
Abstract
Purpose
A personal series of 131 patients with split cord malformation (SCM) operated on is presented.
Methods
Age, gender, symptoms and signs, radiological and operative findings, complications, associated anomalies, outcome, and pathological specimens were analyzed.
Results
There were 88 girls (73 %) and 43 boys (27 %). The female predominance was slightly more remarkable in type I SCMs than in type II SCMs. The presenting symptoms can be summarized as skin lesions, spina bifida aperta, scoliosis or kyphoscoliosis, sphincter disturbance, foot deformities and weakness, and/or atrophy in the lower extremities. The ages of patients with neurological deficits and orthopedic deformities were significantly older than those without deficits (P = 0.030). The duration of symptoms was longer in the patients with neurological deficits and orthopedic deformities than that in those without deficits (P = 0.00095). In six patients, composite SCMs were present. Only one patient with a type II SCM did not have an associated spinal cord lesion. A type I SCM was more frequently encountered in patients with spina bifida (P < 0.0005). Transient postoperative complications were seen in 29 patients (22 %). There was no permanent complication. Retethered cord syndrome developed in five patients with a type I SCM.
Conclusions
The risk of neurological and orthopedic deficits increases with the age of the patient. The risk of permanent deficit after surgery is very low. The whole spine must be examined for additional lesions. All patients should be surgically treated when diagnosed, especially before the development of orthopedic and neurological manifestations, and all associated lesions should also be treated at the same session.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Pang and colleagues [72, 74] proposed an alternative classification to deal with all double spinal cord malformations. They also have proposed a unified theory that explains the embryogenetic mechanisms of all variants of split cord malformations (SCMs). All SCMs originate from one basic error that occurs at approximately the time at which the primitive neurenteric canal closes. This basic error is the formation of an accessory neurenteric canal between the yolk sac and amnion, which is subsequently invested with mesenchyme to form an endomesenchymal tract that splits the notochord and neural plate. Pang et al. [72, 74] defined two types of SCMs. A type I SCM consists of two hemicords; each contained within its own dural tube and separated by a dura-sheathed rigid median septum. A type II SCM consists of two hemicords housed in a single dural tube separated by a nonrigid, fibrous median septum. I have reviewed and analyzed 131 cases of SCMs, operated on by myself.
Clinical material and methods
Patient population
The 131 patients with SCMs who were surgically treated between January 1, 1993 and December 31, 2011 were reviewed. The data regarding patient age and gender, symptoms and signs, radiological and operative findings, complications, associated anomalies, outcome, and pathological specimens were prospectively entered into a database.
Surgical procedures
In patients who had type I SCMs, laminectomy was performed around the attachment of the rigid median septum, and the septum was dissected subperiosteally from its dural sleeve within the dural cleft. The septum was removed by using a rongeur or a high-speed microdrill. After removal of the septum, the dura was opened on both sides of the dural cleft. Fibrous bands or paramedian dorsal roots from the medial aspects of the hemicords, which were adherent to the dural sleeve, were sectioned, and the dural sleeve was resected. Only posterior dural closure was performed.
In patients with type II SCMs, laminotomy was performed near or at the caudal end of the split segment. The dura was opened in the midline. The fibrous septum and/or paramedian dorsal roots were excised. Any associated spinal lesions that might lead to spinal tethering, such as a thick filum terminale, dermal sinus tract, or lipoma, were also surgically treated in both types of SCMs. The filum terminale was explored even if magnetic resonance imaging (MRI) did not show the presence of a thick filum. Split cord malformations were found and treated in some patients who underwent surgical repair of a myelomeningocele shortly after birth. These patients did have postoperative radiological work-up. Follow-up evaluation of all patients ranged from 12 months to 18 years (mean 140.1 months). Fisher’s exact test, chi-square test, t test, and diagnostic test were used in statistical analyses.
Results
Patient age and gender
Patients ranged in age from less than 1 day to 19 years (median 16.5 months). There were 88 girls (73 %) and 43 boys (27 %). The female predominance was slightly more remarkable in type I SCMs than in type II SCMs. When composite-type SCMs were excluded, girls comprised 72 % of the patients with a type I SCM and 63 % of those with type II; however, this difference was not statistically significant. In composite SCMs, there were three girls and three boys. The ages of patients with neurological deficits and orthopedic deformities (average 60.4 months) were significantly older than those without deficits (average 38.6 months) (P = 0.030), when the patients with neural tube defects were not taken into consideration.
Symptoms and signs
The presenting symptoms can be summarized as skin lesions, spina bifida aperta, scoliosis or kyphoscoliosis, sphincter disturbance, foot deformities and weakness, and/or atrophy in the lower extremities (Table 1). Forty-five patients were asymptomatic, and they had MRI scans, because of skin lesions. The duration of symptoms ranged from birth to 12 years (mean 21.7 months). When the patients with myelomeningocele, hemimyelomeningocele, or lipomyelomeningocele were excluded, the duration of symptoms was longer in the patients with neurological deficits and orthopedic deformities (mean 39.9 months) than that in those without deficits (mean 16.6 months). This difference was found to be statistically significant (P = 0.00095). Of the 50 patients with spina bifida cystica, 24 had myelomeningocele, 6 had hemimyelomeningocele, 19 had meningocele, and 1 patient had both meningocele and hemimyelomeningocele. In addition, four patients had lipomyelomeningocele.
Skin stigmata and/or subcutaneous lipoma were present in 76 patients (58 %). Some patients had more than one skin lesion. Hypertrichosis was the most common skin finding and was present in 59 patients. Capillary hemangioma (17 cases), dimple (12 cases), hyperpigmentation (7 cases), and subcutaneous lipoma (6 cases) were other skin stigmata. Fifty-six patients (42 %) had no skin lesions. Twenty of 50 patients with spina bifida cystica exhibited skin lesions, although 52 of 77 patients without spina bifida cystica exhibited these lesions (P = 0.034).
