained from Taconic M&B (Lille Skensved, Denmark). Mice were housed in single cages, allowed water ad libitum and were kept on a normal 12-h light and dark cycle. Starting at an age of 4 weeks mice received Western diet containing 21% saturated fat and 0.15% cholesterol for 11 weeks. At the age of 16 weeks apoE−/−-mice were divided into three groups receiving (1) Western diet, (2) Western diet containing telmisartan (10 mg/kg) and (3) Western diet plus hydralazine (500 mg/l) in drinking water. Telmisartan, provided by Boehringer Ingelheim, was pelleted into the Western diet by ssniff (Soest, Germany). Subsequently, mice remained for 12 weeks on the respective treatments and were killed at the age of 28 weeks with CO2 (Fig. 1a). Initially ten animals were in each group. Because two controls, one hydralazine-treated animal and one telmisartan-treated animal died, n = 8 for controls and n = 9 for both hydralazine and telmisartan. Subsequently, a second experiment was performed including again ten animals in each treatment group. From these animals five aortas were used for mRNA isolation and five aortas were used for Western blot analysis. The exact numbers of animals that were used for the individual analysis are also indicated in the legends of the respective figures. All experiments were performed according to the guidelines for the use of experimental animals as given by “Deutsches Tierschutzgesetz” and according to the Guide for the Care and Use of Laboratory Animals of the US National Institutes of Health.

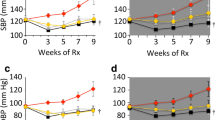
a Experimental design: male apoE−/−-mice were treated with Western diet starting at 4 weeks of age for 24 weeks. At an age of 16 weeks mice were divided into three groups: control, telmisartan (10 mg/kg per day) and hydralazine (500 mg/l). b Systolic blood pressure, measured by a non-invasive tail cuff system. c Total plasma cholesterol and d triglyceride concentration. Data represent mean ± SD, n = 6, * P < 0.05 versus control. Con control, tel telmisartan, hyd hydralazine
Blood pressure measurements
Systolic blood pressure (SBP) was measured by a non-invasive computerized tail cuff system (Vistech System BP-2000, Apex, USA). All animals, six per group, were conditioned to tail cuff measurements over a period of 2 weeks to ensure that the results were not influenced by stress caused by the procedure. SBP values were obtained on three consecutive days at the end of treatment.
Plasma lipid analysis
Blood was collected by heart puncture and anti-coagulated with 100 mM EDTA in isotonic sodium chloride solution. Plasma was prepared via centrifugation at 850 × g for 15 min at 4°C and stored at −20°C for later analysis. Total cholesterol and triglycerides were subsequently quantified using standardized measurement systems used for routine evaluation of human cholesterol and triglyceride levels.
Tissue processing and fixation
The heart and entire aorta were fixed in 4% neutral buffered paraformaldehyde for 2 h and subsequently transferred into 20% sucrose in PBS solution. Hearts were frozen in tissue freezing medium (Leica Nussloch, Bensheim, Germany) in liquid isopentane at −40°C and 14 μm cryosections of the aortic root were prepared for immunohistochemical staining.
Immunohistochemistry
Biglycan and decorin were detected after chondroitinase ABC digestion with polyclonal rabbit antisera against murine biglycan (1:1,000, LF 106) and murine decorin (1:1,000, LF 113) kindly provided by Larry Fisher (National Institute of Dental and Craniofacial Research, National Institutes of Health, Bethesda, MD, USA). Anti-heparin sulphate proteoglycan perlecan was detected with monoclonal rat antisera against murine perlecan (1:50, Seikagaku, Tokyo, Japan). Detection was performed using diaminobenzidine (Zytomed, Berlin, Germany) as a chromogen. Smooth muscle cells (SMC) were stained with a monoclonal mouse anti-α-SM-actin antibody (clone 1A4, 1:1,000, Sigma, Deisenhofen, Germany). Secondary antibody to detect anti-α-SM-actin was obtained from Santa Cruz (Santa Cruz, USA). Affinity histochemistry of hyaluronan was performed with HA binding protein, detected with FITC-labeled streptavidin (2 μg/ml, Calbiochem, Bad Soden, Germany). Macrophages were detected by an antibody against mac2 (1:400, Cedarlane, Burlington, Canada) and a Rhodamine Red-X conjugated goat anti-rat IgG (preabsorbed to rodent, 1:400, Jackson ImmunoResearch, Suffolk, UK) was used as a secondary antibody. ApoB-48 was detected using K23300R (BioDesign, Saco, ME, 1:50) recognizing mouse apoB-48 and human apoB-100 [7].
