
Abstract
Enteroviruses, such as coxsackieviruses of group B (CVB), are able to induce a chronic inflammation of the myocardium, which may finally lead to the loss of functional tissue, remodeling processes and the development of fibrosis, thus affecting the proper contractile function of the heart. In other fibrotic diseases like scleroderma, the prostacyclin agonist iloprost was found to inhibit the extracellular signal-regulated kinase (ERK, p44/42 MAPK), a mitogen-activated protein kinase, and consecutively, the expression of the profibrotic cytokine connective tissue growth factor (CTGF), thereby preventing the development of fibrosis. As CTGF was found to mediate fibrosis in chronic CVB3 myocarditis as well, we evaluated whether the in vivo application of iloprost is capable to reduce the development of ERK/CTGF-mediated fibrosis in enteroviral myocarditis. Unexpectedly, the application of iloprost resulted in a prolonged myocardial inflammation and an aggravated fibrosis and failed to reduce activation of ERK and expression of CTGF at later stages of the disease. In addition, viral replication was found to be increased in iloprost-treated mice. Notably, the expression of cardiac inducible nitric oxide synthase (iNOS), which is known to aggravate myocardial damage in CVB3-infected mice, was strongly enhanced by iloprost. Using cultivated bone marrow macrophages (BMM), we confirmed these results, proving that iloprost potentiates the expression of iNOS mRNA and protein in CVB3-infected and IFN-gamma stimulated BMM. In conclusion, these results suggest a critical reflection of the clinical use of iloprost, especially in patients possibly suffering from an enteroviral myocarditis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Enteroviruses of the Picornaviridae, including coxsackieviruses of group B (CVB), are considered as relevant causes of acute and chronic viral myocarditis [21, 24]. The clinical presentations of this disease vary from mild febrile illness to fulminant myocardial inflammation with congestive heart failure. Following acute infection, susceptible patients may present with ongoing myocardial inflammation on the basis of a virus persistence, finally resulting in chronic dilated cardiomyopathy (DCM) [43], which can be treated effectively only by heart transplantation. Morphological correlates of the late inflammatory disease comprise focally accentuated fibrosis of the myocardium, impairing the contractile function of the heart, thus leading to severe congestive heart failure [43]. Mice of the susceptible strain ABY/SnJ infected with CVB3 proceed to chronic myocarditis, which is based on the persistence of viral RNA and sustained inflammatory infiltrates [23]. Corresponding to findings in human myocarditis, ABY/SnJ mice reveal a loss of functional myocytes in association with an abundant deposition of collagen type I in the course of myocarditis, finally leading to fibrosis [29].
Connective tissue growth factor (CTGF, CCN2) has recently been identified as an important molecule mediating fibrosis in chronic CVB3-induced myocarditis [29]. CTGF is a cysteine-rich 36–38 kDa secreted protein of the CCN-family (CTGF/CYR61/NOV) [6] which is involved in many physiological processes like angiogenesis and cellular differentiation, but has also been proven to play an essential role in fibrogenetic processes of different etiology in the heart [18] as well as lung, liver, kidney, skin and other organs [6]. In vitro, CTGF stimulates the proliferation of fibroblasts, their differentiation toward myofibroblasts and enhances the production of various components of the extracellular matrix (ECM) [12, 13]. In different cell types, including cardiac myocytes and fibroblasts, CTGF was found to act as a downstream mediator of transforming growth factor beta (TGF-β) [5, 6]. In a previous study, we demonstrated a coordinate increase of TGF-β and CTGF in the course of murine CVB3 myocarditis, stressing the relevance of this signaling pathway in the development of fibrosis in the course of chronic inflammatory heart disease [29].
More recently, CTGF has not only been considered a prognostic marker in evolving fibrotic diseases but also as an interesting molecule for establishing new therapeutic antifibrotic approaches [6, 18]. Importantly, by specifically reducing the expression of CTGF, the objectionable consequences of a therapy would be avoided by targeting the comprehensive TGF-β signaling, which has been found to be relevant for numerous physiological and pathophysiological processes in the heart [50]. Meanwhile, there is an increasing evidence that TGF-β dependent regulation of CTGF expression is modulated by a complex network of signaling cascades including the mitogen-activated protein kinases (MAPKs) [19, 36]. In cell culture experiments with human and murine fibroblasts, the MAPK extracellular signal-regulated kinase (ERK, p44/42) was identified as important regulator of CTGF expression [30, 31, 47]. Substances like iloprost, which increase the intracellular level of cAMP, were found to significantly suppress CTGF expression by inhibiting the MEK/ERK cascade [5, 10, 47].
The synthetic prostacyclin (PGI2) analog iloprost was developed in the 1970s. Being more stable than prostacyclin itself, this pharmaceutical proved to optimize the beneficial effects of prostacyclin, which is a potent vasodilator and inhibitor of platelet aggregation. Iloprost is currently used in the therapy of pulmonary arterial hypertension (PAH) and Raynaud’s phenomenon in patients suffering from scleroderma [1, 52]. Iloprost was shown to increase intracellular cAMP levels by stimulating the Gαs-coupled IP prostanoid receptor [22] and to inhibit the Ras/MEK/ERK cascade, thus reducing the CTGF-mediated fibrosis in vitro and in patients suffering from scleroderma [46, 47]. Most recently, the cyclic nucleotide phosphodiesterase 1A-regulated cAMP and cGMP signaling was found to play an important role in the regulation of cardiac fibroblast activation and ECM remodeling in the heart [37].
The aim of this present study was to evaluate the capacity of iloprost to reduce cardiac fibrosis in the murine model of chronic enteroviral myocarditis. Therefore, we evaluated the histological damage, expression of CTGF and procollagen and activation of p44/42 MAPK in the course of enteroviral myocarditis in untreated and iloprost-treated mice. In addition, in vitro experiments using primary isolated cardiac fibroblasts and bone marrow macrophages (BMM) were performed to study the molecular effects of iloprost treatment on CVB3 infection.
Materials and Methods
Virus and mice
Five × 104 plaque-forming units of purified CVB3 Nancy strain [20] were used to intraperitoneally (ip) infect 4-week-old susceptible immunocompetent inbred ABY/SnJ mice (H-2b) as described previously [29]. The experiments were conducted according to the German animal protection law. Treatment with iloprost (1 μg per day) started 1 day prior to infection and ended 12 days post-infection (pi). In addition, one group of animals was treated ip with iloprost from day 12 pi to 22 pi. This additional investigation was done in order to evaluate the effects of iloprost on the hearts at a time point when virus replication is largely abrogated. Treated and untreated animals were sacrificed at days 0 (controls), 4, 8, 12 and 28 pi (n = 6 per time point). One of the treated and none of the untreated animals died during acute infection. Hearts were removed aseptically and tissue was either frozen in liquid nitrogen for reverse transcriptase-polymerase chain reaction (RT-PCR) and Western blot analysis or fixed in 4 % paraformaldehyde for histological analysis, in situ hybridization and immunohistochemistry as previously described [29].
