
Abstract
In this study, changes in plant diversity and aboveground biomass, soil chemical properties, microbial biomass and respiration, microbial composition, and microbial N-cycling potential (represented by the abundance of genes involved in N reaction) were studied after 3 years of urea fertilization (0, 25, 50, and 100 kg N ha−1 year−1) in a semiarid grassland in China. The microbial composition and N-cycling genes were determined using metagenome sequencing. Urea fertilization significantly decreased soil bacterial diversity, possibly through its negative effect on plant diversity, whereas it increased fungal diversity, and microbial biomass and respiration through enhancing aboveground biomass production with increases in the C input into the soil. However, above the threshold N rate of 50 kg N ha−1 year−1, microbial biomass and respiration decreased probably because of a strong N inhibitory effect on aboveground biomass. Further, urea fertilization increased the gene abundances of narH, nrfA, nirB, and napA, which are involved in dissimilatory nitrate reduction, and those of nifH and nifD, involved in N2 fixation, gdh, involved in organic N decomposition, and glnA, involved in glutamine synthesis and ureC. These findings suggested that urea addition has a positive effect on N-turnover potential. Burkholderiales and Rhizobiales play an important role in soil N cycling. Changes in plant community (diversity and biomass) were responsible for the shift in microbial diversity, biomass, and respiration, whereas alterations in inorganic N levels (exchangeable NH4+ and NO3−) potentially affected microbial N cycling. Our results show that N-mediated environmental stresses can play an important role in microbial diversity and functions, which appear to be mediated largely by plant–soil interactions.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Atmospheric nitrogen (N) deposition caused by fossil fuel combustion and by the use of fertilizers has increased three- to fivefold in the past century (Galloway et al. 2008). Intensified N deposition has drastic impacts on terrestrial ecosystems, including soil acidification, loss of biodiversity, and eutrophication (Lu et al. 2014; Stevens et al. 2004). The additional N has potentially important impacts on not only the plant communities, but also the soil microbial communities. Understanding how these additional N inputs affect above- and belowground communities is becoming increasingly important within the context of the global climate change (Liu and Greaver 2010). Not all ecosystems respond in the same way to N deposition, especially in semiarid grassland, where species diversity and composition usually change relatively rapidly (Zhang et al. 2016). The effect of N deposition on semiarid grassland is not fully known, despite the increasing research on this type of ecosystem (Lu et al. 2011).
Soil microbes participate in soil biogeochemical processes and play an essential role in soil carbon (C) and N turnover (Bardgett et al. 2008; Nannipieri et al. 2018; Duan et al. 2018). However, the mechanisms underlying soil microbial feedback in response to N deposition remain unclear. The effect of N enrichment on microbial activity in grassland soil has been reported to be positive (Zhang et al. 2014), negative (Zeng et al. 2016), or neutral (Alster et al. 2013), and is supposed to be driven by various forces. Because the majority of terrestrial ecosystems are N-limited (LeBauer and Treseder 2008), N deposition is expected to enhance aboveground plant growth and thus, primary productivity (DeMalach 2018; Harpole et al. 2011). Therefore, plant-mediation hypotheses predict that N enrichment increases microbial respiration as a result of increased C input into the soil via plant roots, due to enhanced photosynthesis (Zang et al. 2017). However, this effect turns negative when the amount of N added exceeds a threshold, because excess N reduces the allocation of photosynthetic C to the roots (Nakaji et al. 2001). In addition, some studies attribute changes in microbial activities to alternations in soil N availability and pH. Ramirez et al. (2010) reported that decreases in microbial respiration induced by N fertilizer, regardless of soil type and N form, were mainly a direct result of the increase in soil N availability, rather than effects caused by plant community. In contrast, Wang et al. (2018) reported that decreases in microbial activity caused by N fertilizer were most likely mediated via its negative effect on soil pH. N fertilization also causes alterations in soil microbial composition through altering the plant community and soil properties. van der Heijden et al. (2008) reported an increase in microbial diversity because of increased plant diversity, which increased the diversity of C resources, and Fierer et al. (2007) observed a decrease in the abundance of oligotrophs resulting from a decrease in the soil C:N ratio. Overall, the influences of N enrichment on soil microbial activity and diversity may be caused by direct effects of N as a nutrient or by indirect changes in soil and plant properties. Exploring the contribution of each of these factors to the microbial community is needed for understanding the mechanisms underlying these ecological responses and feedbacks.
