
Abstract
Patients with chronic kidney disease (CKD) exhibit a massively increased risk for cardiovascular (CV) events, and traditional strategies to improve CV outcome have largely failed in the context of CKD. This review article summarizes the current understanding of the pathophysiology of CVD in patients with CKD, defines the gaps in knowledge and describes the structure of the German Transregional Research Consortium SFB TRR219 which addresses “Mechanisms of Cardiovascular Complications in Chronic Kidney Disease”.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Patients with chronic kidney disease (CKD) exhibit a massively increased risk for cardiovascular (CV) events. About 50% of patients with CKD stage 4–5 suffer from cardiovascular disease (CVD) [1], and CV mortality accounts for ~ 40–50% of all deaths in patients with CKD stage 4 (eGFR of 16–29 ml/min) as well as in patients with end-stage renal disease (ESRD = CKD stage 5; need for dialysis or transplant, eGFR of 15 ml/min or less), compared with 26% in controls with normal kidney function [2, 3]. In addition to the high risk for fatal atherosclerosis-related complications such as ischemic heart disease and stroke, cardiac deaths also result from heart failure and arrhythmias [2, 4]. In more than 70 studies in non-dialysed subjects with CKD, correction for classical and even less classical CV risk factors, such as hypertension, diabetes mellitus and dyslipidemia, did not neutralize the impact of CKD on CV risk [5]. The underlying pathophysiological processes of CVD in CKD patients obviously differ from the CVD processes in the general population. This underlines the importance of non-traditional, CKD-specific CV risk factors [6] and that CKD itself is an independent risk factor for CV events [7]. This may explain at least in part why traditional strategies to improve CV outcome have largely failed in the context of CKD [6]. In addition, this emphasizes the need to identify pathological mechanisms adversely affecting the CV system in CKD, with the aim to reduce the increased CV mortality in CKD patients through novel therapeutic strategies.
Cardiovascular disease in chronic kidney disease
Chronic kidney disease (CKD)
Via impaired regulation of the acid–base balance, the water and electrolyte balance, blood pressure and the bone metabolism, CKD affects essentially all organs. CKD has developed into a serious health problem. In 2005, the incidence rate of CKD in Europe was 135 per million, the rate in the USA 336 per million [8]. In 2012, the overall prevalence of CKD exceeded 13% in the USA, which is a relative increase with ~ 12% compared to 1994 [4]. This worldwide rise in the prevalence of CKD is reflected in an increase of patients reaching CKD stage 5 (ESRD), necessitating renal replacement therapy. The prevalence of ESRD exceeded 635.000 people in the USA in 2012, which is more than a doubling compared to 1994 [4], and a similar development is seen in Europe. Key reasons are (1) the rapidly aging population with data from the National Health and Nutrition Examination Survey in the USA showing an increase in CKD prevalence to ~ 33% in elderly people, and (2) the rapidly increasing global prevalence of type 2 diabetes mellitus, an important risk factor for CKD [9]. In addition to age and diabetes, hypertension and higher body mass index are also associated with CKD. In these high-risk subpopulations, the prevalence of CKD is more than 50% [10].
Cardiovascular complications in CKD patients
CV death is responsible for ~ 40–50% of all deaths in patients with CKD stage 4–5 [2, 11, 12]. In addition to reduced glomerular filtration rate (GFR), proteinuria, even if mild, is a second key risk factor for CVD [13]. Vice versa, in patients with established CVD, the presence of CKD markedly worsens outcomes [14, 15], resulting in a vicious circle of increased CV injury and renal disease. CKD-related CV death is mostly caused by ischemic heart disease (myocardial infarction, angina pectoris and sudden cardiac death), accounting for ~ 55% of CV deaths in patients with CKD stage 2–4 [2] and up to 75% in patients with CKD stage 5 (ESRD) [4]. In patients with ESRD, there is an annual incidence of myocardial infarction of 10%; sudden cardiac death becomes dominant as CKD progresses and is responsible for 60% of all heart-associated deaths in dialysis patients [4, 16]. Other CV-related deaths in CKD patients are caused by cerebrovascular disease (14% in CDK stage 4), heart failure (including heart failure due to left ventricular hypertrophy; 14% in CKD stage 4) [17,18,19, Miro, 2018 #49] and arrhythmia (7% in CKD stage 4) [2].