Histopathology
Histological analysis was performed on deparaffinized 5 μm thick tissue sections. Staining was performed with hematoxylin/eosin to assess inflammation and myocyte injury. To visualize fibrotic lesions, tissue sections were stained with picrosirius red. To quantify myocardial damage comprising cardiac cell necrosis, inflammation and scarring, we adapted a myocarditis score from 0 to 4 as previously described [48].
In situ hybridization
CVB3 positive-strand RNA, CTGF mRNA and inducible nitric oxide synthase (iNOS) mRNA were visualized in heart tissue using single-stranded 35S-labeled RNA probes as previously described [29, 48]. Pretreatment, hybridization, and washing procedures of dewaxed 5 μm thick paraffin tissue sections were performed as previously described [23]. Slide preparations were subjected to autoradiography, exposed for 3–4 weeks at 4 °C, and counterstained with hematoxylin/eosin. Quantification of CVB3 RNA, CTGF mRNA and iNOS mRNA following in situ hybridization was done using the following score obtained in ten high-power fields (200×): 0 = no positive cells; 1 = a few small foci with some positive cells; 2 = a few foci with ≥100 positive cells; 3 = ≤10 % of the tissue sections contain positive cells; 4 = 10–30 % of the tissue sections contain positive cells.
Immunohistochemistry and immunofluorescence
To localize phospho-p44/42 MAPK protein expression, 5 μm thick heart tissue sections were deparaffinized and incubated with polyclonal rabbit anti-phospho-p44/42 MAPK (Thr202/Tyr204) antibody (Cell Signaling, Beverly, USA). For the detection of IP-10 and CD3+ T cells in the hearts, a monoclonal rabbit anti-CD3 antibody (Neomarkers, Newmarket, UK) and a polyclonal goat anti-IP10 antibody (R&D, Minneapolis, USA) were used, followed by incubation with biotinylated goat anti-rabbit IgG or biotinylated rabbit anti-goat IgG (Vector, Burlingame, USA), respectively. Controls using normal serum were run to exclude nonspecific staining. Slides were further processed using Elite Vectastain ABC Kit (Vector, Burlingame, USA) and DAB (DAKO, Glostrup, Denmark) or Histogreen (Linaris, Dossenheim, Germany) as substrate. Slides were counterstained with hematoxylin.
Identification of pERK expressing cardiac cells was done by immunofluorescence double-labeling experiments. Antibodies were diluted 1:150 (anti-S100A4, Acris Antibodies, Herford Germany), 1:400 (phospho-p44/42 MAPK, Cell Signaling Beverly, USA), 1:100 (anti-desmin, LifeSpan Biosciences, Seattle, USA) in PBS + 1 % BSA and incubated at 4 °C overnight. As secondary antibodies, Alexa-488 or-594-conjugated antibodies (Life Technologies, Paisley, UK) were diluted 1:800 in PBS and incubated for 1 h at room temperature in the dark. After washing in PBS, nuclei were counterstained with DAPI and embedded in mounting media (Dianova, Hamburg, Germany) before microscopic analysis.
For the detection of iNOS in bone marrow-generated macrophages, the cultivated cells were fixed with acetone and incubated with a polyclonal rabbit anti-iNOS antibody (Abcam, Cambridge, UK) followed by incubation with biotinylated goat anti-rabbit IgG (Vector, Burlingame, USA) and Cy3-conjugated streptavidin (Jackson Immunoresearch, West Grove, USA). Slides were mounted with Vectashield mounting medium (Vector, Burlingame, USA).
Cultivation and infection of primary cardiac mouse fibroblasts
For isolation of cardiac mouse fibroblasts, hearts from 8- to 14-week-old ABY/SnJ mice were removed under sterile conditions and dissected in 1 mm pieces. The tissue was incubated for 1.5 h in 1 mg/ml collagenase II (Worthington, Lakewood, USA) at 37 °C and then seeded into culture dishes containing Dulbecco’s modified Eagle’s medium (DMEM, Invitrogen, Heidelberg, Germany) supplemented with 10 % fetal calf serum (FCS) and 1 % penicillin/streptomycin. At passage 5 or 6, 3 × 105 cells were seeded in a culture dish and incubated for 48 h. After further 24 h of serum starving, cells were infected with CVB3 at a multiplicity of infection (MOI) of 10. Iloprost was applied at a concentration of 0.139 μM (50 ng/ml) or 2.774 μM (1,000 ng/ml) 1 h prior to infection. Cells were harvested 6 h pi and activation of p44/42 MAPK was analyzed by Western blot.
Cultivation and infection of bone marrow-generated macrophages
Femora of 8- to 14-week-old ABY/SnJ mice were removed and the bone marrow was seeded in low endotoxin DMEM (Biochrome, Berlin, Germany) supplemented with 10 % FCS, 5 % horse serum, 1 % penicillin/streptomycin, 1 % glutamine, 1 % sodium pyruvate and 30 % (v/v) supernatant of L-929 cells as a source of macrophage-colony stimulating factor (M-CSF) [11]. Cells were cultured for 9 days in ultra-low attachment 6-well plates (Corning Inc., New York, USA) at 37 °C and 10 % CO2. Two hours after seeding, cells were stimulated with 100 U/ml recombinant murine interferon-γ (IFN-γ, BD Biosciences, San Jose, USA). After 12 h of stimulation, cells were infected with CVB3 at a MOI of 10. Iloprost was applied at a concentration of 0.139 μM (50 ng/ml) or 2.774 μM (1,000 ng/ml) 1 h prior to infection. Cells were harvested 6 h pi. The non-infected cells underwent the same changes in media and after the addition of serum in the media as the CVB3-infected cells. Expression of iNOS mRNA and CVB3 RNA were analyzed by quantitative real-time RT-PCR and in situ hybridization. Expression of iNOS protein was visualized by immunofluorescence.
Quantitative real-time RT-PCR
Total RNA of frozen heart tissue and of cultured cells was isolated using TriFast-reagent (Peqlab, Erlangen, Germany) according to the manufacturer’s instructions. Isolated RNA (0.2 μg) was used to perform one-step quantitative real-time RT-PCR (TaqMan One-Step RT-PCR Master Mix Reagents Kit, Applied Biosystems, Foster City, USA). Data analysis of CTGF, procollagen Iα1, and iNOS was performed in relation to the expression of the housekeeping gene hypoxanthine-guanine-phosphoribosyltransferase (HPRT) as an internal standard as described [48]. The number of CVB3 genomes was determined in relation to an external virus standard of known copy numbers using PanEntero-primers and probe as described [38]. Primers and probes were purchased from MWG-Biotech AG (Ebersberg, Germany): CTGF: forward 5′-TCC CGA GAA GGG TCA AGC T-3′, reverse 5′-TCC TTG GGC TCG TCA CAC A-3′; procollagen Iα1: forward 5′-TCC GGC TCC TGC TCC TCT TA-3′, reverse 5′-GTA TGC AGC TGA CTT CAG GGA TGT-3′; iNOS: forward 5′-CAG CTG GGC TGT ACA AAC CTT-3′, reverse 5′-CAT TGG AAG TGA AGC GTT TCG-3′; HPRT: forward 5′-TTT GCC GCG AGC CG-3′, reverse 5′-TAA CCT GGT TCA TCA TCG CTA ATC-3′; PanEntero: forward 5′-TCC TCC GGC CCC TGA-3′, reverse 5′-RAT TGT CAC CAT AAG CAG CCA-3′. Probes used were: CTGF: 5′FAM-CCT GGG AAA TGC TGC AAG GAG TGG-3′TAMRA; procollagen Iα1: 5′FAM-TTC TTG GCC ATG CGT CAG GAG GG-3′TAMRA; iNOS: 5′FAM-CGG GCA GCC TGT GAG ACC TTT GA-3′TAMRA; HPRT: 5′FAM-CGA CCC GCA GTC CCA GCG TC-3′TAMRA; PanEntero: 5′FAM-CGG AAC CGA CTA CTT TGG GTG WCC GT-3′TAMRA.