N cycling is one of the most important soil biogeochemical processes that may constrain plant growth in terrestrial ecosystems (Nannipieri and Paul 2009). The soil N cycle includes several processes, mainly driven by soil microorganisms, and these microbial drivers can be monitored by detecting process-related genes (Vitousek et al. 1997). The abundance of genes involved in N cycling can be used to assess the N-turnover potential in soils (Nelson et al. 2015). In grassland ecosystems, different responses of N-related genes and the associated microbes to N fertilization have been reported. High N addition significantly increased the abundance of the amoB gene of ammonia-oxidizing bacteria due to the enhanced N substrate, whereas it had no impact on the nirS and nirK abundances in an Alpine grassland soil (Tian et al. 2014). Recent studies have suggested that N fertilization increased amoB abundance, but not amoA abundance in temperate grassland soils (Shen et al. 2011), whereas it decreased nifH, nirS, and nosZ abundances due to suppression of acidification and salt toxicity (Ning et al. 2015). Increases in the abundances of nirK- and nirS-type denitrifiers occurred in soils following N addition (Jung et al. 2011). In general, N-cycling genes respond to N addition, and changes in N-cycling potential induced by N fertilizer are inconsistent and likely site-dependent.
This study aimed to determine how N enrichment affects soil microbial diversity and N-cycling potential. To this end, we measured plant diversity and aboveground biomass, soil chemical properties, and microbial biomass, respiration, composition, and genes involved in N reactions in a semiarid grassland in China that had received 3 years of urea fertilizer. Soil microbial composition and the abundances of N-cycling genes were determined using shotgun metagenome sequencing. We hypothesized that (i) urea fertilization would reduce soil bacterial and fungal diversity, and increase microbial biomass and respiration below a certain threshold N rate, above which microbial biomass and respiration would decrease because of the inhibitory effect of excess N on plants; (ii) urea fertilization would increase the N-cycling potential owing to the increase in soil N availability; and (iii) changes in plant community and inorganic N levels caused by N enrichment would be responsible for the variations in soil microbial community and N-cycling potential.
Materials and methods
Site description and experimental design
This study was conducted at the Dunshan field-experimental site at the Ansai Water and Soil Conservation Research Station (36° 51′ 30″ N, 109° 19′ 23″ E) on the northern Loess Plateau of China. The climate in this region is semiarid, with a mean annual precipitation of 500 mm. The mean annual temperature is 8.8 °C, with a minimum of 6.2 °C in January and a maximum of 37.5 °C in August. The soil, derived from wind-blown deposits, is classified as a Huangmian soil (Calcaric Cambisol in the FAO classification).
An N fertilization experiment was established in a grassland dominated by the grass species Bothriochloa ischaemum, Stipa przewalskyi, Stipa grandis, and Leymus secalinus. Four N treatments, with three replicated plots per treatment, were established in 2014: 0 (N0), 25 (N25), 50 (N50), and 100 (N100) kg N ha−1 year (y)−1. Each plot was 3 × 2 m in size and plots were separated by 1-m walkways. Urea was dissolved in deionized water, and the amounts of added N were in line with the global N-deposition rates for grassland reported by Bobbink et al. (2010). Urea fertilizer was applied in three split applications at the beginning of June, July, and August.
Sampling and soil chemical analysis
Plant and soil samples were collected in September 2017. Soil samples were collected from the top 20 cm of the soil profile using an auger (5 cm in diameter and 20 cm long) after the litter layer was removed. Six soil cores were collected at randomly selected locations in each plot and were pooled. Roots, litter, and stones were removed, and the pooled samples were sieved through a 2-mm mesh. The sieved samples were divided into three subsamples. One was immediately frozen at − 80 °C for DNA analysis, another was stored at 4 °C for the measurement of microbial biomass, and the third was air-dried for physicochemical analyses. Soil pH was determined by an automatic titrator (Metrohm 702, Switzerland) in suspensions of 1:2.5 soil/water.