Orthopedic deformities in lower extremities and spinal deformities were seen in 55 and 36 patients, respectively. Some patients had more than one deformity. Scoliosis was the sole presenting symptom in ten children. Paraparesis was present in 25 patients; however, 13 of these patients had a myelomeningocele. Unilateral leg paresis and atrophy were detected in 32 patients, and bladder and/or bowel incontinence was present in 17 patients (Table 1). The proportion of patients with neurological or orthopedic deficits did not significantly vary with the type and level of SCMs.
Radiological findings
Plain X-ray films of the spine were obtained in 113 patients. Magnetic resonance images, computerized tomography (CT) scans, and CT myelograms were available in 131, 22, and 12 patients, respectively. Bifid lamina, a widened interpediculate distance at the level of SCM, scoliosis, a bony spur, hemivertebra, block vertebra, and butterfly vertebra were detected on plain X-rays. Of 60 patients with a type I SCM for whom a plain X-ray film was available, 35 showed a bony spur. CT myelography has not been obtained since 1998, because development in MRI technology made the requirement for obtaining CT myelography unnecessary. MRI has the capability of showing the level and type of SCMs, bone anomalies, associated spinal cord malformations, and the conus level. In composite-type SCMs, MRI missed the second SCM, located in the sacral region. In those cases, axial MRI scan should have been performed not to fail to show the second SCM. In addition, MRI displays the other associated lesions such as hydromyelia, dermoid, epidermoid, lipoma, and tight filum terminale. On MRI scans, hydromyelia was detected in 55 patients. Filum was normal only in one of 65 patients whose MRI scans showed a tight filum. Although the filum was normal on MRI in 63 patients, it was found tight and transected at surgery in 37 patients. Therefore, the specificity of MRI in diagnosing a tight filum is 96.3 %, and sensitivity is 63.4 %. It was very difficult to identify the filum terminale and the level of the conus medullaris, particularly on MR images obtained in children with severe scoliosis.
Length of the split segment
A correlation was not found with the length of the split segment of the spinal cord and the age. The split segment length was significantly longer in a type I SCM (mean 3.1) than in type II (mean 2.3) (P = 0.0336) (Fig. 1).

The graphic showing the length of split segments in types I and II SCMs. Note that the split segment in type I SCM (mean 3.1) is greater than that in type II (mean 2.3) (P = 0.0336)
Surgical findings
Sixty-eight patients had a single type I SCM (Fig. 2), and 57, a single type II SCM (Fig. 3); six patients had composite SCMs. An osteocartilaginous septum was detected in type I SCMs, and a fibrous septum, in type II SCMs. Fibrous bands and/or paramedian dorsal roots from the medial aspects of the hemicords to the dural sleeve were present and cut in all cases of type I SCMs (Fig. 4). All type II SCMs had a fibrous median septum and/or paramedian dorsal roots that tethered the spinal cord, particularly at the caudal end of the split, except for one. Ventral paramedian roots were present in 12 cases (Fig. 5). The hemicords reunited after splitting in 123 patients but did not reunite in 8 patients. Both hemicords were equal in size in 87 patients. In 44 patients, one hemicord was smaller than the other. If a patient has unilateral atrophic or short leg and the hemicords are not equal in size, the possibility of being the atrophic limb and smaller hemicord on the same side was significantly high (P < 0.0001). However, some atrophic legs were associated with symmetrical hemicords.

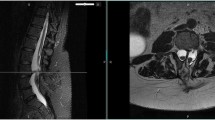
a Coronal, b sagittal, and c axial MRI scans showing a type I SCM

a Sagittal MRI scan showing hydromyelia and tight filum terminale (red arrow). b The patient’s photograph showing lumbar hypertrichosis. c A type II SCM on axial MRI scans

a Sagittal MRI scan showing a bony septum, low-lying conus, tight filum terminale, and intradural lipoma. b Axial MRI scans showing the bony septum (type I SCM), tight filum terminale, and intradural lipoma. Operative photographs showing the removal of bony septum extradurally (c), dorsal paramedian rootlets (d, e; arrow), myelomeningocele manqué (f; arrow), intradural dorsal lipoma (g; arrow), tight filum terminale (h; arrow) after removal of lipoma and sectioning the tight filum (i) and untethered spinal cord (j)

Operative photographs of a patient with a type II SCM showing a dorsal paramedian rootlets (arrow), b aberrant ganglion at the lower part of the cleft and dural attachment (arrow), c ventral paramedian rootlet, and d sectioning of the tight filum terminale
In six patients, composite SCMs were present. In two patients, the dermal sinus tract penetrated the dorsal dura and was connected to a fibrous median septum, attaching the median aspects of the both hemicords. A small bony septum, which had a conical shape and was covered with a dural sheath, was also noted ventrally. The location was at T11 in the first patient and at L3 in the second one. The second patient had also a dorsal lipoma and a dermoid cyst located on the dorsal surfaces of the hemicords at the split level. Four patients had two different SCMs located in tandem, separated by a normal spinal cord. Two patients had types I and II SCMs, one had two type I SCMs, and the other one, two type II SCMs.
Level of SCMs and conus medullaris
The uppermost split segment was between T2 and T4. There were eight SCMs at S1 and below. The majority of SCMs were located in the lumbar region, and there was no SCM located in the cervical region in this series. There were no cervical SCMs in this series. The conus medullaris was located at L1 or L2 in only eight patients (6 %). Both the levels of coni and split segments did not vary significantly (Fig. 6).

Distribution of the lower split segment and the level of coni
Associated lesions
Only one patient with a type II SCM did not have an associated spinal cord lesion. The number of associated spinal cord lesions was one, and more than one in 56 and 76 patients, respectively. The most common associated lesion was a tight filum terminale (113 patients). The lesions can be seen in Table 2. In addition, hydromyelia was detected on MRI of 54 patients. A type I SCM was more frequently encountered in the patients with spina bifida cystica (myelomeningocele, hemimyelomeningocele, and meningocele), and a type II SCM was more common in the patients with spina bifida cystica (P < 0.0005). The bony septum was usually located proximal to myelomeningocele (22 patients). It was at the same level in one patient and distal to neuroplacode in three patients. In all hemimyelomeningoceles, SCM was at the same level.