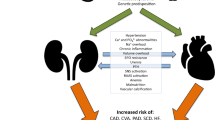
CV complications in CKD are triggered by interactions of the kidney with the circulation and the myocardium. a Important CV pathologies in CKD patients. b Alterations in the circulation as well as in the myocardium crucially contribute to the increased CV risk in patients with CKD. c Pathological mechanisms underlying increased CVD risk in patients with CKD
The impact of typical CV risk factors such as gender, smoking or overweight is markedly reduced in CKD (stage 5) patients compared to the general population [20,21,22]. In more than 70 studies in non-dialysed subjects with CKD, correction for various CV risk factors did not neutralize the impact of CKD on CV risk [5]. Both, clinical as well as experimental data suggest that the underlying processes of CVD in patients suffering from CKD obviously differ from the process of non-CKD patients (reviewed in [23]). Thus, besides the traditional risk factors for CVD, integrated into the traditional Framingham risk score, non-traditional or novel CV risk factors exist in CKD patients [6, 24], and CKD was recently suggested as an independent risk factor for CV events [7], independent of geographic or ethnic factors [11].
The pathological mechanisms leading to CVD in CKD include hyperactivity of the renin–angiotensin–aldosterone system, sodium retention, volume overload, endothelial dysfunction, dyslipidemia, coagulopathy, inflammation, anemia, hyperparathyroidism, hypoalbuminemia, mineral–bone disorders, the uremic state and oxidative stress [25], each leading to histomorphological alterations of the heart, kidneys and vessels. Further pathologic features include reduced elasticity of large arteries [26,27,28,29]. Of course, risk factors for CVD in the general population also contribute to CV risk in CKD. These factors, for example, contribute to the progression of coronary heart disease via atherosclerotic plaques in CKD patients. The presence of coronary heart disease in turn accelerates the loss in kidney function resulting in further risk aggravation for cardiac events. This diversity of risk factors illustrates the complexity of the link between CKD and CVD and many underlying mechanisms are still unknown or poorly understood.
Sudden cardiac death in CKD
In the general population, the risk of sudden cardiac death (SCD) is 1 in 1000 patient-years. This risk increases in CKD patients up to 59 in 1000 patient-years. Conflicting data exist on the occurrence of SCD in relationship to the day and schedule of hemodialysis: some studies show an increased risk on the day after dialysis [30] suggesting that dialysis itself—in addition to the myocardial changes in uremia described above—may represent a risk factor for SCD. Other data point towards an increased mortality after a long interval between dialysis sessions [31, 32], suggesting that alterations in electrolyte levels as well as fluid changes favor the development of cardiac arrhythmias [33].
To date, no established risk score exists to predict cardiac arrhythmias in CKD patients and so far, successful therapeutic strategies to prevent SCD in these patients are lacking.
The circulation and myocardium as main components contributing to increased CV risk in CKD
Our current understanding of the increased CV risk in CKD (Fig. 1a) is based on the interaction of the kidney with the circulation (encompassing the vascular system and blood) and the myocardium (Fig. 1b). This increases CV risk by triggering pathological mechanisms (Fig. 1c).
Circulation
The vascular morphology in CKD exhibits two distinct characteristic but overlapping pathological features, namely accelerated atherosclerosis and arteriosclerosis (mediasclerosis). In contrast to non-CKD patients, most of the uremic vascular damage appears to be degenerative and thus has potential relevance to also understand processes in human aging. Atherosclerosis entails the formation of atherosclerotic lesions in the arterial intima driven by endothelial dysfunction and chronic inflammation of the vessel wall [34]. Upregulation of pro-inflammatory mediators in uremic vessels has been reported [35]. Dialysis patients also exhibit mostly calcified plaques in coronary arteries, whereas plaques of non-uremic patients were mostly fibroatheromatous [29, 36, 37]. Moreover, a lower estimated GFR (eGFR) was associated with increased numbers of newly formed intramural blood vessels and intraplaque hemorrhages [38]. Arteriosclerosis is characterized by fibrosis and thickening of the medial arterial layer. Whereas inflammation is not a major feature of arteriosclerosis, arterial wall calcification associated with an osteoblastic transformation of vascular smooth muscle cells (VSMCs), is an important characteristic of uremic damage [39]. Calcification is frequent in the general population and associated with high CV risk [5, 40, 41]. In patients with CKD calcification is accelerated and amplified, and heralds a poor prognosis in terms of CV morbidity [5, 40, 42, 43]. An important cause of increased calcification in CKD is the lack or dysfunction of calcification inhibitors, with one of the most important ones being the matrix gla protein (MGP), which is activated in the presence of vitamin K. CKD patients are vitamin K deficient [43], and a recent study by members of this Consortium showed that vitamin K2 supplementation in dialysis patients can improve the activation status of MGP [44]. Ongoing studies will determine whether clinically relevant parameters such as the progression of vascular calcification and CV mortality in dialysis patients are affected by vitamin K supplementation.