Histochemistry
Lipid deposition was determined on cryosections of the aortic root by Oil-red-O staining. Collagen accumulation was detected by picrosirius red staining. Qualitative analysis of collagen deposition was performed using polarized light microscopy and birefringence analysis [9].
Plaque burden of the aorta
Atherosclerotic plaques were visualized by Oil-red-O staining of lipid deposits. Subsequently images of en face preparations of the whole mounted aorta were taken and the percentage of plaques in relation to the entire aortic surface calculated as plaque score in percent of total area using ImageJ 1.37v software (NIH).
mRNA analysis
Total mRNA was extracted from the aorta after removal of the adventitial layer using TriReagent according to the manufactures instructions. mRNA was reverse transcribed and quantitative real-time analysis for ECM cross-linking enzymes, prolyl 4-hydroxylase subunit alpha-3 (P4ha3) (forward: 5′-CGACTTGACCAGATTCTAT-GAC-3′, reverse: 5′-GAAGGCAAGTAGAGGATTCAC-3′), tissue transglutaminase 1 (TGM1) (forward: 5′-CCTCAGATGGATCTTCAATGGT-3′, reverse: 5′-CCATTGTGC-CTTATATTGCAGAG-3′), procollagen-lysine 1,2-oxoglutarate 5-dioxygenase 1 lysyl hydroxylase (PLOD1) (forward: 5′-GAGCCTTGGATGAAGTTGTG-3′, reverse: 5′-TA-GTTGCCCAGGTAGTTCAG-3′), performed as described previously [31].
Morphometric plaque analysis
Plaque area was determined at the aortic root level using bright field images taken by ColorViewII and AnalySIS 3.2 (Soft Imaging System; Münster, Germany) and analyzed by ImageJ 1.37v software (NIH).
Western blot analysis
Sulphated proteoglycans were extracted from the total thoracic and abdominal aortas of apoE−/−-mice under dissociative conditions and subsequently purified by anion exchange chromatography, ethanol precipitation and subsequently digested with chondroitinase ABC to expose core proteins as described previously [10]. Western blot analysis of biglycan was performed using the LF159 antibody for biglycan (1:1,000). Quantification was performed using fluorescent secondary antibodies and the Odyssey Infrared Imaging System (LI-COR Biosciences, Lincoln, USA).
Statistical analysis
Data are presented as the mean ± SD. Statistical analysis was performed by one-way ANOVA followed by comparison of selected pairs (Bonferroni). A value of P < 0.05 was considered significant.
Results
Blood pressure and metabolic parameters
ApoE−/−-mice were fed Western type diet from 4 to 16 weeks of age and subsequently drug treatment was initiated. At this starting point of the experiment apoE−/−-mice already had developed atherosclerotic lesions and treatment on Western diet was continued for 12 weeks. Thus, during the experimental period plaque burden further increases and complex atherosclerotic lesions developed (Fig. 1). Systolic blood pressure measured by a non-invasive tail cuff system showed that both telmisartan and hydralazine lowered blood pressure to a similar extent (Fig. 1b). Therefore, the effects of telmisartan that did not occur in response to hydralazine were indicative for pleiotropic effects not due to blood pressure lowering. Plasma was analyzed with regard to cholesterol and triglyceride concentrations (Fig. 1c, d). The total cholesterol level and triglyceride concentration were similar in control and telmisartan-treated animals. A nonsignificant trend towards a reduction of total plasma cholesterol and plasma triglycerides was observed in hydralazine-treated animals. Taken together, telmisartan did not significantly affect metabolic parameters.