The percentage cover of each plant species was estimated visually in each plot (Table S1). Species richness was defined as the number of different species. The Shannon–Wiener index (H = –ΣPilnPi) was used to estimate the diversity of the plant communities, where Pi is the ratio of the number of individuals of each species to the total number of individuals of all species. Aboveground vegetation was sampled by clipping individuals of all plant species at the soil surface, and was dried at 60 °C for 36 h for biomass determination. The soil organic C (OC) content was determined using the Walkley–Black method (Nelson and Sommers 1982), total N (TN) content was determined using the Kjeldahl method (Bremner and Mulvaney 1982), and available P (AP) content was measured by the Olsen method (Olsen and Sommers 1982). The exchangeable NH4+-N and NO3−-N contents were determined after extraction with 2 M KCl for 18 h by colorimetry on an Alpkem Autoanalyzer (OI Analytics, College Station, TX, USA).
Microbial biomass and respiration
Microbial biomass C (MBC) and N (MBN) were determined using a chloroform-fumigation extraction method (Vance et al. 1987). Briefly, 25 g of the oven-dry equivalent of field-moist soil was fumigated with CHCl3 at 25.8 °C for 24 h. The soil was added to 100 ml of 0.5 M potassium sulfate and shaken at 200 rpm for 1 h, and then filtered after fumigant removal. Another 25 g of non-fumigated soil was simultaneously extracted. The OC and TN contents of the extracts were determined using a Liqui TOCII analyzer (Elementar Analysensystem, Hanau, Germany). MBC and MBN concentrations were corrected for unrecovered biomass using extraction efficiency factors of 0.45 and 0.54, respectively (Beck et al. 1997; Brooks et al. 1985).
Soil respiration was measured using the method of Zhong et al. (2015). Briefly, 1 kg of fresh top soil collected as described above was transferred into pre-weighed PVC containers (20 cm diameter and 10 cm length) that were fitted with filter paper on the bottom. All samples were adjusted to 50% water-holding capacity and incubated at 25 °C, and the soil moisture was held constant throughout the incubation. Soil respiration (SR) was measured every 2 days over a 60-day incubation period using an LI-8100A soil respiration analyzer (Li-Cor Biosciences, Lincoln, NE USA).
DNA extraction, library construction, and metagenome sequencing
DNA was extracted from the 0.5 g homogenized soil samples using a Power Soil DNA kit (MoBiol Laboratories, Solana Beach, CA, USA), according to the manufacturer’s instruction. DNA concentration and purity (A260/A280) were determined using a TBS-380 fluorometer (TurnerBioSystems, CA, USA) and a NanoDrop ND-1000 spectrophotometer (NanoDrop, Wilmington, DE, USA), respectively. DNA quality was evaluated by 2% agarose gel electrophoresis. A total of 12 soil samples (three replicates for each N treatment) were DNA extracted for metagenomic sequencing.
The DNA was sheared into fragments of approximately 300 bp using an M220 Focused-ultrasonicator (Covaris, Woburn, MA, USA). Then, a paired-end library was constructed using a TruSeqTM DNA Sample Prep Kit (Illumina, San Diego, CA, USA). Adapters containing the full complement of the sequencing primer hybridization sites were ligated to the blunt end of the fragments. Paired-end sequencing was conducted on an Illumina X-Ten PE150 at Hengchuang Bio-Pharm Technology (Shenzhen, China) according to the manufacturer’s instructions (www.illumina.com). The metagenome sequences have been deposited in the NCBI database under accession number SRP149201.
Sequence quality control and genome assembly
Sequence quality control involved the removal of sequencing adapters, and quality and length filtering (Vestergaard et al. 2017). The 3′- and 5′-adaptors were trimmed using SeqPrep (https://github.com/jstjohn/SeqPrep). Reads with a minimum quality score 20 and a minimum length of 50 bp were maintained for analysis. In total, 128.5 GB of clean reads were obtained. The clean reads were assembled using MEGAHIT (https://github.com/voutcn/megahit). Contigs longer than 300 bp were selected as the final assembly result and were used for gene prediction and annotation.