Postoperative complications
Transient postoperative complications were seen in 29 patients (22 %). All of these complications improved within 3 weeks. The transient complications consisted of cerebrospinal fluid (CSF) leak, transient unilateral leg paresis, paraparesis, wound infection, urinary retention, subcutaneous CSF collection, and neuropathic pain in the legs (Table 3). Dural repair was performed in two out of ten patients with postoperative CSF leak. Complication rates did not vary with the age, SCM type, and associated spinal cord lesions. There was no permanent complication.
Pathological findings
Pathological examinations of the specimens from the median septa revealed fetal renal tissue, tubular epithelia, lymphoid tissue, five dermoid cysts, muscle tissue, ganglion, and blood vessels in addition to bone and cartilage in type I SCMs and fibrous tissue in type II SCMs. A teratoma was resected in one of hemicord in a 9-year-old girl with a type I SCM. She has been symptom free for more than 15 years (Fig. 7). Endodermal, ectodermal, and mesenchymal structures detected within the median septa support the theory of endomesenchymal tract and ectoendodermal adhesion.

a Axial MRI scans showing a type I SCM and a cystic lesion within the right hemicord. b CT scans displaying a bony septum and a widened right-sided spinal canal. c The intramedullary cystic lesion (gray and black arrows) and lipoma (white arrows). d Postoperative sagittal MRI scan showing a total removal of teratoma; intramedullary lipoma was not removed
Patient outcome
There were two deaths in this series. These patients were reported in our previous publication. Ninety-five patients remained stable, and some improvements were detected in 34. Retethered cord syndrome developed in five patients with a type I SCM. Regrowth of the bony spur was detected in one of the patients who had undergone the second surgery for untethering. Although there was no retethering among the patients with a type II SCM, this difference did not reach a statistical significance.
Discussion
The classification of SCMs proposed by Pang et al. [74] was a big step to put an end the chaos in terminology. The terms “type I” and “type II” split spinal cord malformations (SSCMs) have been generally used instead of diastematomyelia and diplomyelia, respectively, since 1992. The female predominance has been reported in many series [1, 9, 28, 29, 40, 54, 72, 81, 82, 86, 91]. Ansari et al. [9] reported that the female predominance was more remarkable in patients with type I SCMs than that in those with type II SCMs in this series, but the difference was not statistically significant. Male to female ratio was 1:09 in the series of Kumar et al. [55]. Presenting symptoms and signs can be categorized into three parts such as skin findings and neurologic and orthopedic manifestations in infants and small children. At least half of our patients had skin stigmata. The most common skin finding was hypertrichosis as reported by many authors [3, 4, 6, 15, 18, 22, 23, 26, 28–30, 39, 40, 50, 54, 61, 62, 72, 74, 79, 80, 85, 87, 90, 91, 98, 100]. Capillary hemangioma [3, 4, 9, 28–30, 39, 54, 72, 74, 80, 91], dimple [3, 4, 13, 15, 22, 26, 28, 34, 39, 47, 51, 54, 56, 67, 72–74, 91], hyperpigmentation [28, 91], subcutaneous lipoma [26, 28, 47, 54, 72, 74, 80, 84, 91, 93], aplasia cutis [45], and neuroectodermal skin appendages [28, 54, 72, 74, 91, 93, 105] are the other skin lesions seen in patients with SCMs. Skin lesions were less common in the patients with meningocele or myelomeningocele than those without. In 1992, Pang [72] reported that the incidence of skin lesions in patients with myelomeningocele was significantly less than that in those without myelomeningocele. Pang [72] assumed that skin lesions such as hypertrichosis, capillary hemangiomas, and dermal sinus tracts represent minor aberrations in the development of surface ectoderm that result from the adverse influence of a dorsal endomesenchymal tract, but that these aberrations might be largely overshadowed by chaotic changes in the surface ectoderm occasioned by the unneurulated neural plate in the case of an associated myelomeningocele. Patients with myelomeningocele or lipomyelomeningocele do usually have neurological deficits. When these patients, older patients, were excluded, significantly more neurological deficits and orthopedic findings were present. The duration of symptoms was also longer in the patient with deficits than that in those without deficits. Progressive deficits were present in both children and adults in the series of Pang [72]. Progressive neurological or orthopedic deficits have been described in patients with SCMs by many authors [15, 23, 42, 55, 71, 73, 80, 81]. Paraparesis, asymmetric or unilateral paresis in lower extremities, sphincter dysfunction, sensory loss, trophic ulcer, autoamputation, and the short and thin left lower limb have been reported [4, 9, 11, 13, 14, 19, 22–24, 30, 37, 38, 47, 50, 51, 55, 59, 64, 65, 67, 71, 78, 80, 83, 85, 92, 97, 98, 100–102, 104, 105].
Spinal deformities such as scoliosis, kyphosis, or kyphoscoliosis are frequently associated with SCMs [1, 6, 9, 15, 19, 24, 28, 30, 34, 37, 42, 44, 50, 52, 54, 55, 58, 59, 68, 72, 76, 78–82, 85, 86, 88–92, 96, 98, 99, 101, 103–106]. Spinal deformity was a presenting symptom in 36 patients in the present series, and all congenital scoliosis associated with SCMs needed corrective surgeries. Neural axis abnormalities occur in up to 40 % of patients with congenital scoliosis [12, 16]. Bradford and colleagues [16] reported a 38 % incidence of neural axis abnormalities in 42 patients: 16 harbored a tethered cord, 7 had SCM (16.6 %) (diastematomyelia in four and diplomyelia in three patients), 4 had a syringomyelia, and 1 had a sacral teratoma. McMaster [63] determined that diastematomyelia was the most common anomaly (16.3 % of patients) in patients with congenital scoliosis. Suh et al. [95] reported that the overall rate of intraspinal anomalies in a nonrandomized group of spinal deformity was 32 % (13/41): tethered cord in 12 patients, syringomyelia in 3 patients, and diastematomyelia in 5 patients (12 %). Scoliosis is usually seen in up to 60 % of patients with SCM [1, 4, 14, 15, 19, 24, 28, 30, 32, 34, 37, 42, 45, 50–52, 54, 55, 58, 59, 72, 78–82, 85, 86, 88, 90–92, 98, 99, 103–106]. Kyphosis or kyphoscoliosis may accompany SCMs [6, 8, 15, 28, 42, 50, 58, 68, 76, 89, 91, 96, 101]. Hilal et al. [41] reported that the risk of scoliosis increases significantly in older children with diastematomyelia. James and Lassman [48] and Guthkelch [36] determined the orthopedic and neurological syndromes in children with diastematomyelia. The orthopedic syndromes consist of foot deformities and dwarfing of one leg with muscle atrophy in addition to spine deformity.