Quantitative analysis of atherosclerosis in the aorta and aortic root
At the starting point at 16 weeks of the experiment, plaque burden was determined to be 4.5 ± 1.1% in controls (Fig. 2b). During the experimental period of 12 weeks, aortic plaque score progressed to 8.2 ± 2.6%, which was prevented by telmisartan. In contrast, hydralazine treatment had no effect on plaque burden (9.1 ± 2.9%, Fig. 2b). To determine the effect of the treatment specifically on the plaque area, morphometric analysis was performed at the aortic root (Fig. 2c), which revealed a significant reduction of plaque size only by telmisartan. Taken together, telmisartan decreased aortic plaque score and plaque size at the aortic root which is likely an effect independent of blood pressure lowering.

a Lipid accumulation in the aorta detected by en face staining with Oil-red-O at 28 weeks. b Plaque score of the aorta at 16 weeks and after treatment at 28 weeks. c Plaque area at the aortic root. Plaque score of the aorta and aortic root were determined using analysis software from Analysis Soft Imaging System. Data represent mean ± SD, n = 8–9, * P < 0.05 versus control. Con control, tel telmisartan, hyd hydralazine
Proteoglycans in plaques of the aorta and of the aortic root
Aortic biglycan expression was analyzed by Western blot analysis performed on aortic extracts. As shown in Fig. 3a only telmisartan treatment resulted in a significant reduction of biglycan protein expression. The extracts used for Western blotting were prepared from the entire aorta, thus representing atherosclerotic and non-atherosclerotic regions. Considering that only 5–10% of the aorta is affected by plaque development (compare to plaque score), the result likely indicates substantial reduction of biglycan in aortic plaques. For specific analysis of biglycan expression in atherosclerotic lesions biglycan accumulation was detected by immunohistochemistry in plaques at the aortic root. Biglycan was already strongly expressed at the beginning of the experimental period at 16 weeks of age and further increased to about 30% area fraction (Fig. 3b) at the end of the study. This increase was prohibited by telmisartan treatment but not by hydralazine suggesting an effect independent of blood pressure. Figure 3c shows the expression pattern of biglycan in the aortic root lesions characterized by strong expression throughout the plaque neointima and to a lesser extent in the media.

a Western blot analysis of biglycan extracted from thoracic aorta of apoE-deficient mice at 28 weeks, quantification was performed using fluorescent secondary antibodies and the Odyssey Infrared Imaging System, n = 5. b Densitometric quantification of biglycan immunohistochemistry at the aortic root at 16 and 28 weeks, n = 8–9. Data represent mean ± SD, * P < 0.05. c Representative aortic root lesions of apoE−/−-mice aged 28 weeks stained for biglycan. Pictures were taken at a 40-fold magnification. Con control, tel telmisartan, hyd hydralazine
To analyze whether also other proteoglycans were affected by the drug treatment decorin, a small leucine-rich proteoglycan related to biglycan, and perlecan, a large heparin sulphate proteoglycan, were detected as well. As shown in Fig. 4, decorin was neither affected by telmisartan nor by hydralazine. Interestingly, perlecan was increased in the group receiving telmisartan (Fig. 4).

