
Abstract
Coxsackieviruses of group B (CVB) are well-known causes of acute and chronic myocarditis. Chronic myocarditis can evolve into dilated cardiomyopathy (DCM) characterized by fibrosis and cardiac remodeling. Interleukin-1β (IL-1β) plays a decisive role in the induction of the inflammatory response as a consequence of viral replication. In this study, we analyzed the effects of IL-1β neutralization on the transition of acute to chronic myocarditis in a mouse model of CVB3 myocarditis. Mice were treated with an anti-murine IL-1β antibody as a surrogate for Canakinumab at different time points post CVB3 infection. Treatment was performed in the early phase (day 1–14 pi, day 3–14 pi) or at a later stage of myocarditis (day 14–28 pi). Subsequently, the hearts were examined histologically, immunohistochemically and by molecular biology. A significant reduction of viral replication, cardiac damage and inflammation was found after administration of the antibody in the early phase and in the later phase of infection. Furthermore, less collagen I deposition and a considerable reduction of fibrosis were found in antibody-treated mice. Using microarray analysis, a significant upregulation of various extracellular matrix and fibrosis-associated molecules was found in CVB3-infected mice, including TGF-β, TIMP-1 and MMP12, as well as diverse matricellular proteins, whereas, these molecules were significantly downregulated in all IL-1β antibody-treated infected mice. Neutralization of IL-1β at different stages of enteroviral infection prevents the development of chronic viral myocarditis by reducing inflammation, interstitial fibrosis and adverse cardiac remodeling. These findings are relevant for the treatment of patients with acute and chronic myocarditis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Myocarditis is an inflammatory disease of the myocardium that can be caused by a large variety of triggers, including infectious agents, systemic immune-mediated diseases, drugs and toxins. Viral infections are considered to represent the most common cause of myocarditis in North America and Europe [4]. Acute cardiac inflammation can evolve into a chronic stage and finally dilated cardiomyopathy (DCM) with DCM being the third most common cause of heart failure and a frequent reason for heart transplantation [26].
The treatment of myocarditis is symptomatic and mainly aims to inhibit arrhythmias and heart failure. The TIMIC study provided evidence that immunosuppressive therapy using prednisone and azathioprine has a beneficial effect in virus-negative myocarditis [14]. The ESC position statement recommends immunosuppressive therapy for the treatment of proven autoimmune myocarditis like giant cell myocarditis and cardiac sarcoidosis [4]. In contrast, in the case of viral myocarditis, immunosuppression represents a double-edged sword as it possibly prevents viral clearance. Prednisolone was even shown to aggravate viral myocarditis in mice [39]. In case of herpesvirus infections, antiviral treatment with acyclovir, ganciclovir or valacyclovir may be considered; whereas, infections with enteroviruses might be treated with interferon-β [18, 19]. Yet, until today, no curative therapy has been approved for the treatment of viral myocarditis [4, 32].
IL-1 is an apical pro-inflammatory cytokine that exists in two isoforms, IL-α and IL-1β. It is released upon myocardial damage and induces the expression and activation of many other inflammatory mediators [10]. Several studies indicate that IL-1 plays an important role in the development of cardiac inflammation. It was shown that IL-1 expression is markedly upregulated in coxsackievirus B3 (CVB3)-induced myocarditis in mice and that the heart of CVB3-infected mice is infiltrated by inflammatory cells that secrete IL-1 [21, 40]. Additionally, in endomyocardial biopsies of patients with CVB3-induced myocarditis and idiopathic DCM, increased IL-1β mRNA levels were found [40, 41]. Treatment of mice infected with CVB3 or encephalomyocarditis virus with an IL-1 receptor antagonist (IL-1Ra) by infusion or in vivo electroporation led to reduced myocardial damage and decreased cellular infiltration of the myocardium [27, 28]. Likewise, local expression of human IL-1Ra in the heart of mice improved mortality and decreased myocardial inflammation in CVB3-induced myocarditis [22]. The IL-1 receptor antagonist Anakinra was recently shown to block the IL-1β-mediated decrease of contractility in Theiler’s murine encephalomyelitis virus-infected rat cardiac lymphatic muscle cells, suggesting a potential role in myocarditis [1]. Furthermore, several case reports illustrate a beneficial effect of IL-1 inhibition using Anakinra in treating life-threatening cases of myocarditis [5, 6, 31]. Canakinumab, a monoclonal IL-1β antibody, was recently tested successfully in the CANTOS trial for the anti-inflammatory therapy of atherosclerotic disease [33].
As these findings suggest that IL-1 inhibition is a promising treatment option for myocarditis, in the present study, we analyzed the effects of IL-1β neutralization on the progression of myocarditis in a mouse model of CVB3-induced myocarditis. Using an anti-murine IL-1β antibody as a surrogate for Canakinumab, IL-1β was blocked at different time points post CVB3 infection to evaluate how IL-1β neutralization influences the acute phase of myocarditis and whether the development of a chronic myocarditis can be circumvented. To our knowledge, this is the first study that specifically inhibits IL-1β. All previous studies concerning IL-1 in the context of myocarditis did not differentiate between IL-1α and IL-1β.
Methods
Mice, infection and antibody treatment
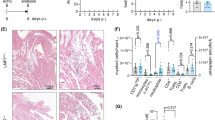
ABY/SnJ mice, originally purchased from The Jackson Laboratory (Bar Harbor, ME, USA), were bred and kept under specific pathogen-free conditions at the animal facility of the Department of Molecular Pathology, University Hospital Tübingen. Experiments were conducted according to the German animal protection law. For this study, a total of 36 mice were used (n = 6 per group). Virus infection of mice at day 0 was performed as described previously [17]. In addition, the mice received the IL-1β antibody intraperitoneally (ip) every third day at a dose of 150 µg per injection. Group one received the antibody from day 1 to 14 post infectionem (pi) and was killed on day 14 pi. Group two was given the antibody from day 3 to 14 pi and group three from day 14 to 28 pi. The mice of groups two and three were killed on day 28 pi. The mice of the respective control groups were infected with CVB3 but not treated with the antibody. In concurrence with the experimental groups, they were killed on day 0, day 14 and day 28 (Fig. 1a). The hearts of all mice were either fixed in 4% paraformaldehyde and embedded in paraffin for histology and immunohistochemistry or snap-frozen in liquid nitrogen and stored at − 80 °C for RNA isolation.