Besides vessel wall pathology, patients with CKD also exhibit alterations of the circulating blood that contribute to their increased CV risk. Thus, patients with CKD display a chronic pro-inflammatory state with increased levels of circulating pro-inflammatory and pro-atherogenic mediators. Furthermore, various studies document altered platelet function in CKD, and CKD patients exhibit a compact clot structure with an impaired fibrinolysis, which is associated with increased mortality [45, 46]. In addition, recent work to a large extent performed by our group, suggests that CKD leads to major changes in lipoproteins: high-density lipoprotein (HDL) is considered atheroprotective in non-renal populations through its anti-inflammatory properties and by promoting endothelial repair. However, independent of its concentration, the vasoprotective functions of HDL are severely reduced in patients with CKD and complex compositional and structural changes cause accumulation of dysfunctional HDL in these patients [47].
Finally, when kidney excretory function fails, a multitude of compounds accumulates in the blood, which under healthy conditions are excreted into the urine [48, 49]. Such uremic toxins, e.g., indoxyl sulfate and p-cresylsulfate, are the compounds that cause a gradual state of endogenous intoxication, termed uremia after the most abundant but relatively inert compound urea. Many uremic toxins have been associated with increased morbidity and mortality in CKD patients, but the underlying pathological mechanisms are only partially understood. Uremic toxins alter platelet function, but also affect the function of other organs including the CV system [23]. Adverse effects of uremic toxins are at least partially mediated through DNA damage [50]. Furthermore, recent data revealed that increased plasma levels of the uremic toxin phenylacetic acid in ESRD inhibit the expression of the atheroprotective protein iNOS [51], and indoxyl sulfate contributes to oxidative stress and endothelial dysfunction in CKD [52]. Uremic toxins also promote phenotypic changes and damage of VSMCs. In addition, clinical and epidemiological data suggest an association of CKD with poor coronary collateral vessel development [53].
Some of the uremia-associated risk factors also seem to mediate CV mortality in aging persons with normal renal function, confirming the long-standing clinical observation that uremia is a state of accelerated (vascular) aging [50]. This suggests that findings and therapeutic strategies targeting increased CV risk in CKD could also be extrapolated to the aging population without CKD, which markedly strengthens the potential implications of our planned research.
Myocardium
Cardiac arrest is responsible for most CV deaths in patients with ESRD. Characteristic changes in the myocardium such as fibrosis and cardiac hypertrophy are of critical importance here and have been previously summarized under the not very well defined term “uremic cardiomyopathy”.
Extracellular matrix protein accumulation in the cardiac interstitium contributes to systolic and diastolic dysfunction in many cardiac pathophysiologic conditions. In addition to activated myofibroblasts, which are the main effector cells of the fibrotic heart, monocytes/macrophages, lymphocytes, mast cells, vascular cells and cardiomyocytes are involved in the fibrotic response by secreting key fibrogenic mediators. Inflammation, oxidative stress, the renin–angiotensin–aldosterone system, serum lipoproteins, mineral and iron homeostasis have all been recognized to influence cardiac fibrosis [54]. However, their specific role in the cardiorenal interaction, and their potential contribution to increased cardiac death in CKD patients, is poorly understood.
During the dialysis procedure, significant hypotensive phases occur in 25% of cases, which in combination with microvascular changes lead to local ischemia. Left ventricular dysfunction can persist after the return to normal perfusion, known as myocardial stunning [55], which is associated with an increased mortality [56]. A further risk factor of sudden cardiac death in CKD patients is left ventricular hypertrophy, resulting from hypertension, left ventricular dilatation, chronic inflammation and an activation of the renin–angiotensin–aldosterone system [57]. However, whereas the progress of left ventricular hypertrophy is prevented or even reversed in non-dialysis patients with strict anti-hypertensive therapy, this is not the case in dialysis patients, suggesting that CKD-associated risk factors contribute to a greater extent to left ventricular hypertrophy than hypertension itself.