a Western blot analysis of decorin extracted from thoracic aorta of apoE-deficient mice, quantification was performed using fluorescent secondary antibodies and the Odyssey Infrared Imaging System, n = 5. b Densitometric quantification of decorin immunohistochemistry at the aortic root. c Densitometric quantification of perlecan immunohistochemistry at the aortic root. d Representative aortic root lesions of apoE−/−-mice stained for decorin. e Representative aortic root lesions of apoE−/−-mice stained for perlecan. Data in b and c represent mean ± SD, n = 8–9, * P < 0.05. Pictures were taken at a 40-fold magnification. Con control, tel telmisartan, hyd hydralazine
Plaque composition of aortic root lesions of apoE −/−-mice
To analyze whether the down regulation of biglycan by telmisartan affects collagen accumulation and collagen fibril packing picrosirius red staining was performed and analyzed by light microscopy and under polarized light. Total collagen was not changed as shown in Fig. 5a, b. Furthermore, no differences were detected in collagen fibril density as shown by birefringence analysis (Fig. 5c, d). Another parameter indicative for collagen stability is collagen cross-linking. Therefore, the aortic mRNA expression of key enzymes involved in collagen cross-linking, prolyl 4-hydroxylase subunit alpha-3 (P4ha3) (Fig. 5e), tissue transglutaminase 1 (TGM1) (Fig. 5f), procollagen-lysine 1,2-oxoglutarate 5-dioxygenase 1 lysyl hydroxylase (PLOD1) (Fig. 5g), was analyzed by quantitative real-time analysis. TGM1 was upregulated and PLOD1 showed a nonsignificant trend towards upregulation in both telmisartan and hydralazine-treated mice, suggesting even increased expression of cross-linking enzymes in response to blood pressure lowering (Fig. 5e, g). Because plaque stability was not directly determined, the results on collagen and collagen cross-linking enzymes may be taken as evidence that the collagen matrix is at least not weakened in response to telmisartan treatment despite of lower biglycan content.

Composition of aortic root lesions of apoE−/−-mice after treatment with telmisartan and hydralazine. a, b Total collagen content of aortic root plaques as indicated by picrosirius red staining observed by light microscopy. c, d Collagen fibril packing analyzed by picrosirius red staining and birefringence using polarized light. Pictures were taken at 40-fold magnification, densitometric quantitation of total collagen and red polarizing collagen indicating tightly packed collagen fibrils is presented, n = 8–9. e–g mRNA expression of ECM cross-linking enzymes performed from total aortic mRNA. e Prolyl 4-hydroxylase subunit alpha-3 (P4ha3). f Tissue transglutaminase 1 (TGM1). g Procollagen-lysine 1,2-oxoglutarate 5-dioxygenase 1 lysylhydroxylase (PLOD1), n = 5, data represent mean ± SD, * P < 0.05 versus control. Con control, tel telmisartan, hyd hydralazine
To complete the analysis of matrix components considered to be critically involved in atherogenesis, hyaluronan accumulation was detected. Hyaluronan (HA) was not changed by telmisartan. Furthermore, the cellular composition was assessed by detection of macrophages (mac2) and SMC (α-SM-actin). However, also the cellular composition was not changed by telmisartan or hydralazine (Fig. 6a–f).

Aortic root lesions of treated apoE−/−-mice. a, b Hyaluronan (HA), visualized by fluorescent HABP staining. c, d VSMC (α-SM-actin) content. e, f Macrophage (mac2) content. Pictures were taken at 100-fold magnification. Densitometric quantitation of HA (b), α-SM-actin (d) and mac2 (f) is presented, n = 8–9, mean ± SD. Con control, tel telmisartan, hyd hydralazine
Lipid retention in aortic root lesions
The data presented above revealed that telmisartan specifically reduced biglycan accumulation whereas it increased perlecan. In contrast, other matrix components and the cellular composition were not affected. To investigate whether the changes in proteoglycan matrix affected the retention of lipids in the plaque matrix Oil-red-O staining indicating total lipid retention and apoB-48 staining indicative for LDL were performed and quantified in aortic root lesions. As shown in Fig. 7 neither total lipid retention nor apoB-48 were affected.