Experimental setup and comparison of myocardial damage and cellular infiltration in CVB3-infected untreated and IL-1β antibody-treated A.BY/SnJ mice. a Schema of the experimental setup. Thirty-six mice were used (n = 6 per group) and virus infection was performed at day 0. The mice received the IL-1β antibody intraperitoneally every third day at a dose of 150 µg per injection. Group one received the antibody from day 1 to 14 pi and was killed on day 14 pi. Group two was given the antibody from day 3 to 14 pi and group three from day 14 to 28 pi. The mice of group two and three were killed on day 28 pi. The mice of the respective control groups were infected with CVB3 (except group four) but not treated with the antibody. In concurrence with the treated animals they were killed on day 0, day 14 and day 28. b HE staining of representative heart tissue sections revealed higher numbers of inflammatory lesions 14 and 28 days pi in CVB3-infected mice compared to antibody-treated mice and non-infected control mice. c Correspondent Masson trichrome staining of representative heart tissue sections
Virus
CVB3 used in this study was derived from the infectious cDNA copy of the cardiotropic Nancy strain. Virus stocks were prepared as described in a preceding publication [17].
IL-1β antibody
The IL-1β antibody (01BSUR, Novartis Pharma AG, Basel, Switzerland) is a monoclonal antibody that recognizes murine IL-1β (IgG2a). Since Canakinumab does not react with murine IL-1β, 01BSUR was used as a surrogate antibody. The antibody was a generous gift from Hermann Gram (Novartis) [11, 29].
Histology
To assess myocardial injury, inflammation and fibrosis paraffin-embedded hearts were cut in 5-μm-thick tissue sections and stained with hematoxylin and eosin (HE), Masson’s trichrome or picrosirius red stain. To quantify myocardial damage comprising cardiac cell necrosis, inflammation, and scarring, we applied a myocarditis score from 0 to 4 as previously described (score 0, no inflammatory infiltrates 1, small foci of inflammatory cells between myocytes 2, larger foci > 100 inflammatory cells 3, ≤ 10% of cross section involved 4, 10–30% of a cross section involved) [38].
RT2 Profiler PCR Array
A RT2 Profiler PCR Array from Qiagen (Extracellular Matrix and Adhesion Molecules, PAMM-013Z) was used to analyze gene expression of genes related to the extracellular matrix and adhesion molecules in murine hearts of infected mice treated with and without the IL-1β antibody. Total cellular RNA was extracted from the murine hearts using the RNeasy Mini Kit (Qiagen, Cat. 74104). For cDNA synthesis and genomic DNA elimination, the RT2 First Strand Kit (Qiagen, Cat. 330401) was used. The cDNA templates were combined with the RT2 SYBR green ROX qPCR mix (25 μL/well), loaded into 96-well array plates coated with 84 predispensed gene-specific primer sets and processed on a 7300 Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) according to the manufacturer’s protocol. Data analysis was performed with Qiagen’s online web analysis tool. All data were normalized to an average of five housekeeping genes Gusb, B2m, Hsp90ab1, Gapdh and Actb.
In situ hybridization (ISH)
To detect CVB3 RNA and IL-6 mRNA, heart tissue sections were hybridized using specific probes for CVB3 resp. IL-6 (ACD, Newark, CA, USA) followed by the RNAscope 2.5 HD Detection Kit Brown (for CVB3) and RNAscope 2.5 HD Detection Kit Red (for IL-6) from ACD (Newark, CA, USA) according to the manufacturer’s protocol. Virus replication was measured in a score of 0–4, in which 4 is the maximum of virus replication in acutely infected mouse hearts (8 days pi, see [17]) and 1 reflects single positive cells.
Immunohistochemistry (IHC)
For immunohistochemistry, tissue sections were deparaffinized, subjected to heat-induced epitope retrieval (in 10 mM citrate buffer) and incubated for 1 h at 25 °C with rat-anti-Mac-3 (Becton Dickonson, Franklin Lakes, NJ, USA), rabbit-anti-CD3 (Thermo Fisher Scientific, Waltham, MA, USA), rabbit-anti-Erk1/2 (Cell Signaling Technology, Danvers, MA, USA), rabbit-anti-collagen Type I (abcam, Cambridge, GB), rabbit-anti-osteopontin (abcam, Cambridge, GB), rabbit-anti-periostin (abcam, Cambridge, GB) and rabbit-anti-tenascin C (abcam, Cambridge, GB). Controls using normal goat, rabbit or rat serum were run to exclude non-specific staining. Subsequently, the sections were processed with the rat-on-mouse HRP-Polymer (for Mac-3) resp. the rabbit-on-rodent-HRP-Polymer (all other antibodies, ZYTOMED Systems, Berlin, Germany). All tissue sections were counterstained with hematoxylin. IHC was graded in a scale 0–4, and macrophages and T cells were also assessed as number of positive cells per mm2 myocardium.
Quantitative RT-PCR (qRT-PCR)
For RNA isolation, hearts were lysed in Trifast (Peqlab, Erlangen, Germany) according to the manufacturer’s protocol. 200 ng of RNA was used to perform one step quantitative real-time reverse transcription PCR (TaqMan RNA-to-CT 1-Step Kit, Applied Biosystems, Foster City, CA, USA) at the appropriate annealing temperature for 40 cycles on a 7300 Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). Data analysis was performed as relative quantification in relation to the expression of the housekeeping gene hypoxanthine-guanine phosphoribosyltransferase (HPRT) as internal standard. Specific primers and probes were purchased from MWG Biotech (Ebersberg, Germany).