Western blot analysis
Proteins from frozen cardiac tissue were isolated as previously described [29]. 30 μg of the proteins per lane were electroblotted, blocked for 1 h in non-fat dry milk and incubated with the following primary antibodies: polyclonal goat anti-CTGF antibody, polyclonal goat anti-GAPDH antibody, HRP-coupled (Santa Cruz Biotechnology, Santa Cruz, USA), polyclonal rabbit anti-phospho-p44/42 MAPK (Thr202/Tyr204) and rabbit anti-p44/42 MAPK antibodies (Cell Signaling, Danvers, USA). Secondary antibodies used were: polyclonal donkey anti-goat antibody, HRP-coupled (Santa Cruz Biotechnology, Santa Cruz, USA), polyclonal goat anti-rabbit antibody, HRP-coupled (Dianova, Hamburg, Germany). Visualization was done using the luminal electrochemiluminescence system according to standard procedures. Densitometric analysis was performed using Aida Image Analyzer (Raytest, Straubenhardt, Germany). Expression of CTGF was normalized to glyceraldehydes-3-phosphate dehydrogenase (GAPDH). The amount of activated p44/42 MAPK (ERK) was determined as ratio of phospho-p44/42 and p44/42.
Statistics
Data are provided as mean ± SEM. n represents the number of independent experiments or the number of animals per trial group. All data were tested for significance with Student’s t-test with Welch’s correction when indicated. P values ≤ 0.05 are considered as significant (*), ≤0.01 as very significant (**) and ≤0.001 as highly significant (***).
Results
Effects of iloprost on myocardial inflammation, expression of procollagen Iα1 and fibrosis
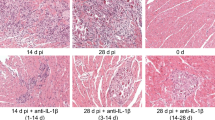
Hearts of infected ABY/SnJ mice as well as of infected animals treated from 1 day prior to infection until 12 days pi with iloprost were evaluated histologically 0, 4, 8, 12 and 28 days post-CVB3 infection. As shown in Fig. 1a–d and g, we noted an increased myocardial injury comprising myocyte necrosis, inflammation and fibrosis in iloprost-treated compared to untreated CVB3-infected animals in the course of infection with significant differences at days 12 and 28 pi (Fig. 1c, d, g). The aggravated myocardial damage was found to result in an increased fibrosis in iloprost-treated mice as shown by picrosirius red staining at day 28 pi (Fig. 1e, f). Consistently, expression of procollagen Iα1 mRNA was found to be significantly enhanced in the treated animals compared to untreated mice at day 12 pi (Fig. 1h). In animals which were treated with iloprost only after the acute phase of infection (days 12–22 pi), no significant differences with regard to the extent of myocardial lesions and procollagen Iα1 mRNA levels were observed at day 28 pi in mice compared to untreated CVB3-infected mice (Fig. 1g, h).

Myocardial injury, inflammation, procollagen mRNA expression, fibrosis, infiltration of CD3+ T lymphocytes and expression of IP-10 in ABY hearts following CVB3 infection. a, b Increased (8 days pi) and c, d significantly increased (day 28 pi) myocardial damage is found in iloprost-treated mice compared to untreated animals. e, f Compared to the untreated animals, iloprost-treated mice show increased fibrosis at day 28 pi. g Significantly enhanced myocardial damage is found in animals treated with iloprost during days 1–12 pi in contrast to animals treated from day 12 to 22 pi. h Expression of procollagen Iα1 mRNA detected by RT-PCR. The peak expression was found 12 days pi in the iloprost-treated animals. i, j Cardiac infiltration by CD3+ T lymphocytes and expression of IP-10 in monocytes (k, l) is not downregulated in iloprost-treated CVB3-infected mice (a–d hematoxylin/eosin staining, e, f picrosirius red staining, i–k immunohistochemistry ×400, d days, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001)
As iloprost was described to interfere with the immune response by directly inhibiting the lymphocytic infiltration in a murine model of bleomycin-induced pulmonary inflammation [57] we compared the infiltration of CD3+ T lymphocytes into hearts of treated and untreated CVB3-infected mice. As exemplarily shown in Fig. 1i and j, no suppression of invading T cells was noted in the infected hearts upon iloprost treatment. In addition, we stained IP-10 in the myocardium as this Th1-related chemokine was found to be downregulated in iloprost-treated monocytes [27]. Also, no reduction of IP-10 expression was observed in cardiac monocytes of iloprost-treated mice compared to untreated CVB3-infected animals (Fig. 1k, l).
Effect of iloprost on CTGF expression in vivo
In order to investigate whether iloprost can suppress the CTGF expression as was implicated by the findings of Stratton et al. [46, 47], we performed in situ hybridization experiments of hearts from treated and untreated CVB3-infected mice in the course of chronic myocarditis (Fig. 2). At days 4 and 8 pi CTGF expression was comparably decreased in cardiac interstitial cells of iloprost-treated animals, but the downregulation was not significant (Fig. 2c, d, g). However unexpectedly, the amount of CTGF-positive interstitial cells was found to be significantly enhanced in iloprost-treated animals at days 12 and 28 pi (Fig. 2e–g). Western blot analysis confirmed an increased cardiac expression of CTGF protein at day 12 pi in treated mice (data not shown). In contrast, animals treated with iloprost from day 12 to 22 pi showed a comparable expression of CTGF mRNA as the untreated mice (Fig. 2g).

Localization of CTGF mRNA as detected by in situ hybridization in CVB3 myocarditis. a, b Non-infected animals reveal very low numbers of CTGF-positive cells. c, d 4 days pi iloprost-treated animals show a slightly decreased number of CTGF mRNA-expressing cells compared to non-treated mice. e, f Comparably increased amounts of CTGF mRNA-positive cells are found in iloprost-treated animals 28 days pi. g Quantitative assessment of CTGF mRNA confirms a higher and prolonged expression of CTGF mRNA in the myocardium after application of iloprost during days 1–12 pi compared to treatment during days 12–22 pi (a–f ×400, d days pi, not inf not infected, *P ≤ 0.05, **P ≤ 0.01)
Activation of p44/42 MAPK by iloprost in the course of infection
In order to evaluate whether iloprost modifies the ERK/p44/42 MAPK signaling pathway, we examined the degree of activation of p44/42 MAPK in the hearts by immunohistochemistry (Fig. 3a–h) and immunofluorescence (Fig. 3i–n) visualizing the phosphorylated form of p44/42 MAPK. As shown in Fig. 3a and b, during the acute phase (8 days pi) of myocarditis, a suppression of p44/42 MAPK was observed in hearts of iloprost-treated mice. These data were confirmed by Western blot analysis of hearts obtained at days 4 and 8 pi supporting the notion that iloprost is generally capable to suppress p44/42 MAPK activation (data not shown). However, in the course of myocarditis (Fig. 3c–f), there was a strong focal enhancement in p44/42 MAPK activation within affected myocytes (Fig. 3i–k) and fibroblasts (Fig. 3l–n) of iloprost-treated animals at days 12 and 28 pi compared to their untreated littermates. In contrast to our findings in hearts of infected mice, activation of p44/42 MAPK is not detectable in cardiac cells of non-infected mice (Fig. 3g). No specific signals are observed in immunohistochemistry when tissue sections (a heart 28 days pi is shown) are stained without applying the first antibody (Fig. 3h).