Gene prediction, taxonomy, and functional assignment
The open reading frames (ORFs) of contigs and annotations in each sample were predicted with MetaGene Annotator (Noguchi et al. 2006). Predicted ORFs longer than 200 bp were translated into amino acid sequences via NCBI ORF finder (Li and Durbin 2009). All predicted genes with a 95% sequence identity (90% coverage) were clustered using CD-HIT (Fu et al. 2012) (http://www.bioinformatics.org/cd-hit/), the longest sequences from each cluster were selected as representative sequences to a construct non-redundant gene catalog. Clean reads were mapped to the representative sequences with 95% identity using SOAPaligner (Li et al. 2008) (http://soap.genomics.org.cn/), and gene abundances in each sample were evaluated. Representative sequences of the non-redundant gene catalog were aligned to the NCBI NR database with an e-value cutoff of 1e−5 using BLASTP (Version 2.2.28+, http://blast.ncbi.nlm.nih.gov/Blast.cgi) for taxonomic annotations. Bacterial and fungal richness were estimated by the number of observed species, and diversity was estimated by the Shannon–Wiener index (H = –ΣPilnPi). KEGG annotation was conducted using BLASTP (Version 2.2.28+) against the Kyoto Encyclopedia of Genes and Genomes database (http://www.genome.jp/keeg/), using an e-value cutoff of 1e−5. Functional annotation of genes was conducted in KOBAS 2.0 (Kanehisa et al. 2016) (KEGG Orthology Based Annotation System). The total number of sequences was normalized to 1 million per sample for further analysis.
Data analysis
One-way analysis of variance (ANOVA) followed by a post hoc test for multiple comparisons was used to evaluate differences between the four treatments and these differences were significant for P < 0.05. Principal coordinate analysis (PCoA) was used to assess differences in plant compositions and microbial communities under the various N treatments based on Bray–Curtis distances, and significance was tested by analysis of similarity (ANOSIM). Partial least squares path modeling (PLS-PM) (Kou et al. 2017; Sanchez 2013) was used to gain a mechanistic understanding of how soil and plant properties mediated alterations in microbial biomass, respiration, and composition under N enrichment conditions. PLS-PM is a data analysis approach for studying observed variables that can be summarized by the use of a latent variable and assumes that linear relationships exist between latent variables (Sanchez 2013). The latent variables in our PLS-PM analysis included bacterial composition, fungal composition, and N-cycling genes. Each latent variable included manifest variables, for example, bacterial composition included the relative abundances of the microbial taxa, and N-cycling genes included the abundances of N-cycling genes that changed significantly under N addition. The first principal components (PC1) of principal component analysis (PCA) (Fig. S1) for the composition of bacterial and fungal communities and N-cycling genes were used in the PLS-PM. Each variable has a relative contribution degree shown in arrow in path diagram. Path coefficients (representing the direction and strength of the linear relationships between latent variables) and explained variability (R2) are estimated in models. Models with different structures are evaluated using the goodness of fit (GOF) statistic, a measure of their overall predictive power, and a GOF > 0.7 is an acceptable value for a PLS partial model (Sanchez 2013). The models were constructed using the function “inner plot” in the R package (plspm). Pearson correlation coefficients were used to test relationships between individual properties, including plants, soils, microbial community, and N-cycling genes. ANOVA, PCoA, PCA, and Pearson’s correlation were conducted with the vegan package in R (version 3.0.2; http://www.r-project.org/).
Results
Plant and soil properties under N fertilization
Three-year urea fertilization significantly improved the aboveground biomass, whereas it lowered plant diversity (Table 1). Specifically, aboveground biomass and soil OC increased with the rate of N addition, peaked at N50, and then decreased. Plant richness and diversity decreased with increasing levels of N addition. Soil TN, exchangeable NH4+-N and NO3−-N contents increased with increasing levels of N addition, whereas pH showed an opposite trend and ranged from 8.46 to 7.02. AP had no response to N additions. The N100 treatment resulted in the lowest C:N ratio.
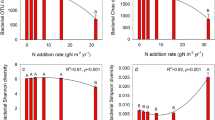
Microbial activity and diversity under N fertilization
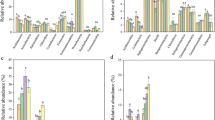
Three-year urea fertilization significantly enhanced microbial biomass, respiration, and fungal richness and diversity, whereas it lowered bacterial richness and diversity (Table 2). Specifically, MBC and MBN, microbial respiration, and fungal richness and diversity increased with the amount of N added, reaching the highest values at N50, and then decreased. Bacterial richness and diversity decreased with increasing levels of N addition. Urea fertilization strongly affected microbial composition. As shown in Fig. 1, Proteobacteria was the most abundant bacterial phylum across the treatments, accounting for 40.2% of all taxa on average, followed by Firmicutes (10.6%), Acidobacteria (10.9%), and Actinobacteria (8.8%) (Fig. 1a). Acidobacteria and Alphaproteobacteria increased with the amount of N added (Fig. 1b), whereas Actinobacteria exhibited an opposite trend. Interestingly, bacterial taxa belonging to the same group did not always respond in a similar manner. For example, Acidimicrobiia, with relatively low abundance, and other bacterial taxa within the phylum Actinobacteria responded oppositely to fertilization.