Primers were:
-
mHPRT: fwd: 5′ TTTGCCGCGAGCCG 3′;
-
rev: 5′ TAACCTGGTTCATCATCGCTAATC3′;
-
probe: 5′ FAM-CGACCCGCAGTCCCAGCGTC-TAM 3′;
-
mMMP12: fwd: 5′ TGTGGAGTGCCCGATGTACA 3′;
-
rev: 5′ AGTGAGGTACCGCTTCATCCAT 3′;
-
probe: 5′ FAM-CATCTTAGAGCAGTGCCCCAGAGGTCA-TAMRA 3′;
-
mTIMP-1: fwd: 5′ TCCTCTTGTTGCTATCACTGATAGCTT 3′;
-
rev: 5′ CGCTGGTATAAGGTGGTCTCGTT 3′;
-
probe: 5′ FAM-TCCTGCAACTCGGACCTGGTCATAAGG-TAMRA 3′.
Statistics
When statistical analysis was performed, data are presented as mean ± SD. Statistical analysis was performed with SPSS 24.0 software. Statistical significance was assessed using student’s t test. A probability of p < 0.05 was regarded as significant.
Results
Neutralization of IL-1β reduces myocardial damage and inflammation
In this study, we analyzed the effects of IL-1β neutralization in a mouse model of CVB3-induced myocarditis. IL-1β was blocked at different time points post CVB3 infection to evaluate the role of IL-1ß in the development and outcome of chronic myocarditis. The detailed experimental setup is represented in Fig. 1a.
Myocyte necrosis and inflammatory cell infiltrates are characteristics of myocarditis. In susceptible mouse strains, CVB3 causes severe cytopathic effects due to viral replication during the acute phase of the infection. As a consequence, the innate and cellular immune response is initiated [16, 17]. Importantly, virus persistence and ongoing inflammation can lead to the chronic phase with myocardial remodeling in susceptible mice and humans.
To assess the effects of IL-1β neutralization on the extent of myocardial damage, the murine hearts were examined histologically using hematoxylin and eosin (HE) and Masson trichrome staining (Figs. 1b, c and 2a). The analysis revealed extensive myocardial damage in the hearts of CVB3-infected mice 14 and 28 days pi (Fig. 2b). Multiple focal lesions and a considerable loss of the regular myocardial structure were found 14 days pi (Fig. 2a). 28 days pi, the lesions revealed less inflammation and more fibrosis (Fig. 2a). Application of the IL-1β antibody led to a significant reduction of myocardial damage in all three antibody-treated groups. The most notable reduction of lesions was achieved when the antibody was administered from day 3 to 14.

Quantification of myocardial damage and cellular infiltration in IL-1β antibody-treated and untreated CVB3-infected A.BY/SnJ mice. a HE and Masson trichrome staining in overviews of CVB3-infected mice hearts revealed extensive myocardial damage and immune cell infiltration 14 and 28 days pi compared to antibody-treated mice and uninfected animals, ×2.5. b Quantification of myocardial damage (score 0–4) revealed a significant reduction of myocardial damage in all antibody-treated groups, *p < 0.05, **p < 0.001
In Fig. 1b, a massive cellular infiltration was observed in the hearts of untreated CVB3-infected mice. Using immunohistochemistry, it was shown that these cellular infiltrates consist of Mac-3+ macrophages and CD3+ T cells as expected (Figs. 3a and 4a). 14 days pi, the myocardium of untreated CVB3-infected mice was massively infiltrated with macrophages and to a lesser extent with T cells. 28 days pi, the number of inflammatory cells was reduced, but both macrophages as well as T cells were still present at high levels. All three antibody-treated groups showed significantly less infiltration by macrophages and T cells (Figs. 3b and 4b).

Immunohistochemical detection of Mac-3 revealed significantly higher numbers of Mac-3+ macrophages (a) 14 and 28 days pi in CVB3-infected mice compared to all antibody-treated CVB3-infected mice and control mice (0 days pi), ×200. b Quantification of Mac-3+ macrophages per mm2 myocardium. The number of Mac-3+ macrophages was significantly reduced in all antibody-treated groups compared to non-treated infected mouse hearts, *p < 0.05, **p < 0.001

Immunohistochemical detection of CD3+ T cells revealed significantly higher numbers of CD3+ T cells (a) 14 and 28 days pi in CVB3-infected mice compared to all antibody-treated CVB3-infected mice and control mice (0 days pi), ×200. b Quantification of CD3+ T cells per mm2 myocardium. The number of Mac-3+ macrophages was significantly reduced in all antibody-treated groups compared to non-treated infected mouse hearts, *p < 0.05, **p < 0.001
Neutralization of IL-1β declines cardiac expression of ERK1/2 and IL-6 in CVB3 myocarditis
Binding of IL-1β to the IL-1-receptor triggers a complex signaling cascade resulting in the expression of numerous target genes. As ERK1/2 are key players of the IL-1β signalling cascade and known to play an important role for CVB3 replication, the influence of IL-1β neutralization on the expression of ERK1/2 was assessed by immunohistochemical staining (Fig. 5a) [23, 42]. High levels of ERK1/2 were found in the heart of CVB3-infected mice, not only during the early but also during the late phase of CVB3-infection. Neutralization of IL-1β resulted in a significant reduction of cardiac ERK1/2 expression in all antibody-treated groups compared to the untreated groups (Fig. 5b) by a score (0–4). ERK1/2 expression was found to be spatially identical with myocardial damage and inflammation (compare Figs. 1b, c and 2b).