A crucial interaction between the kidney, the circulation and the myocardium
The increasing incidence and prevalence as well as the high mortality of CVD require accurate identification and characterisation of involved signaling pathways and mechanisms. Numerous studies indicate a close interaction of the kidney, the heart and the circulation, influencing each other in their function. Worldwide researchers have identified important mediators that play relevant roles in renal, myocardial and vascular pathology. However, their functions regarding the complex interactions between kidney, heart and vessel are overall still poorly understood.
Transregional Research Consortium SFB TRR219
The Transregional Research Consortium SFB TRR219 is composed by two academic German centers with a strong scientific and clinical focus on CV diseases in CKD patients: the University Hospital Aachen (RWTH) and the Saarland University.
We assume that the increased CV risk of patients with CKD is determined by the circulation (blood and vessels) as well as the myocardium, both of which can contribute to cardiac and renal pathology. By collaborative and translational research based on existing interactions between groups of different clinical and methodological expertise, we pursue the overall long-term goal to gain understanding of the renal and CV interactions that may contribute to the development of novel treatment strategies to decrease CV risk in CKD patients. The following specific aims were defined:
-
Characterisation of known and newly identified mediators relevant in CKD, and understanding of the mechanisms underlying their effect on heart and vessels causing CKD-related CVD.
-
Identification of novel mediators relevant in CKD with a strong impact on CVD.
-
Initiate clinical translation by investigating novel mediators as potential biomarkers of CKD-related CVD, by analyzing the effect of novel interventions on CKD-related CV pathology, and by optimizing diagnostic tests to aid its application into clinical practice. This should stimulate the development of novel pharmacological strategies in CKD-associated CVD to reduce CV morbidity and mortality in CKD in the long run.
We are focussing on ischemic heart disease (which includes myocardial infarction) and chronic heart failure, as they account for more than 70% of CV deaths in CKD patients (Fig. 1a). This includes analysis of uremic cardiomyopathy as well as atrial fibrillation, two common causes of heart failure in these patients.
Structure of the Transregional Research Consortium and research strategy to reach the consortium aims
Alterations in the circulation as well as in the myocardium crucially contribute to the increased CV risk in patients suffering from CKD, identifying these alterations as the main, critical components for their high CVD risk. However, the molecular mechanisms as well as the mediators involved are largely unexplored. Therefore, the aim of this Transregional Research Consortium is to analyze in experimental and clinical studies the multi-factorial aspects of CKD-related CV morbidity and mortality caused by:
-
(a)
the circulation, here, we focus on (1) the pathological remodeling of the vessel wall and involved mediators and vascular cells, which results in inflammation, endothelial dysfunction, formation of vascular lesions and calcification; and on (2) components of the blood such as dyslipidemia, thrombogenicity, increased mineral load and uremic toxins, all contributing to CV damage;
-
(b)
the myocardium, comprising pathological myocardial remodeling associated with deranged myocardial metabolism, altered oxidative stress, micro-angiopathy, autonomous neuropathy, and impaired cardiac function.
Future projects in this consortium will focus on the increased stroke risk in patients with CKD as well as changes in the brain vasculature in CKD.
In addition to examining pathological mechanisms affecting the CV system in CKD at the basic science level, we also study translational aspects by analyzing novel interventions and diagnostic tests in the context of CKD-related CV pathology. For example, we investigate in mouse models or/and small CKD patient cohorts whether interventions as renal denervation, vitamin K therapy or application of fibrinogen-binding peptides can modulate CVD (cardiac remodeling, calcification and thrombosis, respectively) in the context of CKD. Furthermore, we search for novel factors contributing to CVD in the context of CKD, to trigger the subsequent development of novel diagnostic and/or predictive tests. In addition, by optimizing a previously developed calcification blood test, we will actively contribute to the application of novel diagnostic tests into clinical practice.
In summary, a better understanding of the pathophysiology of CVD in CKD together with the development of novel diagnostic tests should pave the way for a better risk prediction in patients with CKD and facilitate the development of future therapeutic strategies to reduce CV morbidity and mortality in this high-risk population.