Aortic root lesions of treated apoE−/−-mice. a Oil-red-O staining of plaques of the aortic root. b Quantification of Oil-red-O staining by image analysis. c ApoB-48 immunostaining and d densitometric quantification. Data represent mean ± SD, n = 8–9. Pictures were taken at a 40-fold magnification. Con control, tel telmisartan, hyd hydralazine
Discussion
It is well documented that biglycan binds to LDL particles and thereby supports LDL retention within the vascular ECM [12]. It has been shown that lipid accumulation occurs in human pathological intimal thickening in the same regions and prior to macrophage accumulation [13]. Therefore, inhibition of biglycan expression might be an effective strategy to inhibit the very first step in atherogenesis as detailed in the “response to retention hypothesis”. Furthermore, Tannock and co-workers [7] showed in an elegant study that infusion of Ang II into LDL-receptor deficient mice induces biglycan expression in the intima of atherosclerotic lesions and that LDL is deposited at the sites of high biglycan expression in these fatty streak like lesions.
However, on the other hand it was recently discovered that deficiency of biglycan leads to aortic dissection in mice [6], that biglycan deficiency causes rupture of infarct scars [31] and that low biglycan is associated with aneurysms in humans [25]. Therefore, it appeared important to investigate if telmisartan would indeed affect biglycan in advanced atherosclerotic plaques and if so, does this then cause unfavorable changes in vascular matrix composition and does it affect lipid accumulation.
Thus, the first question addressed in the present study was whether AT1-receptor antagonists can effectively reduce biglycan accumulation in advanced atherosclerosis rather than in the initial phase of atherogenesis. Therefore, we started treatment at the age of 16 weeks in male apoE−/−-mice with pre-existing atherosclerosis and extended the experimental period to the age of 28 weeks. At this time apoE−/−-mice present advanced atherosclerotic plaques. Indeed, telmisartan reduced aortic biglycan expression in atherosclerotic plaques of the aortic root in the present experimental model. This is in line with previous studies showing that AT1-blockade reduces biglycan expression after myocardial infarction in rats and in vitro in cardiac fibroblasts [1, 26]. Thus, it is likely an effect directly due to inhibition of AT1-receptor signaling and independent of blood pressure lowering since hydralazine had no effect on biglycan. The present results thus complement the finding by Huang et al. 2008 [7] that Ang II induces biglycan expression in early atherosclerotic lesions in LDL-receptor negative mice. Considering also that candesartan inhibited renal expression of biglycan in SHR-rats [17], it is very likely that biglycan is indeed a target of Ang II in atherosclerosis. Thus, other AT1-antagonists and ACE-inhibitors may have similar effects. However, this remains to be shown in future studies. Taken together our results are the first indication that pharmacologic down regulation of biglycan can indeed be achieved by the treatment with the AT1-receptor antagonist telmisartan in advanced atherosclerosis.
The second so far unanswered question addressed in this manuscript was whether pharmacologic reduction of biglycan affects the stability of vascular collagen matrix. This question appears very important to us, since several lines of evidence suggest that absence of biglycan will cause undesirable weakening of the collagenous matrix in a variety of organs. The first evidence came from the initial phenotyping of biglycan−/0-mice showing reduced growth and mineralization of bones [36]. Followed by the observation that collagen fibrillogenesis in tendon is impaired accompanied by the occurrence of an osteoarthritis-like phenotype [2]. Recently it was discovered that biglycan−/0-mice developed irregular collagen fibrils also in the adventitia of the aorta which in turn caused aortic aneurysm and aortic dissection [6]. A similarly dramatic result was obtained in biglycan−/0-mice after experimental myocardial infarction, revealing impaired collagen fibril assembly in infarct scars leading to frequent left ventricular ruptures and hemodynamic insufficiency [31]. These effects of biglycan deletion were comparable to the effect on collagen network stability and the biological consequences observed in mice deficient of decorin, a closely related small leucine-rich proteoglycan [4, 30]. Importantly, the present findings clearly indicate that in contrast to the genetic knock out, partial reduction of biglycan in the aorta and in atherosclerotic plaques does not lead to undesirable changes either of the collagenous matrix or the cellular composition of plaques. Furthermore, both telmisartan and hydralazine treatment led to upregulation of ECM cross-linking enzymes which is likely a consequence of blood pressure lowering and may lead to strengthening of the ECM.