Influence of IL-1β neutralization on the IL-1 signaling cascade. a Immunohistochemical detection of ERK1/2 protein expression revealed a high expression of ERK1/2 14 and 28 days pi in CVB3-infected mouse hearts which was considerably reduced in all treated animals. b Quantification of ERK1/2 protein expression in IHC revealed a significantly reduced ERK1/2 expression in hearts of all antibody-treated groups compared to non-treated CVB3-infected mice, *p < 0.05, **p < 0.001
Several cytokines are among the target genes of IL-1β, including the pro-inflammatory cytokine interleukin-6 (IL-6) [2, 42]. To investigate the effects of IL-1β neutralization on the expression of IL-6 mRNA, in situ hybridization (Fig. 6a) and qRT-PCR analysis were performed (Fig. 6b). During acute infection (14 days pi), a marked upregulation of IL-6 mRNA was observed in CVB3-infected mice compared to uninfected animals. Application of the IL-1β antibody from day 1 to 14 led to a clear reduction of IL-6 mRNA expression 14 days pi.

IL-1β neutralization interferes with the IL-1 signaling cascade. a IL-6 mRNA was detected by RNA in situ hybridization. The highest levels of IL-6 mRNA which were found 14 days pi in CVB3-infected mice were largely reduced by IL-1β-neutralization. b IL-6 mRNA expression was analyzed by quantitative RT-PCR. IL-1β neutralization from day 1 to 14 led to a significant reduction of IL-6 mRNA expression 14 days pi in CVB3-infected mice. *p < 0.05
Neutralization of IL-1β does not enhance virus replication
To exclude that viral replication is enhanced due to the anti-inflammatory treatment with the IL-1β antibody, the presence of CVB3 positive-strand RNA in the murine hearts was visualized by RNA in situ hybridization (Fig. 7a). Large areas with infected myocytes were detected in the hearts of CVB3-infected mice 14 days pi. 28 days pi, the number of CVB3 positive cells had decreased, but viral RNA was still present, indicative of a persistent infection. Interestingly, the antibody treatment was accompanied with a reduction of viral RNA positivity in the hearts of all three groups which were treated with the IL-1β antibody. To quantify the amount of virus RNA-positive myocytes, we used a score of 0–4 with the following results: 14 days pi 1.66 ± 0.51, 28 days pi 1.0 ± 0.00 14 days (treated 1–14 d) 0.66 ± 0.51, 28 days (treated 3–14 d) 0.50 ± 0.54, 28 days pi (treated 14–28 d) 0.16 ± 0.41.

Influence of IL-1β neutralization on viral replication and development of cardiac fibrosis in IL-1β antibody-treated and untreated CVB3-infected A.BY/SnJ mice. a To determine the viral replication in the myocardium, CVB3 RNA was localized by RNA in situ hybridization. Virus positive cells were markedly reduced by IL-1β neutralization 14 and 28 days pi, respectively, ×200. b CVB3-infected mice developed extensive fibrosis 28 days pi as shown by picrosirius red staining which was significantly diminished in all antibody-treated groups. c Correspondingly, collagen type I protein expression was markedly reduced in all antibody-treated groups compared to non-treated infected mice, ×200
Fibrosis and cardiac remodeling are attenuated after IL-1β blockade
Ongoing inflammation is the hallmark of chronic myocarditis which is characterized by extensive fibrosis and cardiac remodeling finally resulting in DCM. To study the impact of IL-1β neutralization on the development of fibrosis and cardiac remodeling, picrosirius red (Fig. 7b) and Masson trichrome staining (compare Figs. 1c and 2a) were applied. Both stainings revealed some areas of fibrosis 14 days pi in untreated CVB3-infected mice. In untreated mice, the greatest extent of fibrosis was found after 28 days pi. Administration of the IL-1β antibody led to a significant reduction of fibrosis in all antibody-treated groups compared to non-treated mice. Only minor fibrosis was found in group one and the extent of fibrosis was notably reduced in groups two and three. In accordance with these results, immunohistochemical detection of collagen type I expression revealed abundance of collagen type I in untreated CVB3-infected mice 14 days pi and especially 28 days pi (Fig. 7c). The antibody-treated animals of all three groups showed a considerable reduction of collagen type I protein expression.
The upregulation of extracellular matrix protein mRNA levels is reduced after administration of the IL-1β antibody
As the neutralization of IL-1β was able to reduce inflammation, fibrosis and cardiac remodeling, the underlying mechanisms were further elucidated. As the cardiac extracellular matrix plays an important role in the process of remodeling, a microarray analysis was conducted to investigate the expression of 84 genes encoding extracellular matrix proteins and adhesion molecules. As expected, CVB3-infected mice 14 as well as 28 days pi revealed an upregulation of numerous genes compared to uninfected mice (Fig. 8) like transforming growth factor β (TGF-β), matrix metalloproteinase 12 (MMP12), tissue inhibitor of metalloproteinase 1 (TIMP-1) and the matricellular proteins periostin (POSTN), osteopontin (OPN) and tenascin-C (TN-C). A very high upregulation was found for MMP12 (up to 1200-fold 28 days pi) and OPN (up to 1000-fold 14 days pi). The antibody treatment resulted in a downregulation of the aforementioned proteins in all antibody-treated groups (Fig. 8). The application of the antibody from day 14 to 28 pi in group three resulted in the most notable downregulation of these molecules.

Influence of IL-1β neutralization on the gene expression of extracellular matrix proteins and adhesion molecules. The mRNA levels of numerous extracellular matrix proteins and adhesion molecules were upregulated as found by microarray analysis in the hearts of CVB3-infected mice 14 and 28 days pi. IL-1β neutralization resulted in a downregulation of those proteins in all antibody-treated mice
To further confirm these results, the protein expression of the matricellular proteins osteopontin, tenascin-C and periostin was analyzed using immunohistochemistry. As shown in Fig. 9a–c, the three matricellular proteins are highly expressed in CVB3-infected murine hearts 14 days pi as well as 28 days pi. IL-1β neutralization resulted in a drastic reduction of the expression of all three matricellular proteins in all antibody-treated groups. The extent and spatial localization of the matricellular proteins parallels that of myocardial damage (see Fig. 1b, c). Additionally, MMP12 and TIMP-1 mRNA was quantified by qRT-PCR (Fig. 10a, b). MMP12 was found to be most highly expressed 28 days pi, whereas TIMP-1 expression peaked on day 14 pi in untreated mice. The expression of MMP-12 was significantly reduced in all antibody-treated groups compared to the untreated groups. A diminished TIMP-1 expression was found in group one that was treated with the antibody from day 1 to 14.