References
Stevens PE, O’Donoghue DJ, de Lusignan S, Van Vlymen J, Klebe B, Middleton R, Hague N, New J, Farmer CK (2007) Chronic kidney disease management in the United Kingdom: NEOERICA project results. Kidney Int 72(1):92–99. https://doi.org/10.1038/sj.ki.5002273
Thompson S, James M, Wiebe N, Hemmelgarn B, Manns B, Klarenbach S, Tonelli M (2015) Cause of Death in Patients with Reduced Kidney Function. J Am Soc Nephrol 26(10):2504–2511
Drey N, Roderick P, Mullee M, Rogerson M (2003) A population-based study of the incidence and outcomes of diagnosed chronic kidney disease. Am J Kidney Dis 42(4):677–684
Saran R, Li Y, Robinson B, Ayanian J, Balkrishnan R, Bragg-Gresham J, Chen JT, Cope E, Gipson D, He K, Herman W, Heung M, Hirth RA, Jacobsen SS, Kalantar-Zadeh K, Kovesdy CP, Leichtman AB, Lu Y, Molnar MZ, Morgenstern H, Nallamothu B, O’Hare AM, Pisoni R, Plattner B, Port FK, Rao P, Rhee CM, Schaubel DE, Selewski DT, Shahinian V, Sim JJ, Song P, Streja E, Kurella Tamura M, Tentori F, Eggers PW, Agodoa LY, Abbott KC (2015) US renal data system 2014 annual data report: epidemiology of kidney disease in the United States. Am J Kidney Dis 65(6 Suppl 1):A7
Vanholder R, Argiles A, Baurmeister U, Brunet P, Clark W, Cohen G, De Deyn PP, Deppisch R, Descamps-Latscha B, Henle T, Jorres A, Massy ZA, Rodriguez M, Stegmayr B, Stenvinkel P, Wratten ML (2001) Uremic toxicity: present state of the art. Int J Artif Organs 24(10):695–725
Ortiz A, Covic A, Fliser D, Fouque D, Goldsmith D, Kanbay M, Mallamaci F, Massy ZA, Rossignol P, Vanholder R, Wiecek A, Zoccali C, London GM (2014) Epidemiology, contributors to, and clinical trials of mortality risk in chronic kidney failure. Lancet 383(9931):1831–1843
Tonelli M, Muntner P, Lloyd A, Manns BJ, Klarenbach S, Pannu N, James MT, Hemmelgarn BR (2012) Risk of coronary events in people with chronic kidney disease compared with those with diabetes: a population-level cohort study. Lancet 380(9844):807–814
El Nahas AM, Bello AK (2005) Chronic kidney disease: the global challenge. Lancet 365(9456):331–340. https://doi.org/10.1016/S0140-6736(05)17789-7
Hippisley-Cox J, Coupland C (2010) Predicting the risk of chronic Kidney Disease in men and women in England and Wales: prospective derivation and external validation of the QKidney Scores. BMC Fam Pract 11:49. https://doi.org/10.1186/1471-2296-11-49
Eckardt KU, Coresh J, Devuyst O, Johnson RJ, Kottgen A, Levey AS, Levin A (2013) Evolving importance of kidney disease: from subspecialty to global health burden. Lancet 382(9887):158–169. https://doi.org/10.1016/S0140-6736(13)60439-0
Vanholder R, Massy Z, Argiles A, Spasovski G, Verbeke F, Lameire N (2005) Chronic kidney disease as cause of cardiovascular morbidity and mortality. Nephrol Dial Transpl 20(6):1048–1056
Tonelli M, Wiebe N, Culleton B, House A, Rabbat C, Fok M, McAlister F, Garg AX (2006) Chronic kidney disease and mortality risk: a systematic review. J Am Soc Nephrol 17(7):2034–2047
Hillege HL, Fidler V, Diercks GF, van Gilst WH, de Zeeuw D, van Veldhuisen DJ, Gans RO, Janssen WM, Grobbee DE, de Jong PE (2002) Urinary albumin excretion predicts cardiovascular and noncardiovascular mortality in general population. Circulation 106(14):1777–1782
Anavekar NS, McMurray JJ, Velazquez EJ, Solomon SD, Kober L, Rouleau JL, White HD, Nordlander R, Maggioni A, Dickstein K, Zelenkofske S, Leimberger JD, Califf RM, Pfeffer MA (2004) Relation between renal dysfunction and cardiovascular outcomes after myocardial infarction. N Engl J Med 351(13):1285–1295. https://doi.org/10.1056/NEJMoa041365