Detection of phosphorylated ERK (p44/42 MAPK) by immunohistochemistry and immunofluorescence in CVB3 myocarditis. a, b Whereas at 8 days pi, a minor suppression of ERK is noted in iloprost-treated mice, an enhanced activation of ERK is noted after application of iloprost 12 and 28 days pi compared to the untreated animals in the inflammatory foci (c–f). g ERK is not activated in the myocytes of non-infected mice. h Negative control immunohistochemistry of the heart shown in f. Double labeling of ERK (green) and desmin (red) (i–k) or S100A4 (red) (l–n) reveals the expression of ERK in cardiomyocytes (k) and fibroblasts (n) 12 days pi (a–h ×400, d days pi, not inf not infected, i–n bar = 20 um)
Influence of iloprost on viral replication in the course of infection
To assess the influence of the iloprost treatment on replication rates of CVB3, we performed in situ hybridization targeting CVB3 genomic RNA. Whereas the amount of CVB3-infected cells was comparable in treated and untreated animals at day 4 pi (Fig. 4a, b, i), we detected a significant increase in cardiac viral replication from day 8 pi until the chronic phase of myocarditis (day 28 pi) in animals which received iloprost (Fig. 4c–f, i). In addition, viral load was found to be enhanced in other organs (pancreas, spleen) of iloprost-treated animals compared to non-treated mice (data not shown). The in situ hybridization results were confirmed by RT-PCR quantifying CVB3 transcripts (Fig. 4j). On the other hand, the group of animals that had been treated with iloprost only from day 12 to 22 pi showed a similar viral load as the untreated animals 28 days pi (Fig. 4h–j). Figure 4g illustrates that tissue sections of non-infected mouse hearts which were hybridized with CVB3-specific probes do not show specific silver grains, confirming the high specificity of the in situ hybridization method.

Detection of CVB3 RNA by in situ hybridization in the myocardium of ABY/SnJ mice. a, b reveals a similar affection 4 days pi. c, d At 8 days pi, iloprost-treated animals show an increased replication of viral RNA. e, f Almost no detection in untreated animals, but still foci of viral RNA in the iloprost-treated animals are seen 28 days pi. g Non-infected animals. h No detection of viral RNA 28 days pi in animals treated with iloprost from day 12 to 22 pi. i Assessment of myocardial affection of CVB3 genomic RNA by an in situ hybridization score. Increase in viral RNA replication in the iloprost-treated animals from day 8 to 28 pi. j Detection of CVB3 RNA by RT-PCR (a–h ×400, d days pi, not inf. not infected, ilo 12–22d iloprost treatment from day 12 to 22 pi, *P ≤ 0.05)
Influence of iloprost on activation of p44/42 MAPK in primary cardiac mouse fibroblasts
To gain further insight into the effects exerted by iloprost on the molecular mechanisms underlying the development of fibrosis, we isolated cardiac fibroblasts from hearts of ABY/SnJ mice. In order to compare the levels of p44/42 MAPK activation under the influence of iloprost in cultured fibroblasts, Western blot analyses were performed. Upon infection, a highly significant increase in p44/42 MAPK was noted in fibroblasts compared to uninfected cells with or without iloprost. There was no suppression but a moderate (however, not significant) increase of the p44/42 MAPK in CVB3-infected cells when they were treated with different concentrations of iloprost compared to untreated infected cells (Fig. 5).

Analysis of ERK (p44/42 MAPK) activation by Western blot analysis illustrates a significant upregulation in CVB3-infected cells compared to non-infected cells. However, no suppression of ERK is found in CVB3-infected iloprost-treated cells compared to infected, untreated cells (h hours pi, mean ± SD, **P ≤ 0.01)
Expression of inducible-type nitric oxide (iNOS) mRNA in the course of myocarditis
We have previously shown that iNOS expression in cardiac macrophages exerts deleterious effects in chronic CVB3 myocarditis in susceptible ABY/SnJ mice [48]. In order to evaluate whether iloprost may influence iNOS expression in the course of myocarditis, hearts of untreated and iloprost-treated animals were analyzed by radioactive in situ hybridization for the visualization of iNOS mRNA (Fig. 6). There was no relevant detection of iNOS mRNA-positive macrophages in non-infected mouse hearts (Fig. 6a, b) and in hearts 4 days pi (Fig. 6g). During acute infection, we observed a slightly higher increase of the iNOS mRNA-positive cardiac macrophages upon treatment with iloprost than in non-treated animals as demonstrated in Fig. 6c, d and g (8 days pi). iNOS mRNA expressing macrophages were found to be significantly enhanced at 12 days pi in iloprost-treated mice (Fig. 6g). Even in the late phase of infection (day 28 pi), the hearts of treated mice revealed numerous iNOS mRNA expressing macrophages compared to those of non-treated animals as demonstrated in Fig. 6e–g. Interestingly, as demonstrated before for CTGF, in animals which had been treated from day 12 to 22 pi, no increase in the amount of iNOS mRNA expressing cells was noted compared to their untreated littermates (Fig. 6g).

Expression of iNOS mRNA detected by in situ hybridization in CVB3-infected hearts. a, b Not infected animals. c, d A slight increase of cardiac iNOS mRNA-expressing cells is noted in iloprost-treated mice 8 days pi but a significant increase of iNOS mRNA-positive cells is observed at later stages of infection compared to untreated mice (e, f day 28 pi). g Quantification of iNOS mRNA expression by in situ hybridization. Prolonged and enhanced expression of iNOS mRNA in hearts is noted after application of iloprost during days 1–12 pi but not in animals treated during days 12–22 pi (a–f ×400, d days pi, not inf. not infected, *P ≤ 0.05, **P ≤ 0.01)
Expression of iNOS mRNA and protein in murine bone marrow macrophages
In order to investigate in more detail our findings on iloprost-mediated increase in cardiac iNOS, we generated BMM from ABY/SnJ mice which were stimulated with IFN-γ, infected with CVB3 and treated with iloprost. iNOS mRNA was detected by radioactive in situ hybridization (Fig. 7a–g) and iNOS protein by immunofluorescence microscopy (Fig. 7h–p). Infection of the BMM with CVB3 resulted in a visible but minimal increase in iNOS mRNA and protein expression compared to non-infected cells (Fig. 7a, c, h, i). BMM showed a stronger increase in iNOS mRNA and protein expression when additionally stimulated with IFN-γ (Fig. 7e, j). As shown in Fig. 7d and f, iloprost potentiated the expression of iNOS mRNA in CVB3-infected BMM by a factor of 5–7 [Fig. 7g, at 0.139 μM (50 ng/ml) > 2.774 μM (1,000 ng/ml)]. The observations at the mRNA level were substantiated by immunofluorescence data, illustrating that iNOS protein is significantly higher expressed in infected, IFN-γ-stimulated BMM which were treated with 0.139 μM (50 ng/ml) or 2.774 μM (1,000 ng/ml) concentrations of iloprost compared to infected non-treated cells (Fig. 7l, m, o, p).