Diminished expression of matricellular proteins in IL-1β antibody-treated CVB3-infected mice. Immunohistochemical staining for osteopontin (a), tenascin-C (b) and periostin (c) revealed a high myocardial expression of these three matricellular proteins in CVB3-infected untreated mice which was reduced in all antibody-treated groups

Comparison of MMP12 (a) and TIMP-1 (b) mRNA expression in IL-1β antibody-treated and untreated CVB3-infected mice by quantitative RT-PCR. IL-1β neutralization led to a significant reduction of MMP-12 mRNA expression in all antibody-treated groups. TIMP-1 mRNA expression was most prominent 14 days pi in CVB3-infected mice and significantly reduced by antibody treatment either from day 1 to 14 or from day 14 to 28, *p < 0.05 **, p < 0.001
Discussion
This study shows for the first time that IL-β neutralization using an anti-murine IL-1β antibody as a surrogate antibody for Canakinumab prevents the development of chronic viral myocarditis in a mouse model of CVB3-induced myocarditis. The virus-induced myocardial damage as well as the consecutive immune reaction and ensuing fibrosis were reduced by the treatment with the IL-1β antibody. Importantly, a protective effect of the antibody treatment was observed during acute myocarditis starting 3 days pi but also when the treatment was initiated at later stages (14 days pi), thus preventing severe cardiac remodeling.
In CVB3-induced myocarditis, initial damage to the myocardium is mediated via virus replication [17, 25]. In our experiments, the blockage of IL-1β resulted in reduction of viral replication, which was in concordance with the reduced myocardial damage found in the antibody-treated mice. Neutralization of IL-1β could, therefore, have a direct cardioprotective effect. The investigation of ERK1/2, which are key players of the IL-1β signaling cascade, revealed significantly lower expression of ERK 1/2 in all antibody-treated groups. McManus et al. were able to show that ERK 1/2 activation increased viral replication and infectivity [15, 23, 42]. Their studies correspond to our findings, as neutralization of IL-1β led to a significantly lower expression of ERK1/2, and thus is a plausible explanation for reduced viral replication in treated mice. This is an important finding as most other therapy options cannot be considered in case of viral myocarditis as they bear the risk of preventing viral clearance. Corticosteroids were even shown to aggravate viral myocarditis in mice [39].
IL-1β is known to increase the expression of adhesion molecules such as intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1). The expression of these molecules on the surface of endothelial cells is necessary to promote the infiltration of leukocytes from the circulation into infected tissues [9]. During acute CVB3-induced myocarditis, primarily macrophages and T cells are invading the myocardium [17]. Treatment with the IL-1β antibody led to significantly reduced numbers of Mac-3+ macrophages and CD3+ T cells in the hearts of CVB3-infected mice in all antibody-treated groups. IL-1β neutralization may possibly influence the immune cell infiltration process by downregulating the expression of the necessary adhesion molecules. Indeed, our microarray analysis (Fig. 8) also revealed an upregulation of several integrins, ICAM-1 and VCAM-1 14 and 28 days pi in the heart of infected mice, whereas antibody treatment resulted in a downregulation of these molecules. Importantly, the reduction of inflammation results in an improved heart function in CVB3-infected mice as previously shown [30].
In CVB3 infection several cytokines are among the target genes of IL-1β including the pro-inflammatory cytokine IL-6 [2]. In the heart, IL-6 is predominantly produced by macrophages and fibroblasts [24]. This pathway is thought to be critical in the progression of inflammatory myocarditis to DCM and it was found that IL-6 deficiency reduces angiotensin II-induced cardiac fibrosis [3, 24]. Furthermore, neutralization of IL-6 attenuated fibrosis and improved heart function during chronic cardiac allograft rejection [8]. IL-6 can stimulate the production of TGF-β which promotes the differentiation of cardiac fibroblast into myofibroblasts. Activated myofibroblasts are the main producers of collagen I and, therefore, a dominant factor in the remodeling process of the myocardium [24]. Our results correspond to all these findings, as antibody treatment from day 1 to 14 led to a significantly reduced expression of IL-6 14 days pi and consecutively, collagen type I deposition was diminished in all antibody-treated animals. Our results indicate that virus-induced expression of IL-6 can be influenced beneficially by blockage of IL-1β.
Fibrosis and cardiac remodeling depend on complex changes of the extracellular matrix (ECM) involving a multitude of molecular and cellular events. Matricellular proteins are non-structural proteins of the ECM that modulate cell:cell and cell:matrix interactions. They are minimally expressed in normal hearts but are upregulated following cardiac injury and contribute to cardiac fibrosis under pathologic conditions [13]. For instance, high expression levels of the matricellular protein osteopontin (OPN) were found in acute myocarditis and were associated with consecutive development of extensive fibrosis [37]. Moreover, the matricellular protein tenascin C (TN-C) can accelerate angiotensin II-induced cardiac fibrosis. Furthermore, it was found that deletion of TN-C significantly lessened cardiac fibrosis [35]. In human failing hearts, another matricellular protein, periostin (POSTN), was found to be positively associated with myocardial fibrosis [43]. Using microarray analysis, we were able to demonstrate that IL-1β neutralization suppresses the up-regulation of several genes of the extracellular matrix during CVB3-induced myocarditis. We found that not only OPN but also POSTN and TN-C are highly expressed in the heart of CVB3-infected mice, whereas their expression was reduced in all antibody-treated groups. These findings underline that these matricellular proteins contribute to the development of cardiac fibrosis in viral myocarditis and that this process can be advantageously influenced by IL-1β neutralization. Another important group of ECM proteins is the matrix metalloproteinases (MMPs) which can degrade all components of the ECM [36]. Their activity is regulated by tissue inhibitors of metalloproteinases (TIMPs) and disease entities may result from disturbances of the MMP/TIMP ratio. Previously, it was already shown that ECM remodeling after CVB3 infection involves increased expression and activation of MMPs [7]. Here, we found especially high expression levels of MMP12 28 days pi in the hearts of CVB3-infected mice, indicating that MMP12 contributes to the remodeling process. Importantly, MMP12 expression was significantly decreased by IL-1β neutralization. Altogether, these results show that IL-1β neutralization interferes with the remodeling process in a beneficial manner by influencing numerous target genes of the ECM.