Gibson CM, Dumaine RL, Gelfand EV, Murphy SA, Morrow DA, Wiviott SD, Giugliano RP, Cannon CP, Antman EM, Braunwald E (2004) Association of glomerular filtration rate on presentation with subsequent mortality in non-ST-segment elevation acute coronary syndrome; observations in 13,307 patients in five TIMI trials. Eur Heart J 25(22):1998–2005
Shamseddin MK, Parfrey PS (2011) Sudden cardiac death in chronic kidney disease: epidemiology and prevention. Nat Rev Nephrol 7(3):145–154. https://doi.org/10.1038/nrneph.2010.191
Cotter G, Davison BA, Butler J, Collins SP, Ezekowitz JA, Felker GM, Filippatos G, Levy PD, Metra M, Ponikowski P, Teerlink JR, Voors AA, Senger S, Bharucha D, Goin K, Soergel DG, Pang PS (2018) Relationship between baseline systolic blood pressure and long-term outcomes in acute heart failure patients treated with TRV027: an exploratory subgroup analysis of BLAST-AHF. Clin Res Cardiol 107(2):170–181. https://doi.org/10.1007/s00392-017-1168-0
Bistola V, Simitsis P, Farmakis D, Ikonomidis I, Bakosis G, Triposkiadis F, Hatziagelaki E, Lekakis J, Mebazaa A, Parissis J (2018) Association of mineralocorticoid receptor antagonist use and in-hospital outcomes in patients with acute heart failure. Clin Res Cardiol 107(1):76–86. https://doi.org/10.1007/s00392-017-1161-7
Farmakis D, Simitsis P, Bistola V, Triposkiadis F, Ikonomidis I, Katsanos S, Bakosis G, Hatziagelaki E, Lekakis J, Mebazaa A, Parissis J (2017) Acute heart failure with mid-range left ventricular ejection fraction: clinical profile, in-hospital management, and short-term outcome. Clin Res Cardiol 106(5):359–368. https://doi.org/10.1007/s00392-016-1063-0
Fleischmann EH, Bower JD, Salahudeen AK (2001) Are conventional cardiovascular risk factors predictive of two-year mortality in hemodialysis patients? Clin Nephrol 56(3):221–230
Iseki K, Fukiyama K (2000) Long-term prognosis and incidence of acute myocardial infarction in patients on chronic hemodialysis. The Okinawa Dialysis Study Group. Am J Kidney Dis 36(4):820–825
Kalantar-Zadeh K, Block G, Humphreys MH, Kopple JD (2003) Reverse epidemiology of cardiovascular risk factors in maintenance dialysis patients. Kidney Int 63(3):793–808
Schlieper G, Hess K, Floege J, Marx N (2016) The vulnerable patient with chronic kidney disease. Nephrol Dial Transplant 31(3):382–390
Emrich IE, Zawada AM, Martens-Lobenhoffer J, Fliser D, Wagenpfeil S, Heine GH, Bode-Boger SM (2018) Symmetric dimethylarginine (SDMA) outperforms asymmetric dimethylarginine (ADMA) and other methylarginines as predictor of renal and cardiovascular outcome in non-dialysis chronic kidney disease. Clin Res Cardiol 107(3):201–213. https://doi.org/10.1007/s00392-017-1172-4
McCullough PA (2003) Why is chronic kidney disease the “spoiler” for cardiovascular outcomes? J Am Coll Cardiol 41(5):725–728
Blacher J, Guerin AP, Pannier B, Marchais SJ, Safar ME, London GM (1999) Impact of aortic stiffness on survival in end-stage renal disease. Circulation 99(18):2434–2439
Gelev S, Spasovski G, Dzikova S, Trajkovski Z, Damjanovski G, Amitov V, Sikole A (2008) Vascular calcification and atherosclerosis in hemodialysis patients: what can we learn from the routine clinical practice? Int Urol Nephrol 40(3):763–770
Mizobuchi M, Towler D, Slatopolsky E (2009) Vascular calcification: the killer of patients with chronic kidney disease. J Am Soc Nephrol 20(7):1453–1464