The bone deformities seen on plain X-ray films, CT, or MRI of patients with SCMs are many; scoliosis, bifid lamina, widened interpediculate distance, hemivertebra, bifid vertebra, fused vertebra, and narrowing of the intervertebral disc space have all been observed [3–6, 8, 11, 13, 23, 24, 28, 30, 34, 37, 38, 52, 54, 55, 58, 68, 69, 72–74, 76, 82, 84, 85, 88, 91, 92, 99, 104, 106]. The spinous process is prominent in the area of the bone spur, especially if there is an intersegmental fusion of adjacent laminae [41]. Unsegmented bar [37], sacral dysgenesis [38, 76], and fused ribs [8, 28, 30, 55, 74, 91] have also been reported.
All patients except for one had at least one associated spinal cord lesion in this series. The most common associated lesion was a tight filum terminale (86 %). Some patients had more than one lesion. The rate of associated lesion in our previous publication was 85 %. Pang [72] found at least one unrelated tethering lesion in all lumbosacral and lower thoracic SCMs and in a much smaller number of cervical SCMs. When you look for an associated spinal cord lesion, the probability of finding it will be high. The reported accompanying spinal cord lesions are as follows: tight filum terminale [1, 4, 5, 7, 10, 15, 18, 21, 23, 24, 28–30, 33, 37, 38, 44, 51–54, 57–59, 62, 66, 70, 71, 73, 74, 78–81, 84, 89, 91, 92, 98, 100–102, 104], myelomeningocele [5, 9, 10, 15, 32, 39, 40, 44, 51, 54, 55, 69, 70, 72, 86, 91], lipomyelomeningocele [47, 53, 72, 84, 93, 105, 106], intradural lipoma [3, 4, 30, 34, 49, 54, 55, 57, 59, 70, 72, 76, 80, 91, 92], hemimyelomeningocele [9, 44, 73, 82, 85, 89], meningocele [9, 22, 66, 67, 71, 72, 85, 91, 92], dermoid [3, 15, 20, 42, 50, 54, 55, 59, 70, 72, 78, 91], epidermoid cyst [15, 59, 72, 91], dermal sinus [1, 3, 13, 15, 22, 26, 51, 53, 54, 59, 67, 69, 72, 73, 84, 91], neurenteric cyst [1, 3, 4, 11, 13, 14, 42, 44, 55, 59, 72, 92, 94], myelomeningocele manqué [3, 29, 72–74, 91], teratoma [20, 24, 37, 44, 50, 58, 61, 65, 66, 68, 69, 89, 96, 97, 100], anterior sacral meningocele [21], intrasacral meningocele [52], arachnoid cyst and neurofibroma [55], and Wilms' tumor [31, 90]. The incidence of SCMs in patients with a myelomeningocele has been reported to be 78 % by Emery and Lendon [25]. The rate of SCMs in patients with spinal dysraphism was 5 % in the series reported by Pang [72]. It was 13 % in our previous experience. Campbell and coworkers [17] reported that the occurrence of SCMs in infants with open spinal dysraphism is 36 %. A type I SCM was more commonly associated with a myelomeningocele than a type II SSCM. The myelomeningocele is usually located below the bony septum [9, 10, 17, 40, 43, 44, 69, 72]. Asymmetric symptoms in the lower limbs caused by asymmetric hemicords are usually associated hemimyelomeningocele [72, 85]. In the series of Iskandar et al. [44], six patients had SCM at the level of myelomeningocele, and all six had hemimyelomeningocele, and the others had SCM above the neuroplacode. We found that SCM was at the same level in three and seven patients with myelomeningocele and hemimyelomeningocele, respectively. In only one patient, the bony septum was distal to myelomeningocele and was above the level of myelomeningocele in 22 patients.
Hydromyelia in varying degrees was present on MRI scans of 55 patients. Hydromyelia was considered as a part of radiological signs in tethered cord syndrome, and no surgical intervention was done for hydromyelia. Syringohydromyelia has also been described in patients with SCM [1, 10, 15, 22, 26, 28, 30, 40, 46, 47, 50, 53, 55, 70, 76, 80, 82, 84, 85, 89, 91–93, 101, 104]. Gan et al. [33] reported that there was no change in syringomyelia after untethering the spinal cord in 8 out 15 patients with a type I SCM. They concluded that syringomyelia in patients with a type I SCM very rarely contributes to the neurological symptoms, and the management of the syringomyelia should therefore be conservative at the first instance with regular monitoring.
Composite SCMs are uncommon [1, 4, 28, 51, 72, 78, 84, 86, 101, 103]. Six patients had composite SCMs. In two patients, the dermal sinus tract penetrated the dorsal dura and was connected to a fibrous median septum, attaching the median aspects of the both hemicords. A small bony septum, which had a conical shape and was covered with a dural sheath, was also noted ventrally. This results from two separate loci of ectoendodermal adhesions and endomesenchymal tracts, according to Pang [72]. A similar case has also been reported by Vaishya and Kumarjain [101]. Four patients had two different SCMs located in tandem, separated by a normal spinal cord. Two patients had types I and II SCMs, one had two type I SCMs, and the other one, two type II SCMs. Akay et al. [4] had a patient who had three splitting lesions at three different levels. Multiple accessory neurenteric canals may form two or more noncontiguous SCMs [4, 72, 101]. Mahapatra and Gupta [60] and Gupta and Mahapatra [35] propose a new subclassification of a type I SCM based on the location of the bony spur. In type Ia, the bony spur is in the center of dural cleft; in type Ib, located at the superior pole with no space above; in type Ic, located at the lower pole with a large duplicated spinal cord above; and in type Id, the bony spur is straddling the bifurcation with no space above or below the spur. They concluded that type Ia has the best prognosis, and type Id, the worst prognosis of the four subtypes of SCMs [35]. A smaller hemicord was almost always associated with an atrophic leg on the same side in cases with asymmetrical hemicords. However, all asymmetrical legs were not associated with a smaller hemicord. The hemicords are usually reunited after splitting but rarely do not rejoin [41]. In 11 of our patients, the hemicords did not rejoin after splitting. A bony spur situated dorsally in patients with SCM has been described by some authors [2, 18, 46, 102]. In my opinion, such cases should be a type II SCM, unless they have two different dural tubes [27].
