
Abstract
Vascular and/or valvular calcifications in patients with chronic kidney disease (CKD) appear to indicate a poor prognosis in terms of overall survival and cardiovascular morbidity and mortality. Inflammation and oxidative stress represent new features of the arterial and/or valvular calcification process. However, only limited observational and epidemiological data are available in these areas. Therefore, the link between inflammation, oxidation and vascular and/or valvular calcifications deserves careful consideration in CKD patients, since they may become targets for the development of new therapeutic strategies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Vascular and/or valvular calcifications are frequent in the general population and are associated with high cardiovascular risk [1, 2, 3]. Calcifications of the thoracic and abdominal aorta are associated with an increased risk of cardiovascular morbidity and mortality [4, 5, 6]. Moreover, degenerative calcified aortic valve stenosis is currently the most frequent form of valvular heart disease in industrialized countries, with a prevalence of 2% in a North American population over the age of 65 years [7]. This valvular disease has various clinical presentations, ranging from benign asymptomatic aortic valve sclerosis to tight calcified aortic stenosis, which is often poorly tolerated and requires valve replacement. In 1999, the Cardiovascular Health Study prospectively followed 5,621 patients over the age of 65 years and demonstrated the negative prognostic value of aortic sclerosis [1]. With a follow-up of 5 years, patients with aortic sclerosis had nearly 50% higher risk of cardiovascular mortality and myocardial infarction, after adjustment for age, gender, other cardiovascular risk factors, and the presence of coronary heart disease [1].
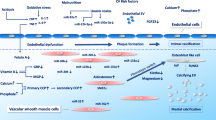
The process of vascular and valvular calcification is accelerated and amplified in patients with chronic kidney disease (CKD) [8, 9]. As in the general population, the calcifications observed in CKD patients also appear to indicate a poor prognosis in terms of overall survival and cardiovascular morbidity [10, 11, 12]. Chronic micro-inflammation and oxidative stress are also commonly observed in patients with CKD [13, 14]. Inflammation and, to a lesser degree, oxidative stress have been shown to predict overall and cardiovascular mortality [13, 14]. Therefore, vascular or valvular calcifications might be one mechanism among several by which inflammation and oxidation influence the chance for survival in CKD patients (Fig. 1).

Schematic link between inflammation and oxidation, vascular or valvular calcifications, and survival in patients with chronic kidney disease (TNFα tumor necrosis factor-α, IL1 interleukin-1)
Cardiovascular calcifications are accompanied by mineralized protein matrix deposition in vessel walls or valves. These calcium-phosphate deposits are essentially localized in the media of vessel walls, but are also present on subintimal atherosclerotic plaques [15, 16]. Inflammation and oxidative stress are now recognized as integral components of subintimal atherosclerotic lesions. Moreover, recent data suggest an active cellular process leading to the deposition of an osteogenic type of extracellular matrix in the media of vessel walls [17]. Several experimental studies have demonstrated the capacity of arterial smooth muscle cells to dedifferentiate into an osteoblastic phenotype able to produce an extracellular matrix related to bone matrix [17]. It has been shown that inflammatory and/or oxidative stress molecules can amplify the smooth muscle cell dedifferentiation process. For example, in vitro studies have shown that oxidized low-density lipoproteins (LDL), certain pro-inflammatory cytokines [e.g., tumor necrosis factor-α (TNFα), and interleukin-1 (IL1)] stimulate dedifferentiation of smooth muscle cells into a bone phenotype [18, 19, 20, 21, 22, 23]. Parhami et al. [18] have shown a strong, dose-dependent positive effect of minimally modified oxidized LDL and different lipid peroxidation products on osteoblastic differentiation of vascular cells. It is possible that this effect is produced not only by lipid peroxidation products, but also by any oxidative stress. Mody et al. [22] found that hydrogen peroxide or xanthine/xanthine oxidase increased intracellular oxygen radicals and enhanced osteoblastic differentiation of vascular cells, and that this effect was blocked by exogenous antioxidants. Advanced glycation end products (glyco-oxidation markers) can induce osteoblastic differentiation of pericytes and could contribute to the development of vascular calcification [23]. Inversely, high-density lipoproteins (HDL) inhibit this dedifferentiation process [24]. The inhibitory effects of HDL were mimicked by lipids extracted from HDL but not by HDL-associated apolipoproteins or reconstituted HDL [24]. Interestingly, oxidation of HDL rendered them pro-osteogenic [24].