Schwarz U, Buzello M, Ritz E, Stein G, Raabe G, Wiest G, Mall G, Amann K (2000) Morphology of coronary atherosclerotic lesions in patients with end-stage renal failure. Nephrol Dial Transpl 15(2):218–223
Perl J, Chan CT (2006) Timing of sudden death relative to the hemodialysis procedure. Nat Clin Pract Nephrol 2(12):668–669. https://doi.org/10.1038/ncpneph0345
Bleyer AJ, Russell GB, Satko SG (1999) Sudden and cardiac death rates in hemodialysis patients. Kidney Int 55(4):1553–1559. https://doi.org/10.1046/j.1523-1755.1999.00391.x
Foley RN, Gilbertson DT, Murray T, Collins AJ (2011) Long interdialytic interval and mortality among patients receiving hemodialysis. N Engl J Med 365(12):1099–1107. https://doi.org/10.1056/NEJMoa1103313
Legrand M, Ludes PO, Massy Z, Rossignol P, Parenica J, Park JJ, Ishihara S, AlHabib KF, Maggioni A, Miro O, Sato N, Cohen-Solal A, Fairman E, Lassus J, Harjola VP, Mueller C, Peacock FW, Choi DJ, Plaisance P, Spinar J, Kosiborod M, Mebazaa A, Gayat E (2018) Association between hypo- and hyperkalemia and outcome in acute heart failure patients: the role of medications. Clin Res Cardiol 107(3):214–221. https://doi.org/10.1007/s00392-017-1173-3
Weber C, Noels H (2011) Atherosclerosis: current pathogenesis and therapeutic options. Nat Med 17(11):1410–1422
Koleganova N, Piecha G, Ritz E, Schirmacher P, Muller A, Meyer HP, Gross ML (2009) Arterial calcification in patients with chronic kidney disease. Nephrol Dial Transpl 24(8):2488–2496
Yoshida H, Yokoyama K, Yaginuma T, Ohkido I, Yamamoto H, Utsunomiya Y, Kawakami M, Hosoya T (2011) Difference in coronary artery intima and media calcification in autopsied patients with chronic kidney disease. Clin Nephrol 75(1):1–7
Leskinen Y, Lehtimaki T, Loimaala A, Lautamatti V, Kallio T, Huhtala H, Salenius JP, Saha H (2003) Carotid atherosclerosis in chronic renal failure—the central role of increased plaque burden. Atherosclerosis 171(2):295–302
Nakano T, Ninomiya T, Sumiyoshi S, Onimaru M, Fujii H, Itabe H, Nakashima Y, Sueishi K, Tsuruya K, Oda Y, Kitazono T, Kiyohara Y (2013) Chronic kidney disease is associated with neovascularization and intraplaque hemorrhage in coronary atherosclerosis in elders: results from the Hisayama study. Kidney Int 84(2):373–380
Ketteler M, Rothe H, Kruger T, Biggar PH, Schlieper G (2011) Mechanisms and treatment of extraosseous calcification in chronic kidney disease. Nat Rev Nephrol 7(9):509–516
Massy ZA, Maziere C, Kamel S, Brazier M, Choukroun G, Tribouilloy C, Slama M, Andrejak M, Maziere JC (2005) Impact of inflammation and oxidative stress on vascular calcifications in chronic kidney disease. Pediatr Nephrol 20(3):380–382
Duranton F, Cohen G, De Smet R, Rodriguez M, Jankowski J, Vanholder R, Argiles A (2012) Normal and pathologic concentrations of uremic toxins. J Am Soc Nephrol 23(7):1258–1270. https://doi.org/10.1681/ASN.2011121175
Giachelli CM (2004) Vascular calcification mechanisms. J Am Soc Nephrol 15(12):2959–2964. https://doi.org/10.1097/01.ASN.0000145894.57533.C4
London GM, Guerin AP, Marchais SJ, Metivier F, Pannier B, Adda H (2003) Arterial media calcification in end-stage renal disease: impact on all-cause and cardiovascular mortality. Nephrol Dial Transpl 18(9):1731–1740
Krueger T, Schlieper G, Schurgers L, Cornelis T, Cozzolino M, Jacobi J, Jadoul M, Ketteler M, Rump LC, Stenvinkel P, Westenfeld R, Wiecek A, Reinartz S, Hilgers RD, Floege J (2013) Vitamin K1 to slow vascular calcification in haemodialysis patients (VitaVasK trial): a rationale and study protocol. Nephrol Dial Transpl. https://doi.org/10.1093/ndt/gft459