Expression of iNOS mRNA detected by in situ hybridization in bone marrow-generated macrophages (BMM) upon CVB3 infection and iloprost treatment (a–f) and g by quantitative RT-PCR. h–p iNOS protein detected by immunofluorescence in BMM. a, b not infected animals. c–g Application of iloprost (0.139 > 2.774 μM) potentiates the expression of iNOS mRNA in BMM after CVB3 infection and stimulation with IFN-γ. h–p Increased production of iNOS protein in CVB3-infected and IFN-γ-stimulated BMMs after application of iloprost (a–f and h–p, ×400, not inf. not infected, IFNg interferon gamma, ilo 2.774 μM (1,000 ng/ml), Ilo 0.139 μM (50 ng/ml), **P ≤ 0.01)
Discussion
In fibrotic diseases like scleroderma and pulmonary hypertension, the antifibrotic prostacyclin (PGI2) analog iloprost was found to suppress the expression of CTGF, a potent mediator of fibrosis, by inhibiting the p44/42 MAPK (ERK) pathway in a PKA-dependent manner [46, 47]. It was suggested that, mainly by this mechanism, the development of fibrosis can be reduced in these patients. However, our results clearly demonstrate that this approach does not work in the setting of coxsackievirus B3-induced myocarditis, although here the development of fibrosis has been proven to be dependent on CTGF as well [29]. In contrast, CVB3-infected ABY/SnJ mice that have been treated with iloprost show increased myocardial damage with aggravated fibrosis and elevated levels of ERK activation and CTGF expression after the peak of virus replication, compared to their untreated littermates. It is important to note that ERK signaling is not only relevant in myocarditis but is also involved in many other cardiopathologic processes like apoptosis, hypertrophy and ischemia/reperfusion injury [4, 36].
In order to gain insight into the molecular mechanisms which determine the deleterious effects of iloprost in viral myocarditis, we performed in vitro experiments using CVB3-infected murine cardiac fibroblasts. Interestingly, in our study, in the presence of enteroviruses, iloprost was not found to reduce the activation of p44/42 MAPK in cardiac fibroblasts. As the difference of the experimental scenario compared to that of Stratton et al. are the direct virus-associated processes, we hypothesized that virus replication may counteract the described inhibition of p44/42 MAPK by iloprost. In fact, CVB3 replication is known to be dependent on the activation of the p44/42 MAPK in the host cell and, vice versa, also promotes the activation of the MAPK in the sense of a self-amplification cycle [17, 34, 39]. The activation of p44/42 MAPK by CVB3 is dependent on the small G-protein p21ras (Ras), which is known to stimulate the MEK/ERK signaling cascade by recruiting the serine/threonine kinase C-Raf (Raf-1) to the plasma membrane [26], allowing the enzyme to phosphorylate further proteins, among which are the members of MEK/ERK cascade [36]. Previously we have shown that in the course of CVB3 infection, p21ras GTPase-activating protein (RasGAP) is inactivated by cleavage by the viral proteinase 3CDpro [17], resulting in the accumulation of Ras and further activation of ERK. Obviously, the inhibitory effects of iloprost on MEK/ERK signaling induced by elevating the intracellular levels of cAMP and consecutive Raf-1 inhibition by phosphorylation on its serine-259 site [7–9] are less effective than the opposite mechanisms induced by the virus. Another possible explanation how the viral replication counteracts the inhibitory effects of iloprost is the activation of Raf-1 by Src-kinases, a family of tyrosine kinases [35]. Src-kinases have been found to be activated in the course of CVB3 infection in HeLa and Vero cells 4–6 h pi [16]. From these results it can be concluded that the activating mechanisms induced by the interaction of virus with Ras/MEK/ERK signaling and consecutive CTGF expression are stronger than the inhibitory effects exerted by iloprost.
A major finding in our study is the fact that iloprost-treated mice reveal increased myocardial damage at later stages of myocarditis compared to non-treated mice. One known determinant influencing the extent of myocardial injury in chronic CVB3-myocarditis is nitric oxide (NO) which is produced from l-arginine by inducible nitric oxide synthase (iNOS, NOS-2) by invading macrophages [48]. In context with ischemic heart disease, NO is known as regulator of endothelial function, and deficiency of NO is associated with endothelial dysfunction, which may finally lead to atherosclerosis and cardiac ischemia [55]. However, the role of NO in cardiac CVB3 infection is rather ambivalent. On the one hand, in acute infection in vitro, NO has been shown to inhibit the CVB3 proteinases 2A and 3C, thereby reducing the viral replication and preventing the cleavage of many cellular proteins that ensure proper cellular functions [54]. Also, CVB3-infected iNOS−/−-mice were found to develop a more severe myocarditis with increased viral replication, indicating that NO plays a pivotal role in the host immune response to CVB3 infection [53]. On the other hand, there is a multitude of detrimental effects induced by NO and reactive nitrogen intermediates (RNIs) such as peroxynitrite that have been discovered during the last decades (reviewed in [40]). In neonatal rat cardiomyocytes, NO mediates apoptosis in a cGMP-dependent way [44], and peroxynitrite (ONOO−), a strong oxidant evolving from the reaction of NO with superoxide anion (O2 −), triggers cardiomyocyte apoptosis in vitro and in a myocardial ischemia–reperfusion in vivo model [32]. Recently, ONOO− has been identified as a suppressor of cardiac contractile function by nitration of myofibrillar proteins in an in vitro motility assay [45]. Accordingly, neutralizing ONOO− in endotoxin-stimulated rats improved cardiac contractility [28]. Experiments in ischemic pig hearts suggest that inhibition of excessive iNOS activity may prove to be beneficial in maintaining the contractile function during sustained moderate ischemia. [15]. Regarding CVB3 myocarditis, susceptible SWR mice which were infected with an attenuated CVB3 revealed a significantly lower expression of iNOS and fewer and smaller inflammatory lesions of the myocardium [3]. As previously reported, susceptible ABY/SnJ mice, but not resistant C57BL/6 mice, show a delayed and prolonged iNOS expression, causing ongoing immunopathology in the heart [48]. Intriguingly, in our study, when iloprost-treated mice were investigated, a significant enhancement of iNOS mRNA expression was detected in interstitial macrophages at later stages of myocarditis compared to untreated CVB3-infected animals, suggesting that RNI may contribute to an enhanced and prolonged cardiac injury in treated animals. To gain further insight in these processes, we isolated BMM of ABY/SnJ mice and monitored the expression of iNOS RNA and protein after treatment with different concentrations of iloprost. According to our findings in vivo, the expression of iNOS RNA and protein was potentiated in IFN-γ-stimulated CVB3-infected BMM after treatment with iloprost.