Monocytes/macrophages, which are inflammatory cells present in the calcified vascular wall, could accentuate stimulation of smooth muscle cell dedifferentiation to a bone phenotype via cell interaction and/or production of soluble factors such as TNFα [25, 26], thereby promoting vascular or valvular calcifications. Systemic inflammation may also be involved. Low concentrations of fetuin-A, a negative acute-phase protein, are associated with both an impaired capacity to inhibit calcium-phosphate precipitation in vitro and severe calcification of various organs in mice, which are fetuin-A deficient and on a mineral- and vitamin D-rich diet [27].
Conflicting data have been reported regarding the correlation between C-reactive protein (CRP) and coronary artery calcification in CKD patients [28, 29], as well as in the general population [30, 31, 32, 33, 34]. Serum CRP was inversely related to fetuin-A, and low fetuin-A concentrations in sera were associated with enhanced all-cause and cardiovascular mortality in chronic hemodialysis patients [35]. It is also possible that other inflammatory markers (e.g., TNFα, and IL1) might better correlate with coronary artery calcification than CRP. In a recent preliminary study, we observed that TNFα and IL1, but not CRP, are associated with the progression of coronary calcification in chronic hemodialysis patients (Drozdz et al., unpublished data). A direct effect of TNFα and IL1 on fetuin A production remains possible, since it has been shown that both cytokines decrease the rate of synthesis of phosphofetuin in adult rat hepatocytes in primary culture [36]. Therefore, fetuin-A, as an extracellular calcium regulatory protein, could play a role linking chronic inflammation and vascular calcification (Fig. 1).
The relationship between oxidative stress and cardiovascular calcification has not yet been firmly established in CKD patients. Observational and epidemiological data are lacking. Recently we observed no relationship between lipid and protein oxidation products and the progression of coronary calcification in chronic hemodialysis patients (Drozdz et al., unpublished data). In a recent study, pentosidine (glycooxidation marker) staining and calcified deposits were co-localized in the media of cadaveric atherosclerosis-free aorta of non-diabetic hemodialysis patients [37]. Moreover, the mean medial contents of both elastin-associated pentosidine and calcium were significantly higher in hemodialysis patients than in controls [37].
In conclusion, inflammation and oxidative stress represent new features of the arterial and/or valvular calcification process. They deserve careful consideration in CKD patients, since they may become targets for the development of new therapeutic strategies.
References
Otto CM, Lind BK, Kitzman DW, Gersh BJ, Siscovick DS (1999) Association of aortic-valve sclerosis with cardiovascular mortality and morbidity in the elderly. N Engl J Med 341:142–147
Rosenhek R, Binder T, Porenta G, Lang I, Christ G, Schemper M, Maurer G, Baumgartner H (2000) Predictors of outcome in severe, asymptomatic aortic stenosis. N Engl J Med 343:611–617
Kondos GT, Hoff JA, Sevrukov A, Daviglus ML, Garside DB, Devries SS, Chomka EV, Liu K (2003) Electron-beam tomography coronary artery calcium and cardiac events: a 37-month follow-up of 5635 initially asymptomatic low- to intermediate-risk adults. Circulation 107:2571–2576
Iribarren C, Sidney S, Sternfeld B, Browner WS (2000) Calcification of the aortic arch: risk factors and association with coronary heart disease, stroke, and peripheral vascular disease. JAMA 283:2810–2815