Limitations of the study
Currently, Canakinumab is approved for the treatment of periodic fever syndromes, juvenile idiopathic arthritis and gouty arthritis [34]. Although the use of IL-1β antibodies for treatment of myocarditis is not yet approved, it represents a promising therapy option in non-infectious myocarditis but also in viral heart disease. In other cardiovascular manifestations, such as arrhythmias and heart failure, IL-1 blockers have already been proven to be successful [12]. Our study suggests a very positive influence of IL-1β treatment with reduced inflammation, fibrosis and cardiac remodeling in this mouse model of inflammatory heart disease; however, long-term observations are lacking. These data are not directly applicable on humans but provide firm evidence for a positive result of this new treatment as the murine model of acute and chronic CVB3-induced myocarditis closely resembles the outcome of myocarditis in humans. A clinical study in humans would be the logical next step to evaluate whether morbidity and mortality of patients with viral and non-infectious myocarditis will be positively affected over the long term.
One important message of our experimental settings is provided by the fact that the suppression of IL-1β in acute CVB3 myocarditis goes along with diminished inflammation and fibrosis at later stages of the disease even in the presence of persistent virus RNA. Clinical studies suggest that CVB3 persistence has deleterious consequences in the long-term prognosis [20]. Unfortunately, no specific antiviral therapy has been approved to treat enteroviral myocarditis. However, type I interferons have been shown to reduce virus replication, resulting in an improved outcome of enteroviral heart disease [19]. But it is not known whether a patient spontaneously eliminates the virus, which occurs in 50% of enterovirus infections, or develops virus persistence after acute infection [20]. Importantly, as shown in our study, the persistence of viral RNA does not lead to an enhanced or reactivated cardiac inflammation when IL-1ß is downregulated in acute infection.
This information is highly relevant for the clinicians. Even if there is an underlying enterovirus infection in the diagnosed myocarditis and the patient is treated with IL-1β inhibitory drugs, it is unlikely that a pathogenic immune reaction evolves, even when this anti-inflammatory treatment is terminated. This is different to prednisone, which is used for the treatment of patients with virus-negative myocarditis [14], as prednisolone was found to aggravate the course of viral myocarditis in mice [39].
References
Al-Kofahi M, Omura S, Tsunoda I, Sato F, Becker F, Gavins FNE, Woolard MD, Pattillo C, Zawieja D, Muthuchamy M, Gashev A, Shihab I, Ghoweba M, Von der Weid PY, Wang Y, Alexander JS (2018) IL-1beta reduces cardiac lymphatic muscle contraction via COX-2 and PGE2 induction: Potential role in myocarditis. Biomed Pharmacother 107:1591–1600. https://doi.org/10.1016/j.biopha.2018.08.004
Althof N, Goetzke CC, Kespohl M, Voss K, Heuser A, Pinkert S, Kaya Z, Klingel K, Beling A (2018) The immunoproteasome-specific inhibitor ONX 0914 reverses susceptibility to acute viral myocarditis. EMBO Mol Med 10:200–218. https://doi.org/10.15252/emmm.201708089
Baldeviano GC, Barin JG, Talor MV, Srinivasan S, Bedja D, Zheng D, Gabrielson K, Iwakura Y, Rose NR, Cihakova D (2010) Interleukin-17A is dispensable for myocarditis but essential for the progression to dilated cardiomyopathy. Circ Res 106:1646–1655. https://doi.org/10.1161/circresaha.109.213157
Caforio AL, Pankuweit S, Arbustini E, Basso C, Gimeno-Blanes J, Felix SB, Fu M, Helio T, Heymans S, Jahns R, Klingel K, Linhart A, Maisch B, McKenna W, Mogensen J, Pinto YM, Ristic A, Schultheiss HP, Seggewiss H, Tavazzi L, Thiene G, Yilmaz A, Charron P, Elliott PM (2013) Current state of knowledge on aetiology, diagnosis, management, and therapy of myocarditis: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J 34:2636–2648. https://doi.org/10.1093/eurheartj/eht210
Cavalli G, Foppoli M, Cabrini L, Dinarello CA, Tresoldi M (2017) Dagna L interleukin-1 receptor blockade rescues myocarditis-associated end-stage heart failure. Front Immunol 8:131. https://doi.org/10.3389/fimmu.2017.00131
Cavalli G, Pappalardo F, Mangieri A, Mangieri A, Dinarello CA, Dinarello CA, Dagna L, Dagna L, Tresoldi M (2016) Treating life-threatening myocarditis by blocking interleukin-1. Crit Care Med 44:e751–e754. https://doi.org/10.1097/ccm.0000000000001654
Cheung C, Luo H, Yanagawa B, Leong HS, Samarasekera D, Lai JC, Suarez A, Zhang J, McManus BM (2006) Matrix metalloproteinases and tissue inhibitors of metalloproteinases in coxsackievirus-induced myocarditis. Cardiovasc Pathol 15:63–74. https://doi.org/10.1016/j.carpath.2005.11.008
Diaz JA, Booth AJ, Lu G, Wood SC, Pinsky DJ, Bishop DK (2009) Critical role for IL-6 in hypertrophy and fibrosis in chronic cardiac allograft rejection. Am J Transplant 9:1773–1783. https://doi.org/10.1111/j.1600-6143.2009.02706.x