Pang has indicated that the length of the split segment was greater in patients diagnosed after reaching 1 year of age than that in patients diagnosed before reaching 1 year of age. This difference was not found to be significant in the present series. The mean split length was greater in type I than that in type II SCMs. Pang [72, 74] has hypothesized that the neural tube is firmly transfixed by a rigid derivative of the type I endomesenchymal tract and that its subsequent ascent results in a long cleavage in the spinal cord. The upper level of septa was at T4, and the lowest was at S4. The majority of septa were localized at the lumbar region, as in other series [3, 4, 6, 7, 9, 15, 18, 22, 26, 28–30, 37, 38, 40, 45, 47, 50, 52–54, 59, 61, 62, 64, 65, 71, 79–82, 84, 89–91, 93, 97–102, 104–106]. Cervical and sacral locations are extremely rare [13–15, 21, 23, 28, 35, 46, 67, 72, 73, 101]. The conus medullaris was located below L2 in 122 patients (83 %) [3, 4, 18, 21, 23, 29, 30, 46, 53, 55, 64, 65, 76, 78, 81, 86, 98, 104].
Pang [73] found a ventral fibrous septum tethering the hemicords to the ventral dura in 11 (21 %) of 52 patients with a type II SCM, and four categories of ventral septa were noted: (1) pure ventral intradural septa (three patients), (2) complete dorsoventral intradural septa (three patients), (3) dorsoventral septa continuous with a dermal sinus tract (two patients), and (4) ventral or complete septa continuous with ventral intestinal bands causing intestinal malrotation or diverticulum (three patients). There was a ventral fibrous septum in only one of our patients. Pang [73] reported that the only another positive predictor of ventral tethering is the association of dermal sinus tract and intestinal malformations. He recommended a thorough exploration of the ventral aspects of the hemicords during surgery for a type II SCM.
Pathological examinations of specimens of median septa disclosed interesting results. Fetal renal tissue, lymphoid tissue, and tubular epithelia have been found within the septa in two cases of SSCM that were previously reported [11]. A teratoma within one of the hemicords was also detected in one of the patients. Blood vessels, muscle tissue, and dermoid cysts were microscopically present in the septa in other cases, as in the series reported by Pang et al. [74]. Pluripotential cells of the endomesenchymal tract could develop into a variety of tissues. Cases of SCM associated with Wilms' tumor [31, 90] and ectopic renal tissues [47, 110] have been reported. Poeze et al. [77] reviewed the literature up to 1999 and found that SCM was observed in 11.7 % of teratomas. Sharma et al. [89] found SCM in ten patients (37.3 %), and the majority of them were children (89 %). Teratomas should be taken into consideration in differential diagnosis of intramedullary lesions associated with SCM [65]. The whole spine must be examined for additional dysraphic lesions.
Immediate postoperative neurological deterioration, transient worsening of sensory function, cerebrospinal fluid leak, subcutaneous CSF collection, wound dehiscence, meningitis, urinary retention, and neuropathic pain in lower extremities have been encountered in patients following surgery, and they are usually transient [4, 28, 33, 51, 53–55, 59, 65, 72, 80, 91, 103]. Similar transient postoperative permanent deficits and deaths are very rare [11, 59]. Retethering of the spinal cord is uncommon [59, 80]. Regrowth of the bony spur has also been reported [34, 75]. In five patients with a type I SCM, retethering was developed, and a second surgery was needed. Regrowth of the bony spur was detected in one of the five patients. Retethering was never been seen in the patients with a type II SCM.
Conclusions
The female predominance was slightly more remarkable in type I SCMs than that in type II SCMs. The ages of patients with neurological deficits and orthopedic deformities were significantly older than those without deficits. The duration of symptoms was longer in the patients with neurological deficits and orthopedic deformities than that in those without deficits. These findings support the prophylactic surgery in patients with SCMs. Skin lesions are less common in patients with spina bifida cystica than in those without. The length of split segment was longer in type I SCM than type II SCM. The incidence of associated spinal cord lesions is very high in SCMs. Therefore, the whole spine should be screened by MRI, and the filum terminale should be explored at surgery, if a low-lying conus is present on MRI. Postoperative complications are not major and are usually manageable. All SCMs should be operated on by experienced surgeons. Patients with spinal deformity should also be closely followed-up after surgery.