Li J, Galvin HK, Johnson SC, Langston CS, Sclamberg J, Preston CA (2002) Aortic calcification on plain chest radiography increases risk for coronary artery disease. Chest 121:1468–1471
Wilson PW, Kauppila LI, O’Donnell CJ, Kiel DP, Hannan M, Polak JM, Cupples LA (2001) Abdominal aortic calcific deposits are an important predictor of vascular morbidity and mortality. Circulation 103:1529–1534
Stewart BF, Siscovick D, Lind BK, Gardin JM, Gottdiener JS, Smith VE, Kitzman DW, Otto CM (1997) Clinical factors associated with calcific aortic valve disease. Cardiovascular Health Study. J Am Coll Cardiol 29:630–634
Braun J, Oldendorf M, Moshage W, Heidler R, Zeitler E, Luft FC (1996) Electron beam computed tomography in the evaluation of cardiac calcification in chronic dialysis patients. Am J Kidney Dis 27:394–401
Perkovic V, Hunt D, Griffin SV, Plessis M du, Becker GJ (2003) Accelerated progression of calcific aortic stenosis in dialysis patients. Nephron Clin Pract 94:c40–c45
Wang AY, Woo J, Wang M, Sea MM, Ip R, Li PK, Lui SF, Sanderson JE (2001) Association of inflammation and malnutrition with cardiac valve calcification in continuous ambulatory peritoneal dialysis patients. J Am Soc Nephrol 12:1927–1936
Blacher J, Guerin AP, Pannier B, Marchais SJ, London GM (2001) Arterial calcifications, arterial stiffness, and cardiovascular risk in end-stage renal disease. Hypertension 38:938–942
London GM, Guerin AP, Marchais SJ, Metivier F, Pannier B, Adda H (2003) Arterial media calcification in end-stage renal disease: impact on all-cause and cardiovascular mortality. Nephrol Dial Transplant 18:1731–1740
Kaysen GA (2001) The microinflammatory state in uremia: causes and potential consequences. J Am Soc Nephrol 12:1549–1557
Massy Z, Nguyen-Khoa T (2002) Oxidative stress and chronic renal failure: markers and management. J Nephrol 15:336–341
Schinke T, Karsenty G (2000) Vascular calcification—a passive process in need of inhibitors. Nephrol Dial Transplant 15:1272–1274
Tomson C (2003) Vascular calcification in chronic renal failure. Nephron Clin Pract 93:c124–c130
Giachelli CM, Jono S, Shioi A, Nishizawa Y, Mori K, Morii H (2001) Vascular calcification and inorganic phosphate. Am J Kidney Dis 38 [Suppl 1]:S34–S37
Parhami F, Morrow AD, Balucan J, Leitinger N, Watson AD, Tintut Y, Berliner JA, Demer LL (1997) Lipid oxidation products have opposite effects on calcifying vascular cell and bone cell differentiation. A possible explanation for the paradox of arterial calcification in osteoporotic patients. Arterioscler Thromb Vasc Biol 17:680–687
Tintut Y, Patel J, Parhami F, Demer LL (2000) Tumor necrosis factor-alpha promotes in vitro calcification of vascular cells via the cAMP pathway. Circulation 102:2636–2642
Jono S, McKee MD, Murry CE, Shioi A, Nishizawa Y, Mori K, Morii H, Giachelli CM (2000) Phosphate regulation of vascular smooth muscle cell calcification. Circ Res 87:E10–E17
Chen NX, O’Neill KD, Duan D, Moe SM (2002) Phosphorus and uremic serum up-regulate osteopontin expression in vascular smooth muscle cells. Kidney Int 62:1724–1731
Mody N, Parhami F, Sarafian TA, Demer LL (2001) Oxidative stress modulates osteoblastic differentiation of vascular and bone cells. Free Radic Biol Med 31:509–519
Yamagishi S, Fujimori H, Yonekura H, Tanaka N, Yamamoto H (1999) Advanced glycation endproducts accelerate calcification in microvascular pericytes. Biochem Biophys Res Commun 258:353–357
Parhami F, Basseri B, Hwang J, Tintut Y, Demer LL (2002) High-density lipoprotein regulates calcification of vascular cells. Circ Res 91:570–576
Tintut Y, Patel J, Territo M, Saini T, Parhami F, Demer LL (2002) Monocyte/macrophage regulation of vascular calcification in vitro. Circulation 105:650–655