Dinarello CA (2009) Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol 27:519–550. https://doi.org/10.1146/annurev.immunol.021908.132612
Dinarello CA (2011) Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood 117:3720–3732. https://doi.org/10.1182/blood-2010-07-273417
EMEA (2009) CHMP assessment report for Ilaris. London. https://www.ema.europa.eu/documents/assessment-report/ilaris-epar-public-assessment-report_en.pdf. Accessed 23 July 2009
Emmi G, Urban ML, Imazio M, Gattorno M, Maestroni S, Lopalco G, Cantarini L, Prisco D, Brucato A (2018) Use of Interleukin-1 blockers in pericardial and cardiovascular diseases. Curr Cardiol Rep 20:61. https://doi.org/10.1007/s11886-018-1007-6
Frangogiannis NG (2012) Matricellular proteins in cardiac adaptation and disease. Physiol Rev 92:635–688. https://doi.org/10.1152/physrev.00008.2011
Frustaci A, Russo MA, Chimenti C (2009) Randomized study on the efficacy of immunosuppressive therapy in patients with virus-negative inflammatory cardiomyopathy: the TIMIC study. Eur Heart J 30:1995–2002. https://doi.org/10.1093/eurheartj/ehp249
Huber M, Watson KA, Selinka HC, Carthy CM, Klingel K, McManus BM, Kandolf R (1999) Cleavage of RasGAP and phosphorylation of mitogen-activated protein kinase in the course of coxsackievirus B3 replication. J Virol 73:3587–3594
Klingel K, Fabritius C, Sauter M, Goldner K, Stauch D, Kandolf R, Ettischer N, Gahlen S, Schonberger T, Ebner S, Makrigiannis AP, Belanger S, Diefenbach A, Polic B, Pratschke J, Kotsch K (2014) The activating receptor NKG2D of natural killer cells promotes resistance against enterovirus-mediated inflammatory cardiomyopathy. J Pathol 234:164–177. https://doi.org/10.1002/path.4369
Klingel K, Hohenadl C, Canu A, Albrecht M, Seemann M, Mall G, Kandolf R (1992) Ongoing enterovirus-induced myocarditis is associated with persistent heart muscle infection: quantitative analysis of virus replication, tissue damage, and inflammation. Proc Natl Acad Sci U S A 89:314–318. https://doi.org/10.1073/pnas.89.1.314
Krueger GR, Ablashi DV (2003) Human herpesvirus-6: a short review of its biological behavior. Intervirology 46:257–269. https://doi.org/10.1159/000073205
Kuhl U, Pauschinger M, Schwimmbeck PL, Seeberg B, Lober C, Noutsias M, Poller W, Schultheiss HP (2003) Interferon-beta treatment eliminates cardiotropic viruses and improves left ventricular function in patients with myocardial persistence of viral genomes and left ventricular dysfunction. Circulation 107:2793–2798. https://doi.org/10.1161/01.cir.0000072766.67150.51
Kuhl U, Pauschinger M, Seeberg B, Lassner D, Noutsias M, Poller W, Schultheiss HP (2005) Viral persistence in the myocardium is associated with progressive cardiac dysfunction. Circulation 112:1965–1970. https://doi.org/10.1161/circulationaha.105.548156
Lane JR, Neumann DA, Lafond-Walker A, Herskowitz A, Rose NR (1993) Role of IL-1 and tumor necrosis factor in coxsackie virus-induced autoimmune myocarditis. J Immunol 151:1682–1690
Lim BK, Choe SC, Shin JO, Ho SH, Kim JM, Yu SS, Kim S, Jeon ES (2002) Local expression of interleukin-1 receptor antagonist by plasmid DNA improves mortality and decreases myocardial inflammation in experimental coxsackieviral myocarditis. Circulation 105:1278–1281. https://doi.org/10.1161/01.CIR.0000012492.06971.88
Luo H, Yanagawa B, Zhang J, Luo Z, Zhang M, Esfandiarei M, Carthy C, Wilson JE, Yang D, McManus BM (2002) Coxsackievirus B3 replication is reduced by inhibition of the extracellular signal-regulated kinase (ERK) signaling pathway. J Virol 76:3365–3373. https://doi.org/10.1128/JVI.76.7.3365-3373.2002
Ma F, Li Y, Jia L, Han Y, Cheng J, Li H, Qi Y, Du J (2012) Macrophage-stimulated cardiac fibroblast production of IL-6 is essential for TGF β/Smad activation and cardiac fibrosis induced by angiotensin II. PLoS One 7:e35144. https://doi.org/10.1371/journal.pone.0035144
Marchant DJ, McManus BM (2010) Regulating viral myocarditis: allografted regulatory T cells decrease immune infiltration and viral load. Circulation 121:2609. https://doi.org/10.1161/CIRCULATIONAHA.110.960054
Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, Moss AJ, Seidman CE, Young JB (2006) Contemporary definitions and classification of the cardiomyopathies. Circulation 113:1807. https://doi.org/10.1161/CIRCULATIONAHA.106.174287
Nakano A, Matsumori A, Kawamoto S, Tahara H, Yamato E, Sasayama S, Miyazaki JI (2001) Cytokine gene therapy for myocarditis by in vivo electroporation. Hum Gene Ther 12:1289–1297. https://doi.org/10.1089/104303401750270940
Neumann DA, Lane JR, Allen GS, Herskowitz A, Rose NR (1993) Viral myocarditis leading to cardiomyopathy: do cytokines contribute to pathogenesis? Clin Immunol Immunopathol 68:181–190. https://doi.org/10.1006/clin.1993.1116
Osborn O, Brownell SE, Sanchez-Alavez M, Salomon D, Gram H, Bartfai T (2008) Treatment with an interleukin 1 beta antibody improves glycemic control in diet-induced obesity. Cytokine 44:141–148. https://doi.org/10.1016/j.cyto.2008.07.004