In addition, the current experimental design allowed to answer the question whether telmisartan treatment of pre-existing, advanced lesions will reduce lipid retention within the matrix. The present results revealed that this was not the case. This may be due to the fact that the lipid accumulation was already advanced at the time of treatment and not responsive anymore to subsequently induced changes in matrix composition. In addition, we found that perlecan was upregulated. Because perlecan is also thought to mediate lipid retention in the plaque matrix and to serve pro-atherogenic functions [27], the accumulation of perlecan might have compensated the downregulation of biglycan with respect to lipid retention. The upregulation of perlecan was unexpected in the light of the findings by Huang et al. [7] but in line with a study by van Det et al. [29]. In addition, perlecan might be upregulated as part of a compensatory response to the reduction of biglycan accumulation since both proteoglycans are thought to have partially overlapping functions with in the plaque matrix [11].
The macrophage and smooth muscle content were not affected, although it has been shown before that telmisartan reduced the number of inflammatory cells in murine atherosclerosis. The macrophage influx occurs early in the present model peaking between 10 and 14 weeks of age (data not shown). Thus, in our study the treatment was initiated after the peak of the inflammatory response and carried on until inflammation subsided even in controls. In contrast, other studies demonstrated reduced macrophage content in response to telmisartan in the past. However, these studies started the treatment much earlier [5, 24], used a by far higher daily dose of telmisartan [3] or a Western type diet with 1.5% cholesterol [5]. Taken together the previous studies used different timing, dosing and diets, which may explain the detection of anti-inflammatory effects of telmisartan that were not evident in the present experimental design.
In conclusion, telmisartan treatment specifically affects proteoglycan matrix composition of advanced atherosclerotic lesions independent of blood pressure lowering and without disturbance of the architecture of the collagenous plaque matrix and without effects on lipid retention.
References
Ahmed MS, Oie E, Vinge LE, Yndestad A, Andersen GG, Andersson Y, Attramadal T, Attramadal H (2003) Induction of myocardial biglycan in heart failure in rats—an extracellular matrix component targeted by AT(1) receptor antagonism. Cardiovasc Res 60:557–568
Ameye L, Aria D, Jepsen K, Oldberg A, Xu T, Young MF (2002) Abnormal collagen fibrils in tendons of biglycan/fibromodulin-deficient mice lead to gait impairment, ectopic ossification, and osteoarthritis. FASEB J 16:673–680
Blessing E, Preusch M, Kranzhofer R, Kinscherf R, Marx N, Rosenfeld ME, Isermann B, Weber CM, Kreuzer J, Grafe J, Katus HA, Bea F (2008) Anti-atherosclerotic properties of telmisartan in advanced atherosclerotic lesions in apolipoprotein E deficient mice. Atherosclerosis 199:295–303
Danielson KG, Baribault H, Holmes DF, Graham H, Kadler KE, Iozzo RV (1997) Targeted disruption of decorin leads to abnormal collagen fibril morphology and skin fragility. J Cell Biol 136:729–743
Grothusen C, Bley S, Selle T, Luchtefeld M, Grote K, Tietge UJ, Drexler H, Schieffer B (2005) Combined effects of HMG-CoA-reductase inhibition and renin–angiotensin system blockade on experimental atherosclerosis. Atherosclerosis 182:57–69