Shioi A, Katagi M, Okuno Y, Mori K, Jono S, Koyama H, Nishizawa Y (2002) Induction of bone-type alkaline phosphatase in human vascular smooth muscle cells: roles of tumor necrosis factor-alpha and oncostatin M derived from macrophages. Circ Res 91:9–16
Schafer C, Heiss A, Schwarz A, Westenfeld R, Ketteler M, Floege J, Muller-Esterl W, Schinke T, Jahnen-Dechent W (2003) The serum protein alpha 2-Heremans-Schmid glycoprotein/fetuin-A is a systemically acting inhibitor of ectopic calcification. J Clin Invest 112:357–366
Oh J, Wunsch R, Turzer M, Bahner M, Raggi P, Querfeld U, Mehls O, Schaefer F (2002) Advanced coronary and carotid arteriopathy in young adults with childhood-onset chronic renal failure. Circulation 106:100–105
Stompor T, Pasowicz M, Sullowicz W, Dembinska-Kiec A, Janda K, Wojcik K, Tracz W, Zdzienicka A, Klimeczek P, Janusz-Grzybowska E (2003) An association between coronary artery calcification score, lipid profile, and selected markers of chronic inflammation in ESRD patients treated with peritoneal dialysis. Am J Kidney Dis 41:203–211
Wang TJ, Larson MG, Levy D, Benjamin EJ, Kupka MJ, Manning WJ, Clouse ME, D’Agostino RB, Wilson PW, O’Donnell CJ (2002) C-reactive protein is associated with subclinical epicardial coronary calcification in men and women: the Framingham Heart Study. Circulation 106:1189–1191
Redberg RF, Rifai N, Gee L, Ridker PM (2000) Lack of association of C-reactive protein and coronary calcium by electron beam computed tomography in postmenopausal women: implications for coronary artery disease screening. J Am Coll Cardiol 36:39–43
Park R, Detrano R, Xiang M, Fu P, Ibrahim Y, LaBree L, Azen S (2002) Combined use of computed tomography coronary calcium scores and C-reactive protein levels in predicting cardiovascular events in nondiabetic individuals. Circulation 106:2073–2077
Hunt ME, O’Malley PG, Vernalis MN, Feuerstein IM, Taylor AJ (2001) C-reactive protein is not associated with the presence or extent of calcified subclinical atherosclerosis. Am Heart J 141:206–210
Reilly MP, Wolfe ML, Localio AR, Rader DJ; Study of Inherited Risk of Coronary Atherosclerosis (2003) C-reactive protein and coronary artery calcification: The Study of Inherited Risk of Coronary Atherosclerosis (SIRCA). Arterioscler Thromb Vasc Biol 23:1851–1856
Ketteler M, Bongartz P, Westenfeld R, Wildberger JE, Mahnken AH, Bohm R, Metzger T, Wanner C, Jahnen-Dechent W, Floege J (2003) Association of low fetuin-A (AHSG) concentrations in serum with cardiovascular mortality in patients on dialysis: a cross-sectional study. Lancet 361:827–833
Ohnishi T, Nakamura O, Arakaki N, Miyazaki H, Daikuhara Y (1994) Effects of cytokines and growth factors on phosphorylated fetuin biosynthesis by adult rat hepatocytes in primary culture. Biochem Biophys Res Commun 200:598–605
Sakata N, Noma A, Yamamoto Y, Okamoto K, Meng J, Takebayashi S, Nagai R, Horiuchi S (2003) Modification of elastin by pentosidine is associated with the calcification of aortic media in patients with end-stage renal disease. Nephrol Dial Transplant 18:1601–1609
Author information
Authors and Affiliations
Corresponding author
Additional information
This work was presented in part at the IPNA Seventh Symposium on Growth and Development in Children with Chronic Kidney Disease: The Molecular Basis of Skeletal Growth, 1–3 April 2004, Heidelberg, Germany
Rights and permissions
About this article
Cite this article
Massy, Z.A., Mazière, C., Kamel, S. et al. Impact of inflammation and oxidative stress on vascular calcifications in chronic kidney disease. Pediatr Nephrol 20, 380–382 (2005). https://doi.org/10.1007/s00467-004-1623-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-004-1623-9