Pappritz K, Savvatis K, Miteva K, Kerim B, Dong F, Fechner H, Muller I, Brandt C, Lopez B, Gonzalez A, Ravassa S, Klingel K, Diez J, Reinke P, Volk HD, Van Linthout S, Tschope C (2018) Immunomodulation by adoptive regulatory T-cell transfer improves Coxsackievirus B3-induced myocarditis. Faseb J. https://doi.org/10.1096/fj.201701408r
Parisi F, Paglionico A, Varriano V, Ferraccioli G, Gremese E (2017) Refractory adult-onset still disease complicated by macrophage activation syndrome and acute myocarditis: a case report treated with high doses (8 mg/kg/d) of anakinra. Med (Baltim) 24:e6656. https://doi.org/10.1097/md.0000000000006656
Pollack A, Kontorovich AR, Fuster V, Dec GW (2015) Viral myocarditis–diagnosis, treatment options, and current controversies. Nat Rev Cardiol 12:670–680. https://doi.org/10.1038/nrcardio.2015.108
Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, Kastelein JJP, Cornel JH, Pais P, Pella D, Genest J, Cifkova R, Lorenzatti A, Forster T, Kobalava Z, Vida-Simiti L, Flather M, Shimokawa H, Ogawa H, Dellborg M, Rossi PRF, Troquay RPT, Libby P, Glynn RJ (2017) Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med 377:1119–1131. https://doi.org/10.1056/NEJMoa1707914
Rondeau J-M, Ramage P, Zurini M, Gram H (2015) The molecular mode of action and species specificity of canakinumab, a human monoclonal antibody neutralizing IL-1β. MAbs 7:1151–1160. https://doi.org/10.1080/19420862.2015.1081323
Shimojo N, Hashizume R, Kanayama K, Hara M, Suzuki Y, Nishioka T, Hiroe M, Yoshida T, Imanaka-Yoshida K (2015) Tenascin-C may accelerate cardiac fibrosis by activating macrophages via the integrin alphaVbeta3/nuclear factor-kappaB/interleukin-6 axis. Hypertension 66:757–766. https://doi.org/10.1161/hypertensionaha.115.06004
Sternlicht MD, Werb Z (2001) How matrix metalloproteinases regulate cell behavior. Annu Rev Cell Dev Biol 17:463–516. https://doi.org/10.1146/annurev.cellbio.17.1.463
Szalay G, Sauter M, Haberland M, Zuegel U, Steinmeyer A, Kandolf R, Klingel K (2009) Osteopontin: a fibrosis-related marker molecule in cardiac remodeling of enterovirus myocarditis in the susceptible host. Circ Res 104:851–859. https://doi.org/10.1161/circresaha.109.193805
Szalay G, Sauter M, Hald J, Weinzierl A, Kandolf R, Klingel K (2006) Sustained nitric oxide synthesis contributes to immunopathology in ongoing myocarditis attributable to interleukin-10 disorders. Am J Pathol 169:2085–2093. https://doi.org/10.2353/ajpath.2006.060350
Tomioka N, Kishimoto C, Matsumori A, Kawai C (1986) Effects of prednisolone on acute viral myocarditis in mice. J Am Coll Cardiol 7:868–872. https://doi.org/10.1016/S0735-1097(86)80349-7
Tschope C, Muller I, Xia Y, Savvatis K, Pappritz K, Pinkert S, Lassner D, Heimesaat MM, Spillmann F, Miteva K, Bereswill S, Schultheiss HP, Fechner H, Pieske B, Kuhl U, Van Linthout S (2017) NOD2 (nucleotide-binding oligomerization domain 2) is a major pathogenic mediator of Coxsackievirus B3-induced myocarditis. Circ Heart Fail 10:12. https://doi.org/10.1161/circheartfailure.117.003870
Vanderheyden M, Paulus WJ, Voss M, Knuefermann P, Sivasubramanian N, Mann D, Baumgarten G (2005) Myocardial cytokine gene expression is higher in aortic stenosis than in idiopathic dilated cardiomyopathy. Heart 91:926–931. https://doi.org/10.1136/hrt.2004.035733
Weber A, Wasiliew P, Kracht M (2010) Interleukin-1 (IL-1) pathway. Sci Signal 3:cm1. https://doi.org/10.1126/scisignal.3105cm1
Zhao S, Wu H, Xia W, Chen X, Zhu S, Zhang S, Shao Y, Ma W, Yang D, Zhang J (2014) Periostin expression is upregulated and associated with myocardial fibrosis in human failing hearts. J Cardiol 63:373–378. https://doi.org/10.1016/j.jjcc.2013.09.013
Acknowledgements
We acknowledge Sandra Bundschuh for excellent technical assistance and Hermann Gram (Novartis Pharma AG, Basel, Switzerland) for providing the murine IL-1β antibody.
Funding
This work was partially funded by the German Research Foundation (DFG) KL 595/2-3 to KK. TE was supported by a MD scholarship (Interdisziplinäres Zentrum für Klinische Forschung (IZKF)-Promotionskolleg) provided by the University Hospital Tübingen.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
KK received the IL-1β antibody as a gift and research funding from Novartis AG. The other authors declare that they have no conflict of interest.
Additional information
Lisa Kraft, Tugs Erdenesukh, Martina Sauter and Karin Klingel takes responsibility for all aspects of the reliability and freedom from bias of the data presented and their discussed interpretation.
Rights and permissions
About this article

Cite this article
Kraft, L., Erdenesukh, T., Sauter, M. et al. Blocking the IL-1β signalling pathway prevents chronic viral myocarditis and cardiac remodeling. Basic Res Cardiol 114, 11 (2019). https://doi.org/10.1007/s00395-019-0719-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00395-019-0719-0