References
Ackerman LL, Menezes AH, Follett KA (2002) Cervical and thoracic dermal sinus tracts. A case series and review of the literature. Pediatr Neurosurg 37:137–147
Akay KM, Izci Y, Baysefer A (2002) Dorsal bony septum: a split cord malformation variant. Pediatr Neurosurg 36:225–228
Akay KM, Izci Y, Baysefer A, Timurkaynak E (2004) Split cord malformation in adults. Neurosurg Rev 27:99–105
Akay KM, Izci Y, Baysefer A, Timurkaynak E (2005) Composite type of split cord malformation: two different types at three different levels: case report. J Neurosurg 102(4 Suppl):436–438
Akiyama K, Nishiyama K, Yoshimura J, Mori H, Fujii Y (2007) A case of split cord malformation associated with myeloschisis. Childs Nerv Syst 23:577–580
Al Kaissi A, Chehida FB, Ghachem MB, Grill F, Klaushofer K (2006) Progressive non-infectious anterior vertebral fusion, split cord malformation and situs inversus visceralis. BMC Musculoskelet Disord 5:94
Allen LM, Silverman RK (2000) Prenatal ultrasound evaluation of fetal diastematomyelia: two cases of type I split cord malformation. Ultrasound Obstet Gynecol 15:78–82
Andro C, Pecquery R, De Vries P, Forlodou P, Fenoll B (2009) Split cervical spinal cord malformation and vertebral dysgenesis. Orthop Traumatol Surg Res 95:547–550
Ansari S, Nejat F, Yazdani S, Dadmehr M (2007) Split cord malformation associated with myelomeningocele. J Neurosurg 107(4 Suppl):281–285
Bademci G, Evliyaoglu C, Keskil S (2005) Proximally situated osseous septum in complex spina bifida. Case report. J Neurosurg 102(1 Suppl):92–95
Bale PM (1973) A congenital intraspinal gastroenterogenous cyst in diastematomyelia. J Neurol Neurosurg Psychiatry 36:1011–1017
Basu PS, Elsebaie H, Noordeen MH (2002) Congenital spinal deformity: a comprehensive assessment at presentation. Spine (Phila Pa 1976) 27:2255–2259
Becker GW, Battersby RD (2005) Spinal neurenteric cyst presenting as recurrent midline sebaceous cysts. Ann R Coll Surg Engl 87:W1–W4
Birch BD, McCormick PC (1996) High cervical split cord malformation and neurenteric cyst associated with congenital mirror movements: case report. Neurosurgery 38:813–815
Borkar SA, Mahapatra AK (2012) Split cord malformations: a two years experience at AIIMS. Asian J Neurosurg 7:56–60
Bradford DS, Heithoff KB, Cohen M (1991) Intraspinal abnormalities and congenital spine deformities: a radiographic and MRI study. J Pediatr Orthop 11:36–41
Campbell LR, Dayton DH, Sohal GS (1986) Neural tube defects: a review of human and animal studies on the etiology of neural tube defects. Teratology 34:171–187
Chandra PS, Kamal R, Mahapatra AK (1999) An unusual case of dorsally situated bony spur in a lumbar split cord malformation. Pediatr Neurosurg 31:49–52
Chern JJ, Gordon AS, Naftel RP, Tubbs RS, Oakes WJ, Wellons JC 3rd (2011) Intradural spinal endoscopy in children. J Neurosurg Pediatr 8:107–111
Conti P, Tenenbaum R, Capozza M, Mouchaty H, Conti R (2010) Diastematomyelia and tumor in adults: report of two cases and literature review. Spine (Phila Pa1976) 35:E1438–E1443
Dias MS, Azizkhan RG (1998) A novel embryogenetic mechanism for Currarino’s triad: inadequate dorsoventral separation of the caudal eminence from hindgut endoderm. Pediatr Neurosurg 28:223–229
Düz B, Gocmen S, Secer HI, Basal S, Gönül E (2008) Tethered cord syndrome in adulthood. J Spinal Cord Med 31:272–278
Dwarakanath S, Suri A, Garg A, Mahapatra AK, Mehta VS (2005) Adult complex spinal dysraphism with situs inversus totalis: a rare association and review. Spine (Phila Pa 1976) 15:E225–E228
Elmaci I, Dagcinar A, Ozgen S, Ekinci G, Pamir MN (2001) Diastematomyelia and spinal teratoma in an adult. Case report. Neurosurg Focus 10:ecp2
Emery JL, Lendon RG (1973) The local cord lesion in neurospinal dysraphism myelomeningocele. J Pathol 110:83–96
Emmez H, Güven C, Kurt G, Kardes O, Dogulu F, Baykaner K (2004) Terminal syringomyelia: is it as innocent as it seems?—Case report. Neurol Med Chir (Tokyo) 44:558–561
Erşahin Y (2000) An unusual split cord malformation. Pediatr Neurosurg 32:109
Erşahin Y, Demirtaş E, Mutluer S, Tosun AR, Saydam S (1998) Split cord malformations: report of three unusual cases. Pediatr Neurosurg 24:155–159
Erşahin Y, Kitiş O, Oner K (2002) Split cord malformation in two sisters. Pediatr Neurosurg 37:240–244
Etus V, Ceylan S, Ceylan S (2003) Association of spondylocostal dysostosis and type I split cord malformation. Neurol Sci 24:134–137
Fernbach SK, Naidich TP, McLone DG, Leestma JE (1984) Computed tomography of primary intrathecal Wilms tumor with diastematomyelia. J Comput Assist Tomogr 8:523–528
Flitman HP, Stanek J, Hsu HW, de Courten-Myers GM (1999) Anomalous ependyma inducing split cord and meningomyelocele? J Child Neurol 14:473–477
Gan YC, Sgouros S, Walsh AR, Hockley AD (2007) Diastematomyelia in children: treatment outcome and natural history of associated syringomyelia. Childs Nerv Syst 23:515–519
Gupta DK, Ahmed S, Garg K, Mahapatra AK (2010) Regrowth of septal spur in split cord malformation. Pediatr Neurosurg 46:242–244
Gupta DK, Mahapatra AK (2006) Proposal for a new clinicoradiological classification of type I split-cord malformations: a prospective study of 25 cases. Pediatr Neurosurg 42:341–346
Guthkelch AN (1985) Diastematomyelia. In: Wilkins RH, Rengachary SS (eds) Neurosurgery, vol 3. McGraw-Hill, New York, pp 2058–2061
Hader WJ, Steinbok P, Poskitt K, Hendson G (1999) Intramedullary spinal teratoma and diastematomyelia. Case report and review of the literature. Pediatr Neurosurg 30:140–145