Regarding the underlying mechanisms of myocardial damage induced by RNIs, it was described that, for example, ONOO− may act as a mediator of diverse cell signal transduction pathways [33]. For example, ONOO− was found to activate the MEK/ERK signaling cascade in H9C2 cardiomyocytes by activating Raf-1, independent of the growth factor receptor stimulation [41]. The same effect has been reported for lung myofibroblasts in rats [56] and rat-1 fibroblasts, a non-transformed rat embryo cell line [2]. There is evidence that these mechanisms may also be relevant for the detrimental effects of iloprost in our model system of inflammatory heart disease. An enhanced and ongoing ONOO−-mediated ERK activation can contribute to the increased viral load we detected at the later stages of infection, considering the fact that replication of CVB3 is dependent on the activation of ERK signaling [17, 34, 39] as mentioned above.
Altogether these results provide evidence that iloprost is counteracting its primary inhibitory effects on p44/42 MAPK signaling by increasing iNOS expression of infiltrating macrophages. In turn, the excessive production of NO and RNI, especially ONOO−, enhances p44/42 MAPK activation which promotes viral replication and expression of CTGF, finally leading to an increased fibrosis in iloprost-treated mice. It is worth noting that iloprost exerts adversive effects only in the time period of virus replication (up to 12 days pi) but not if iloprost is applied from day 12 to 28 pi when infectious virus is not present any more, substantiating the interference of molecular mechanisms induced by virus replication and those of iloprost.
Finally, it remains the question of the relevance of these findings in patients with heart disease. Tissue samples taken from patients with DCM revealed that 67 % of the hearts expressed iNOS in all four chambers [14]. Moreover, in heart tissue specimen from patients who died from viral myocarditis, a significantly increased amount of nitrotyrosine residues (a marker for the presence of ONOO−) was detectable in the myocardium and endocardium [25], suggesting an etiopathogenic role for RNIs in inflammation-associated myocardial dysfunction. Most importantly and completely congruent to our results, human peripheral blood monocytes induced with LPS/IFN-γ show a potentiated (9.4-fold) expression of iNOS and production of NO when treated with cicaprost, another selective prostacyclin receptor agonist [42].
The development of fibrosis and the degree of cardiac remodeling are associated with the extent and modulatory capacity of the inflammatory response in viral myocarditis [48, 51] and other cardiopathologic settings like myocardial infarction [49]. Most recently, it was shown that in human monocytes which have been stimulated with LPS, iloprost impairs the Th1/Th2-related chemokine expression via epigenetic regulation. In fact, iloprost was found to suppress IP-10, thus probably increasing the Th2 recruitment [27]. In contrast, in a model of bleomycin-induced pulmonary inflammation IP-10 was found to be upregulated by iloprost [57]. In our in vivo system, we did not observe a downregulation of IP-10 or of CD3+ T cells in the hearts of our treated animals, suggesting that iloprost does not interfere with the known Th1-mediated inflammation in susceptible CVB3-infected mice.
Taken together, our data stress the complexity of interactions between the immune system and viral replication in the pathogenesis of chronic myocarditis in view of potential therapeutic drug interventions. Further studies are needed to gain deeper insight into the impact of iloprost or other prostacyclin receptor agonists on inflammatory processes of heart disease, especially in virus-infected patients. However, so far our results obtained in the in vivo model of CVB3 myocarditis implicate a critical reflection of the clinical use of iloprost in patients possibly suffering from an enterovirus infection.
References
Badesch DB, McLaughlin VV, Delcroix M, Vizza CD, Olschewski H, Sitbon O, Barst RJ (2004) Prostanoid therapy for pulmonary arterial hypertension. J Am Coll Cardiol 43(12 Suppl S):56S–61S. doi:10.1016/j.jacc.2004.02.036
Bapat S, Verkleij A, Post JA (2001) Peroxynitrite activates mitogen-activated protein kinase (MAPK) via a MEK-independent pathway: a role for protein kinase C. FEBS Lett 499:21–26. doi:10.1016/S0014-5793(01)02511-X
Bevan AL, Zhang H, Li Y, Archard LC (2001) Nitric oxide and coxsackievirus B3 myocarditis: differential expression of inducible nitric oxide synthase in mouse heart after infection with virulent or attenuated virus. J Med Virol 64:175–182. doi:10.1002/jmv.1033
Cao W, Xie YH, Li XQ, Zhang XK, Chen YT, Kang R, Chen X, Miao S, Wang SW (2011) Burn-induced apoptosis of cardiomyocytes is survivin dependent and regulated by PI3K/Akt, p38 MAPK and ERK pathways. Basic Res Cardiol 106:1207–1220. doi:10.1007/s00395-011-0199-3
Chen MM, Lam A, Abraham JA, Schreiner GF, Joly AH (2000) CTGF expression is induced by TGF-beta in cardiac fibroblasts and cardiac myocytes: a potential role in heart fibrosis. J Mol Cell Cardiol 32:1805–1819. doi:10.1006/jmcc.2000.1215
Daniels A, van Bilsen M, Goldschmeding R, van der Vusse GJ, van Nieuwenhoven FA (2009) Connective tissue growth factor and cardiac fibrosis. Acta Physiol (Oxf) 195:321–338. doi:10.1111/j.1748-1716.2008.01936.x
Dhillon AS, Meikle S, Yazici Z, Eulitz M, Kolch W (2002) Regulation of Raf-1 activation and signalling by dephosphorylation. EMBO J 21:64–71. doi:10.1093/emboj/21.1.64
Dhillon AS, Pollock C, Steen H, Shaw PE, Mischak H, Kolch W (2002) Cyclic AMP-dependent kinase regulates Raf-1 kinase mainly by phosphorylation of serine 259. Mol Cell Biol 22:3237–3246. doi:10.1128/MCB.22.10.3237-3246.2002
Dumaz N, Marais R (2005) Integrating signals between cAMP and the RAS/RAF/MEK/ERK signalling pathways. Based on the anniversary prize of the Gesellschaft für Biochemie und Molekularbiologie Lecture delivered on 5 July 2003 at the Special FEBS Meeting in Brussels. FEBS J 272:3491–3504. doi:10.1111/j.1742-4658.2005.04763.x