Alzahrani SH, Hess K, Price JF, Strachan M, Baxter PD, Cubbon R, Phoenix F, Gamlen T, Ariens RA, Grant PJ, Ajjan RA (2012) Gender-specific alterations in fibrin structure function in type 2 diabetes: associations with cardiometabolic and vascular markers. J Clin Endocrinol Metab 97(12):E2282-2287. https://doi.org/10.1210/jc.2012-2128
Hess K, Ajjan R, Phoenix F, Dobo J, Gal P, Schroeder V (2012) Effects of MASP-1 of the complement system on activation of coagulation factors and plasma clot formation. PLoS One 7(4):e35690. https://doi.org/10.1371/journal.pone.0035690
Speer T, Owala FO, Holy EW, Zewinger S, Frenzel FL, Stahli BE, Razavi M, Triem S, Cvija H, Rohrer L, Seiler S, Heine GH, Jankowski V, Jankowski J, Camici GG, Akhmedov A, Fliser D, Luscher TF, Tanner FC (2014) Carbamylated low-density lipoprotein induces endothelial dysfunction. Eur Heart J 35(43):3021–3032. https://doi.org/10.1093/eurheartj/ehu111
Cheung AK, Sarnak MJ, Yan G, Dwyer JT, Heyka RJ, Rocco MV, Teehan BP, Levey AS (2000) Atherosclerotic cardiovascular disease risks in chronic hemodialysis patients. Kidney Int 58(1):353–362
Vanholder R, De Smet R (1999) Pathophysiologic effects of uremic retention solutes. J Am Soc Nephrol 10(8):1815–1823
Shanahan CM (2013) Mechanisms of vascular calcification in CKD-evidence for premature ageing? Nat Rev Nephrol 9(11):661–670
Jankowski J, van der Giet M, Jankowski V, Schmidt S, Hemeier M, Mahn B, Giebing G, Tolle M, Luftmann H, Schluter H, Zidek W, Tepel M (2003) Increased plasma phenylacetic acid in patients with end-stage renal failure inhibits iNOS expression. J Clin Invest 112(2):256–264. https://doi.org/10.1172/JCI15524
Yu M, Kim YJ, Kang DH (2011) Indoxyl sulfate-induced endothelial dysfunction in patients with chronic kidney disease via an induction of oxidative stress. Clin J Am Soc Nephrol 6(1):30–39
Passauer J, Pistrosch F, Bussemaker E, Lassig G, Herbrig K, Gross P (2005) Reduced agonist-induced endothelium-dependent vasodilation in uremia is attributable to an impairment of vascular nitric oxide. J Am Soc Nephrol 16(4):959–965. https://doi.org/10.1681/asn.2004070582
Kong P, Christia P, Frangogiannis NG (2014) The pathogenesis of cardiac fibrosis. Cell Mol Life Sci 71(4):549–574. https://doi.org/10.1007/s00018-013-1349-6
McIntyre CW (2010) Haemodialysis-induced myocardial stunning in chronic kidney disease—a new aspect of cardiovascular disease. Blood Purif 29(2):105–110. https://doi.org/10.1159/000245634
Burton JO, Jefferies HJ, Selby NM, McIntyre CW (2009) Hemodialysis-induced cardiac injury: determinants and associated outcomes. Clin J Am Soc Nephrol 4(5):914–920. https://doi.org/10.2215/CJN.03900808
Amann K, Rychlik I, Miltenberger-Milteny G, Ritz E (1998) Left ventricular hypertrophy in renal failure. Kidney Int 68:S78-85
Acknowledgements
This work was supported by the German Research foundation (DFG) Grant SBF TRR219 Projects C01, C04, C08, M01, M02, M03, M05, and Z01.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Marx, N., Noels, H., Jankowski, J. et al. Mechanisms of cardiovascular complications in chronic kidney disease: research focus of the Transregional Research Consortium SFB TRR219 of the University Hospital Aachen (RWTH) and the Saarland University. Clin Res Cardiol 107 (Suppl 2), 120–126 (2018). https://doi.org/10.1007/s00392-018-1260-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00392-018-1260-0