Heegaard AM, Corsi A, Danielsen CC, Nielsen KL, Jorgensen HL, Riminucci M, Young MF, Bianco P (2007) Biglycan deficiency causes spontaneous aortic dissection and rupture in mice. Circulation 115:2731–2738
Huang F, Thompson JC, Wilson PG, Aung HH, Rutledge JC, Tannock LR (2008) Angiotensin II increases vascular proteoglycan content preceding and contributing to atherosclerosis development. J Lipid Res 49:521–530
Iozzo RV (1999) The biology of the small leucine-rich proteoglycans. Functional network of interactive proteins. J Biol Chem 274:18843–18846
Karim MA, Miller DD, Farrar MA, Eleftheriades E, Reddy BH, Breland CM, Samarel AM (1995) Histomorphometric and biochemical correlates of arterial procollagen gene expression during vascular repair after experimental angioplasty. Circulation 91:2049–2057 (see comments)
Kinsella MG, Wight TN (1988) Isolation and characterization of dermatan sulfate proteoglycans synthesized by cultured bovine aortic endothelial cells. J Biol Chem 263:19222–19231
Kunjathoor VV, Chiu DS, O’Brien KD, LeBoeuf RC (2002) Accumulation of biglycan and perlecan, but not versican, in lesions of murine models of atherosclerosis. Arterioscler Thromb Vasc Biol 22:462–468
Little PJ, Osman N, O’Brien KD (2008) Hyperelongated biglycan: the surreptitious initiator of atherosclerosis. Curr Opin Lipidol 19:448–454
Nakashima Y, Fujii H, Sumiyoshi S, Wight TN, Sueishi K (2007) Early human atherosclerosis: accumulation of lipid and proteoglycans in intimal thickenings followed by macrophage infiltration. Arterioscler Thromb Vasc Biol 27:1159–1165
Newby AC, Southgate KM, Davies M (1994) Extracellular matrix degrading metalloproteinases in the pathogenesis of arteriosclerosis. Basic Res Cardiol 89(Suppl 1):59–70
O’Brien KD, Lewis K, Fischer JW, Johnson P, Hwang JY, Knopp EA, Kinsella MG, Barrett PH, Chait A, Wight TN (2004) Smooth muscle cell biglycan overexpression results in increased lipoprotein retention on extracellular matrix: implications for the retention of lipoproteins in atherosclerosis. Atherosclerosis 177:29–35
Ross R (1999) Atherosclerosis is an inflammatory disease. Am Heart J 138:S419–S420
Sasamura H, Shimizu-Hirota R, Nakaya H, Saruta T (2001) Effects of AT1 receptor antagonist on proteoglycan gene expression in hypertensive rats. Hypertens Res 24:165–172
Schaefer L, Babelova A, Kiss E, Hausser HJ, Baliova M, Krzyzankova M, Marsche G, Young MF, Mihalik D, Gotte M, Malle E, Schaefer RM, Grone HJ (2005) The matrix component biglycan is proinflammatory and signals through Toll-like receptors 4 and 2 in macrophages. J Clin Invest 115:2223–2233
Schaefer L, Beck KF, Raslik I, Walpen S, Mihalik D, Micegova M, Macakova K, Schonherr E, Seidler DG, Varga G, Schaefer RM, Kresse H, Pfeilschifter J (2003) Biglycan, a nitric oxide-regulated gene, affects adhesion, growth, and survival of mesangial cells. J Biol Chem 278:26227–26237
Schönherr E, Järveläinen HT, Kinsella MG, Sandell LJ, Wight TN (1993) Platelet-derived growth factor and transforming growth factor-b1 differentially affect the synthesis of biglycan and decorin by monkey arterial smooth muscle cells. Arterioscler. Thromb. 13:1026–1036
Shimizu-Hirota R, Sasamura H, Mifune M, Nakaya H, Kuroda M, Hayashi M, Saruta T (2001) Regulation of vascular proteoglycan synthesis by angiotensin II type 1 and type 2 receptors. J Am Soc Nephrol 12:2609–2615
Skalen K, Gustafsson M, Rydberg EK, Hulten LM, Wiklund O, Innerarity TL, Boren J (2002) Subendothelial retention of atherogenic lipoproteins in early atherosclerosis. Nature 417:750–754