Duncan MR, Frazier KS, Abramson S, Williams S, Klapper H, Huang X, Grotendorst GR (1999) Connective tissue growth factor mediates transforming growth factor beta-induced collagen synthesis: down-regulation by cAMP. FASEB J 13:1774–1786
Flesch IEA, Kaufmann SHE (1991) Mechanisms involved in mycobacterial growth inhibition by gamma-interferon-activated bone-marrow-macrophages: role of reactive nitrogen intermediates. Infect Immun 159:3213–3218
Frazier K, Williams S, Kothapalli D, Klapper H, Grotendorst GR (1996) Stimulation of fibroblast cell growth, matrix production, and granulation tissue formation by connective tissue growth factor. J Invest Dermatol 107:404–411
Grotendorst GR, Duncan MR (2005) Individual domains of connective tissue growth factor regulate fibroblast proliferation and myofibroblast differentiation. FASEB J 19:729–738. doi:10.1096/fj.04-3217com
Haywood GA, Tsao PS, von der Leyen HE, Mann MJ, Keeling PJ, Trindade PT, Lewis NP, Byrne CD, Rickenbacher PR, Bishopric NH, Cooke JP, McKenna WJ, Fowler MB (1996) Expression of inducible nitric oxide synthase in human heart failure. Circulation 93:1087–1094. doi:10.1161/01.CIR.93.6.1087
Heinzel FR, Gres P, Boengler K, Duschin A, Konietzka I, Rassaf T, Snedovskaya J, Meyer S, Skyschally A, Kelm M, Heusch G, Schulz R (2008) Inducible nitric oxide synthase expression and cardiomyocyte dysfunction during sustained moderate ischemia in pigs. Circ Res 103:1120–1127. doi:10.1161/CIRCRESAHA.108.186015
Huber M, Selinka HC, Kandolf R (1997) Tyrosine phosphorylation events during coxsackievirus B3 replication. J Virol 71:595–600
Huber M, Watson KA, Selinka HC, Carthy CM, Klingel K, McManus BM, Kandolf R (1999) Cleavage of RasGAP and phosphorylation of mitogen-activated protein kinase in the course of coxsackievirus B3 replication. J Virol 73:3587–3594
Ihm SH, Chang K, Kim HY, Baek SH, Youn HJ, Seung KB, Kim JH (2010) Peroxisome proliferator-activated receptor-gamma activation attenuates cardiac fibrosis in type 2 diabetic rats: the effect of rosiglitazone on myocardial expression of receptor for advanced glycation end products and of connective tissue growth factor. Basic Res Cardiol 105:399–407. doi:10.1007/s00395-009-0071-x
Javelaud D, Mauviel A (2005) Crosstalk mechanisms between the mitogen-activated protein kinase pathways and Smad signaling downstream of TGF-β: implications for carcinogenesis. Oncogene 24:5742–5750. doi:10.1038/sj.onc.1208928
Kandolf R, Hofschneider PH (1985) Molecular cloning of the genome of a cardiotropic Coxsackie B3 virus: full-length reverse-transcribed recombinant cDNA generates infectious virus in mammalian cells. Proc Natl Acad Sci USA 82:4818–4822
Kim KS, Hufnagel G, Chapman N, Tracy S (2001) The group B coxsackieviruses and myocarditis. Rev Med Virol 11:355–368. doi:10.1002/rmv.326
Kiriyama M, Ushikubi F, Kobayashi T, Hirata M, Sugimoto Y, Narumiya S (1997) Ligand binding specificities of the eight types and subtypes of the mouse prostanoid receptors expressed in Chinese hamster ovary cells. Br J Pharmacol 122:217–224. doi:10.1038/sj.bjp.0701367
Klingel K, Hohenadl C, Canu A, Albrecht M, Seemann M, Mall G, Kandolf R (1992) Ongoing enterovirus-induced myocarditis is associated with persistent heart muscle infection: quantitative analysis of virus replication, tissue damage, and inflammation. Proc Natl Acad Sci USA 89:314–318
Klingel K, Sauter M, Bock CT, Szalay G, Schnorr JJ, Kandolf R (2004) Molecular pathology of inflammatory cardiomyopathy. Med Microbiol Immunol 193:101–107. doi:10.1007/s00430-003-0190-1
Kooy NW, Lewis SJ, Royall J, Ye YZ, Kelly D, Beckman JS (1997) Extensive tyrosine nitration in human myocardial inflammation: evidence for the presence of peroxynitrite. Crit Care Med 25:812–819
Kubicek M, Pacher M, Abraham D, Podar K, Eulitz M, Baccarini M (2002) Dephosphorylation of Ser-259 regulates Raf-1 membrane association. J Biol Chem 277:7913–7919. doi:10.1074/jbc.M108733200
Kuo CH, Ko YC, Yang SN, Chu YT, Wang WL, Huang SK, Chen HN, Wei WJ, Jong YJ, Hung CH (2011) Effects of PGI2 analogues on Th1- and Th2-related chemokines in monocytes via epigenetic regulation. J Mol Med 89:29–41. doi:10.1007/s00109-010-0694-2
Lancel S, Tissier S, Mordon S, Marechal X, Depontieu F, Scherpereel A, Chopin C, Neviere R (2004) Peroxynitrite decomposition catalysts prevent myocardial dysfunction and inflammation in endotoxemic rats. Am Coll Cardiol 42:2348–2358. doi:10.1016/j.jacc.2004.01.047
Lang C, Sauter M, Szalay G, Racchi G, Grassi G, Rainaldi G, Mercatanti A, Lang F, Kandolf R, Klingel K (2008) Connective tissue growth factor: a crucial cytokine-mediating cardiac fibrosis in ongoing enterovirus myocarditis. J Mol Med 86:49–60. doi:10.1007/s00109-007-0249-3
Leask A, Holmes A, Black CM, Abraham DJ (2003) Connective tissue growth factor gene regulation. Requirements for its induction by transforming growth factor-β2 in fibroblasts. J Biol Chem 278:13008–13015. doi:10.1074/jbc.M210366200
Leivonen SK, Häkkinen L, Liu D, Kähäri VM (2005) Smad3 and extracellular signal-regulated kinase 1/2 coordinately mediate transforming growth factor-β-induced expression of connective tissue growth factor in human fibroblasts. J Invest Dermatol 124:1162–1169. doi:10.1111/j.0022-202X.2005.23750.x
Levrand S, Vannay-Bouchiche C, Pesse B, Pacher P, Feihl F, Waeber B, Liaudet L (2006) Peroxynitrite is a major trigger of cardiomyocyte apoptosis in vitro and in vivo. Free Radic Biol Med 41:886–895. doi:10.1016/j.freeradbiomed.2006.04.034
Liaudet L, Vassalli G, Pacher P (2009) Role of peroxynitrite in the redox regulation of cell signal transduction pathways. Front Biosci 14:4809–4814. doi:10.2741/3569
Luo H, Yanagawa B, Zhang J, Luo Z, Zhang M, Esfandiarei M, Carthy C, Wilson JE, Yang D, McManus BM (2002) Coxsackievirus B3 replication is reduced by inhibition of the extracellular signal-regulated kinase (ERK) signaling pathway. J Virol 76:3365–3373. doi:10.1128/JVI.76.7.3365-3373.2002