Hamasaki T, Makino K, Morioka M, Hasegawa S, Kurino M, Kuratsu J (2006) Histological study of paramedian dorsal root ganglia in an infant with split cord malformation. Case report. J Neurosurg 104:415–418
Has R, Yuksel A, Buyukkurt S, Kalelioglu I, Tatli B (2007) Prenatal diagnosis of diastematomyelia: presentation of eight cases and review of the literature. Ultrasound Obstet Gynecol 30:845–849
Higashida T, Sasano M, Sato H, Sekido K, Ito S (2010) Myelomeningocele associated with split cord malformation type I: three case reports. Neurol Med Chir (Tokyo) 50:426–430
Hilal SK, Marton D, Pollack E (1974) Diastematomyelia in children. Radiographic study of 34 children. Radiology 112:609–621
Hui H, Tao HR, Jiang XF, Fan HB, Yan M, Luo ZJ (2012) Safety and efficacy of 1-stage surgical treatment of congenital spinal deformity associated with split spinal cord malformation. Spine (Phila Pa 1976) 37:2104–2113
Humphreys RP, Hendrick EB, Hoffman HJ (1982) Diastematomyelia. Clin Neurosurg 23:436–456
Iskandar BJ, McLaughlin C, Oakes WJ (2000) Split cord malformations in myelomeningocele patients. Br J Neurosurg 14:200–203
Izci Y, Gönül M, Secer HI, Gönül E (2007) Aplasia cutis congenita: a rare cutaneous sign of split cord malformations. Int J Dermatol 46:1031–1035
Izci Y, Gurkanlar D, Gönül E (2007) An unusual type of split cord malformation. J Clin Neurosci 14:383–386
Jain D, Sharma MC, Sarkar C, Rishi A, Suri V, Garg A, Mahapatra AK (2007) Spinal teratoma with pulmonary differentiation: a report of rare case and review of literature. Fetal Pediatr Pathol 26:185–191
James CCM, Lassman LP (1958) Diastematomyelia. Arch Dis Child 33:536–539
Jankowitz BT, Albright AL (2007) Cutaneous manifestations of split cord malformations. Report of three cases. J Neurosurg 107(3 Suppl):240–243
Jindal A, Kansal S, Mahapatra AK (2000) Split cord malformation with partial eventration of the diaphragm. Case report. J Neurosurg 93(2 Suppl):309–311
Khandelwal A, Tandon V, Mahapatra AK (2011) An unusual case of 4 level spinal dysraphism: multiple composite type 1 and type 2 split cord malformation, dorsal myelocystocele and hydrocephalous. J Pediatr Neurosci 6:58–61
Kiliçkesmez O, Barut Y, Tasdemiroglu E (2004) Expanding occult intrasacral meningocele associated with diastematomyelia and multiple vertebral anomalies. Case report. J Neurosurg 101(1 Suppl):108–111
Kim SK, Chung YS, Wang KC, Cho BK, Choi KS, Han DH (1994) Diastematomyelia—clinical manifestation and treatment outcome. J Kor Med Sci 9(2):135–144
Kumar R, Bansal KK, Chhabra DK (2002) Occurrence of split cord malformation in meningomyelocele: complex spina bifida. Pediatr Neurosurg 36:119–127
Kumar R, Singh SN, Bansal KK, Singh V (2005) Comparative study of complex spina bifida and split cord malformation. Indian J Pediatr 72:109–115
Kutuk MS, Ozgun MT, Tas M, Poyrazoglu HG, Yikilmaz A (2012) Prenatal diagnosis of split cord malformation by ultrasound and fetal magnetic resonance imaging: case report and review of the literature. Childs Nerv Syst 28:2169–2172
Lee GY, Paradiso G, Tator CH, Gentili F, Massicotte EM, Fehlings MG (2006) Surgical management of tethered cord syndrome in adults: indications, techniques, and long-term outcomes in 60 patients. J Neurosurg Spine 4:123–131
Leung YL, Buxton N (2005) Combined diastematomyelia and hemivertebra: a review of the management at a single centre. J Bone Joint Surg Br 87:1380–1384
Mahapatra AK (2011) Split cord malformation—a study of 300 cases at AIIMS. 1990-2006. J Pediatr Neurosci 6(Suppl 1):S41–S45
Mahapatra AK, Gupta DK (2005) Split cord malformations: a clinical study of 254 patients and a proposal for a new clinical-imaging classification. J Neurosurg 103(6 Suppl):531–536
Maiti TK, Bhat DI, Devi BI, Sampath S, Mahadevan A, Shankar SK (2010) Teratoma in split cord malformation: an unusual association: a report of two cases with a review of the literature. Pediatr Neurosurg 46:238–241
Mastroyianni SD, Kontopoulos E (2002) Split-cord malformation in a girl with Angelman syndrome: a mere coincidence? Am J Med Genet 111:57–60
McMaster MJ (1984) Occult intraspinal anomalies and congenital scoliosis. J Bone Joint Surg Am 66:588–601
Moriya J, Kakeda S, Korogi Y, Soejima Y, Urasaki E, Yokota A (2006) An unusual case of split cord malformation. AJNR Am J Neuroradiol 27:1562–1564
Mut M, Shaffrey ME, Bourne TD, Jagannathan J, Shaffrey CI (2007) Unusual presentation of an adult intramedullary spinal teratoma with diplomyelia. Surg Neurol 67:190–194
Muthukumar N, Arunthathi J, Sundar V (2000) Split cord malformation and neurenteric cyst—case report and a theory of embryogenesis. Br J Neurosurg 14:488–492
Myles LM, Steers AJ, Minns R (2002) Cervical cord tethering due to split cord malformation at the cervico-dorsal junction presenting with self-mutilation of the fingers. Dev Med Child Neurol 44:844–888
Naik V, Mahapatra AK, Gupta C, Suri V (2010) Complex split cord malformation with mediastinal extension of a teratoma and simultaneous ventral and dorsal bony spur splitting the cord. Pediatr Neurosurg 46:368–372
Ozer H, Yüceer N (1999) Myelomeningocele, dermal sinus tract, split cord malformation associated with extradural teratoma in a 30-month-old girl. Acta Neurochir (Wien) 141:1123–1124
Ozturk E, Sonmez G, Mutlu H, Sildiroglu HO, Velioglu M, Basekim CC, Kizilkaya E (2008) Split-cord malformation and accompanying anomalies. J Neuroradiol 35:150–156
Pallatroni HF, Ball PA, Duhaime AC (2004) Split cord malformation as a cause of tethered cord syndrome in a 78-Year-old female. Pediatr Neurosurg 40:80–83