Stary HC (1994) Changes in components and structure of atherosclerotic lesions developing from childhood to middle age in coronary arteries. Basic Res Cardiol 89(Suppl 1):17–32
Takaya T, Kawashima S, Shinohara M, Yamashita T, Toh R, Sasaki N, Inoue N, Hirata K, Yokoyama M (2006) Angiotensin II type 1 receptor blocker telmisartan suppresses superoxide production and reduces atherosclerotic lesion formation in apolipoprotein E-deficient mice. Atherosclerosis 186:402–410
Theocharis AD, Karamanos NK (2002) Decreased biglycan expression and differential decorin localization in human abdominal aortic aneurysms. Atherosclerosis 165:221–230
Tiede K, Stoter K, Petrik C, Chen WB, Ungefroren H, Kruse ML, Stoll M, Unger T, Fischer JW (2003) Angiotensin II AT(1)-receptor induces biglycan in neonatal cardiac fibroblasts via autocrine release of TGFbeta in vitro. Cardiovasc Res 60:538–546
Tran-Lundmark K, Tran PK, Paulsson-Berne G, Friden V, Soininen R, Tryggvason K, Wight TN, Kinsella MG, Boren J, Hedin U (2008) Heparan sulfate in perlecan promotes mouse atherosclerosis: roles in lipid permeability, lipid retention, and smooth muscle cell proliferation. Circ Res 103:43–52
Ungefroren H, Krull NB (1996) Transcriptional regulation of the human biglycan gene. J Biol Chem 271:15787–15795
van Det NF, Tamsma JT, van den Born J, Verhagen NA, van den Heuvel LP, Lowik CW, Berden JH, Bruijn JA, Daha MR, van der Woude FJ (1996) Differential effects of angiotensin II and transforming growth factor beta on the production of heparan sulfate proteoglycan by mesangial cells in vitro. J Am Soc Nephrol 7:1015–1023
Weis SM, Zimmerman SD, Shah M, Covell JW, Omens JH, Ross J Jr, Dalton N, Jones Y, Reed CC, Iozzo RV, McCulloch AD (2005) A role for decorin in the remodeling of myocardial infarction. Matrix Biol 24:313–324
Westermann D, Mersmann J, Melchior A, Freudenberger T, Petrik C, Schaefer L, Lullmann-Rauch R, Lettau O, Jacoby C, Schrader J, Brand-Herrman SM, Young MF, Schultheiss HP, Levkau B, Baba HA, Unger T, Zacharowski K, Tschope C, Fischer JW (2008) Biglycan is required for adaptive remodeling after myocardial infarction. Circulation 117:1269–1276
Wight TN (1995) The extracellular matrix and atherosclerosis. Curr Opin Lipidol 6:326–334
Wight TN, Merrilees MJ (2004) Proteoglycans in atherosclerosis and restenosis: key roles for versican. Circ Res 94:1158–1167
Williams KJ, Tabas I (1995) The response-to-retention hypothesis of early atherogenesis. Arterioscler Thromb Vasc Biol 15:551–561
Woods A, Couchman JR (1992) Heparan sulfate proteoglycans and signalling in cell adhesion. Adv Exp Med Biol 313:87–96
Xu T, Bianco P, Fisher LW, Longenecker G, Smith E, Goldstein S, Bonadio J, Boskey A, Heegaard AM, Sommer B, Satomura K, Dominguez P, Zhao C, Kulkarni AB, Robey PG, Young MF (1998) Targeted disruption of the biglycan gene leads to an osteoporosis-like phenotype in mice. Nat Genet 20:78–82
Zernecke A, Weber C (2005) Inflammatory mediators in atherosclerotic vascular disease. Basic Res Cardiol 100:93–101
Zhang C (2008) The role of inflammatory cytokines in endothelial dysfunction. Basic Res Cardiol 103:398–406
Acknowledgments
Financial support by Boehringer Ingelheim Pharma GmbH & Co. KG and scientific contact by Dr. C. Teutsch (Boehringer Ingelheim) is acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Nagy, N., Melchior-Becker, A. & Fischer, J.W. Long-term treatment with the AT1-receptor antagonist telmisartan inhibits biglycan accumulation in murine atherosclerosis. Basic Res Cardiol 105, 29–38 (2010). https://doi.org/10.1007/s00395-009-0051-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00395-009-0051-1