Mason CS, Springer CJ, Cooper RG, Superti-Furga G, Marshall CJ, Marais R (1999) Serine and tyrosine phosphorylations cooperate in Raf-1, but not B-Raf activation. EMBO J 18:2137–2148. doi:10.1093/emboj/18.8.2137
Michel MC, Li Y, Heusch G (2001) Mitogen-activated protein kinases in the heart. Naunyn Schmiedebergs Arch Pharmacol 363:245–266. doi:10.1007/s002100000363
Miller CL, Cai Y, Oikawa M, Thomas T, Dostmann WR, Zaccolo M, Fujiwara K, Yan C (2011) Cyclic nucleotide phosphodiesterase 1A: a key regulator of cardiac fibroblast activation and extracellular matrix remodeling in the heart. Basic Res Cardiol 106:1023–1039. doi:10.1007/s00395-011-0228-2
Nijhuis M, van Maarseveen N, Schuurman R, Verkuijlen S, de Vos M, Hendriksen K, van Loon AM (2002) Rapid and sensitive routine detection of all members of the genus enterovirus in different clinical specimens by real-time PCR. J Clin Microbiol 40:3666–3670
Opavsky MA, Martino T, Rabinovitch M, Penninger J, Richardson C, Petric M, Trinidad C, Butcher L, Chan J, Liu PP (2002) Enhanced ERK 1/2 activation in mice susceptible to coxsackievirus-induced myocarditis. J Clin Invest 109:1561–1569. doi:10.1172/JCI13971
Pacher P, Beckman JS, Liaudet L (2007) Nitric oxide and peroxynitrite in health and disease. Physiol Rev 87:315–424. doi:10.1152/physrev.00029.2006
Pesse B, Levrand S, Feihl F, Waeber B, Gavillet B, Pacher P, Liaudet L (2005) Peroxynitrite activates ERK via Raf-1 and MEK, independently from EGF receptor and p21Ras in H9C2 cardiomyocytes. J Mol Cell Cardiol 38:765–775. doi:10.1016/j.yjmcc.2005.02.020
Plum J, Huang C, Grabensee B, Schrör K, Meyer-Kirchrath J (2002) Prostacyclin enhances the expression of LPS/INF-γ-induced nitric oxide synthase in human monocytes. Nephron 91:391–398
Rutschow S, Li J, Schultheiss HP, Pauschinger M (2006) Myocardial proteases and matrix remodeling in inflammatory heart disease. Cardiovasc Res 69:646–656. doi:10.1016/j.cardiores.2005.12.009
Shimojo T, Hiroe M, Ishiyama S, Ito H, Nishikawa T, Marumo F (1999) Nitric oxide induces apoptotic death of cardiomyocytes via a cyclic-GMP-dependent pathway. Exp Cell Res 247:38–47. doi:10.1006/excr.1998.4310
Snook JH, Li J, Helmke BP, Guilford WH (2008) Peroxynitrite inhibits myofibrillar protein function in an in vitro assay of motility. Free Rad Biol Med 44:14–23. doi:10.1016/j.freeradbiomed.2007.09.004
Stratton R, Shiwen X, Martini G, Holmes A, Leask A, Haberberger T, Martin GR, Black CM, Abraham D (2001) Iloprost suppresses connective tissue growth factor production in fibroblasts and in the skin of scleroderma patients. J Clin Invest 108:241–250. doi:10.1172/JCI12020
Stratton R, Rajkumar V, Ponticos M, Nichols B, Shiwen X, Black CM, Abraham DJ, Leask A (2002) Prostacyclin derivatives prevent the fibrotic response to TGF-β by inhibiting the Ras/MEK/ERK pathway. FASEB J 16:1949–1951. doi:10.1096/fj.02-0204fje
Szalay G, Sauter M, Hald J, Weinzierl A, Kandolf R, Klingel K (2006) Sustained nitric oxide synthesis contributes to immunopathology in ongoing myocarditis attributable to interleukin-10 disorders. Am J Pathol 169:2085–2093. doi:10.2353/ajpath.2006.060350
Tang TT, Yuan J, Zhu ZF, Zhang WC, Xiao H, Xia N, Yan XX, Nie SF, Liu J, Zhou SF, Li JJ, Yao R, Liao MY, Tu X, Liao YH, Cheng X (2012) Regulatory T cells ameliorate cardiac remodeling after myocardial infarction. Basic Res Cardiol 107:232. doi:10.1007/s00395-011-0232-6
Wenzel S, Henning K, Habbig A, Forst S, Schreckenberg R, Heger J, Maxeiner H, Schlüter KD (2010) TGF-beta1 improves cardiac performance via up-regulation of laminin receptor 37/67 in adult ventricular cardiomyocytes. Basic Res Cardiol 105:621–629. doi:10.1007/s00395-010-0108-1
Xu Z, Desai M, Philip J, Sivsubramanian N, Bowles NE, Vallejo JG (2011) Conditional transgenic expression of TIR-domain-containing adaptor-inducing interferon-β (TRIF) in the adult mouse heart is protective in acute viral myocarditis. Basic Res Cardiol 106:1159–1171. doi:10.1007/s00395-011-0226-4
Zandman-Goddard G, Tweezer-Zaks N, Shoenfeld Y (2005) New therapeutic strategies for systemic sclerosis—a critical analysis of the literature. Clin Dev Immunol 12:165–173. doi:10.1080/17402520500233437
Zaragoza C, Ocampo C, Saura M, Leppo M, Wie XQ, Quick R, Moncada S, Liew FY, Lowenstein CJ (1998) The role of inducible nitric oxide synthase in the host response to coxsackievirus myocarditis. Proc Natl Acad Sci USA 95:2469–2474
Zell R, Markgraf R, Schmidtke M, Görlach M, Stelzner A, Henke A, Sigusch HH, Glück B (2003) Nitric oxide donors inhibit the coxsackievirus B3 proteinases 2A and 3C in vitro, virus production in cells, and signs of myocarditis in virus-infected mice. Med Microbiol Immunol 193:91–100. doi:10.1007/s00430-003-0198-6
Zhang C (2008) The role of inflammatory cytokines in endothelial dysfunction. Basic Res Cardiol 103:398–406. doi:10.1007/s00395-008-0733-0
Zhang P, Wang YZ, Kagan E, Bonner JC (2000) Peroxynitrite targets the epidermal growth factor receptor, Raf-1, and MEK independently to activate MAPK. J Biol Chem 275:22479–22486. doi:10.1074/jbc.M910425199
Zhu Y, Liu Y, Zhou W, Xiang R, Jiang L, Huang K, Xiao Y, Guo Z, Gao J (2010) A prostacyclin analogue, iloprost, protects from bleomycin-induced pulmonary fibrosis in mice. Respir Res 11:34. doi:10.1186/1465-9921-11-34
Acknowledgments
We thank Sandra Bundschuh for excellent technical assistance. This work was supported by the DFG (SFB-TR19 TP Z4 to KK), and the BMBF (01EZ0817 to KK and RK) and the Interfakultäres Zentrum für Klinische Forschung (IZKF), University Hospital Tuebingen, Germany (to SG and KK).
Conflict of interest
There is no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Gruhle, S., Sauter, M., Szalay, G. et al. The prostacyclin agonist iloprost aggravates fibrosis and enhances viral replication in enteroviral myocarditis by modulation of ERK signaling and increase of iNOS expression. Basic Res Cardiol 107, 287 (2012). https://doi.org/10.1007/s00395-012-0287-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00395-012-0287-z