Pang D (1992) Split cord malformation: part II: clinical syndrome. Neurosurgery 31:481–500
Pang D (2001) Ventral tethering in split cord malformation. Neurosurg Focus 10:e6
Pang D, Dias MS, Ahab-Barmada M (1992) Split cord malformation: part I: a unified theory of embryogenesis for double spinal cord malformations. Neurosurgery 31:451–480
Pang D, Parrish RG (1983) Regrowth of diastematomyelic bone spur after extradural resection. Case report. J Neurosurg 59:887–890
Parmar H, Patkar D, Shah J, Maheshwari M (2003) Diastematomyelia with terminal lipomyelocystocele arising from one hemicord: case report. Clin Imaging 27:41–43
Poeze M, Herpers MJ, Tjandra B, Freling G, Beuls EA (1999) Intramedullary spinal teratoma presenting with urinary retention: case report and review of the literature. Neurosurgery 45:379–385
Porensky P, Muro K, Ganju A (2007) Adult presentation of spinal dysraphism and tandem diastematomyelia. Spine J 7:622–626
Proctor MR, Bauer SB, Scott RM (2000) The effect of surgery for split spinal cord malformation on neurologic and urologic function. Pediatr Neurosurg 32:13–19
Proctor MR, Scott RM (2000) Long-term outcome for patients with split cord malformation. Neurosurg Focus 10:e5
Quinones-Hinojosa A, Gadkary CA, Mummaneni PV, Rosenberg WS (2004) Split spinal cord malformation in an elderly patient: case report. Surg Neurol 61:201–203
Rowley VB, Johnson AJ (2009) Lumbar split cord malformation with lateral hemimyelomeningocele and associated Chiari II malformation and other visceral and osseous anomalies: a case report. J Comput Assist Tomogr 33:923–923
Rustamzadeh E, Graupman PC, Lam CH (2006) Basicranial diplomyelia: an extension of the split cord malformation theory. Case report. J Neurosurg 104(5 Suppl):362–365
Salunke P, Kovai P, Malik V, Sharma M (2011) Mixed split cord malformation: are we missing something? Clin Neurol Neurosurg 113:774–778
Sato K, Yoshida Y, Shirane R, Yoshimoto T (2002) A split cord malformation with paresis of the unilateral lower limb: case report. Surg Neurol 58:406–409
Schijman E (2003) Split spinal cord malformations: report of 22 cases and review of the literature. Childs Nerv Syst 19:96–103
Schropp C, Sörensen N, Collmann H, Krauss J (2006) Cutaneous lesions in occult spinal dysraphism—correlation with intraspinal findings. Childs Nerv Syst 22:125–131
Scotti G, Musgrave MA, Harwood-Nash DC, Fitz CR, Chuang SH (1980) Diastematomyelia in children: metrizamide and CT metrizamide myelography. AJR Am J Roentgenol 135:1225–1232
Sharma MC, Jain D, Sarkar C, Suri V, Garg A, Singh M, Mahapatra AK, Sharma BS (2009) Spinal teratomas: a clinico-pathological study of 27 patients. Acta Neurochir (Wien) 151:245–252
Sharma MC, Sarat Chandra P, Goel S, Gupta V, Sarkar C (2005) Primary lumbosacral Wilms tumor associated with diastematomyelia and occult spinal dysraphism. A report of a rare case and a short review of literature. Childs Nerv Syst 21:240–243
Sinha S, Agarwal D, Mahapatra AK (2006) Split cord malformations: an experience of 203 cases. Childs Nerv Syst 22:3–7
Skalej M, Duffner F, Stefanou A, Petersen D (1999) 3D spiral CT imaging of bone anomalies in a case of diastematomyelia. Eur J Radiol 29:262–265
Snelling CM, Ellis PM, Smith RM, Rossiter JP (2008) Lipomatous lumbar mass with an attached digit and associated split cord malformation. Can J Neurol Sci 35:250–254
Soni TV, Pandya C, Vaidya JP (2004) Split cord malformation with neurenteric cyst and pregnancy. Surg Neurol 61:556–558
Suh SW, Sarwark JF, Vora A, Huang BK (2001) Evaluating congenital spine deformities for intraspinal anomalies with magnetic resonance imaging. J Pediatr Orthop 21:525–531
Suri A, Ahmad FU, Mahapatra AK, Mehta VS, Sharma MC, Gupta V (2006) Mediastinal extension of an intradural teratoma in a patient with split cord malformation: case report and review of literature. Childs Nerv Syst 22:444–449
Tsitsopoulos P, Rizos C, Isaakidis D, Liapi G, Zymaris S (2006) Coexistence of spinal intramedullary teratoma and diastematomyelia in an adult. Spinal Cord 44:632–635
Tubbs RS, Salter EG, Oakes WJ (2007) Split spinal cord malformation. Clin Anat 20:15–18
Tubbs RS, Wellons JC 3rd, Oakes WJ (2005) Split cord malformation and situs inversus totalis: case report and review of the literature. Childs Nerv Syst 21:161–164
Uzüm N, Dursun A, Baykaner K, Kurt G (2005) Split-cord malformation and tethered cord associated with immature teratoma. Childs Nerv Syst 21:77–80
Vaishya S, Kumarjain P (2001) Split cord malformation: three unusual cases of composite split cord malformation. Childs Nerv Syst 17:528–530
van Aalst J, Beuls EA, Vles JS, Cornips EM, van Straaten HW (2005) The intermediate type split cord malformation: hypothesis and case report. Childs Nerv Syst 21:1020–1024
Venkataramana NK (2006) Split cord malformations. J Pediatr Neurosci 1:5–9
Vitarbo EA, Sultan A, Wang D, Morcos JJ, Levi AD (2005) Split cord malformation with associated type IV spinal cord perimedullary arteriovenous fistula. Case report. J Neurosurg Spine 3:400–404
Yamada S, Mandybur GT, Thompson JR (1996) Dorsal midline proboscis associated with diastematomyelia and tethered cord syndrome. Case report. J Neurosurg 85:709–712
Yiğit H, Ozdemir HM, Yurduseven E (2013) Duplication of spine with hemi-lipomyelomeningocele. Eur Spine J (in press)
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Erşahin, Y. Split cord malformation types I and II: a personal series of 131 patients. Childs Nerv Syst 29, 1515–1526 (2013). https://doi.org/10.1007/s00381-013-2115-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00381-013-2115-7