Relative abundance of bacterial phyla (a) and classes (b) under urea fertilization. Values are means ± standard error (n = 3). Different letters indicate significant differences (P < 0.05) between the treatments. ns, not significant. Only the taxa with abundance > 1% was shown
Ascomycota was the dominant fungal phylum (49.8%), followed by Basidiomycota (12.7%), Glomeromycota (1.8%), and Zygomycota (1.6%) (Fig. 2a). The relative abundance of Ascomycota increased, whereas the abundance of Glomeromycota decreased with increasing levels of N addition. Taxonomical shifts were significant in the classes Sordariomycetes and Glomeromycota (Fig. 2b). PCoA (Fig. 3) and ANOSIM (Table S2) clearly indicated large shifts in the bacterial and fungal community compositions with increasing levels of N addition, and changes in bacterial and fungal community composition were similar to those in the plant community.

Relative abundance of fungal phyla (a) and classes (b) under urea fertilization. Values are means ± standard error (n = 3). Different letters indicate significant differences (P < 0.05) between the treatments. ns, not significant. Only the taxa with abundance > 1% was shown

Ordination of principal coordinate analysis (PCoA) based on the Bray–Curtis distance for the compositions of bacterial (a), fungal (b), and plant (c) communities. Bacterial and fungal community was calculated based on the abundance of the species, and plant (c) community was calculated based on the cover of species in Table S1
Effects of N fertilization on microbial N processes and related gene families
In total, 9 pathways and 33 gene families involved in N reaction were investigated in this study (Table 3). The illustration of the genes investigated in N cycling is shown in Fig. 4, and detailed functional descriptions are presented in Table S3. Overall, urea addition increased the gene abundances of amoA and amoB involved in nitrification, narH, nrfA, nirB, and napA involved in dissimilatory nitrate reduction, nifH and nifD involved in N2 fixation, gdhA and gdh2 involved in organic N decomposition, glnA involved in glutamine synthesis, and ureC involved in urea hydrolysis. In contrast, the gene abundances of nirS and nirK involved in denitrification and hzsA involved in anammox decreased with N added.

An overall schema illustrating the N-cycling processes and the functional genes in soil grassland ecosystem
At the taxonomical level (Fig. 5), nearly all microbial N genes belonged to Burkholderiales and Rhizobiales. Organic N decomposition was taxonomically the most diverse process, with only five orders with abundances > 3%, but 520 species in total. N-cycling processes were driven by different microbial species (Table S4), including Methyloversatilis discipulorum and Nitrospira moscoviensis accounting for nitrification, Chthoniobacter flavus for denitrification, Candidatus Solibacter for dissimilatory nitrate reduction, Chthoniobacter flavus for anammox, and Burkholderiales bacterium for N2 fixation. Interestingly, the ammonia-oxidizers participating in nitrification were dominated by Nitrosospirales and Nitrososphaerales (Fig. S2). The relative abundance of Nitrosospirales increased, whereas that of Nitrososphaerales decreased, with increasing amount of N added.

Relative abundances of the microbial groups involved in N cycling in the a N0, b N25, c N50, and d N100 treatments. Only the orders with relative abundances > 3% are presented. The numbers above each bar indicate the number of species in the corresponding processes
Dependence of microbial communities and N-cycling genes on environmental variables
PLS-PM was implemented to reveal possible pathways influencing the microbial community and N-cycling genes following N addition (Fig. 6, Table S5). This analysis provided the best fit to our data according to the respective indices of model fit (GOF = 0.73), and the model accounted for 96% of the variation in pH, 88% and 91% in NO3−-N and exchangeable NH4+-N, 95% and 82% in plant diversity and aboveground biomass, and 97%, 77%, and 88% in bacterial composition, fungal composition, and N-cycling genes, respectively. Overall, N addition significantly affected the compositions both plant and microbial communities due to increased NO3−-N and exchangeable NH4+-N and decreased pH. Although a significant difference occurred in soil pH among N treatments and its changes explained variations of bacterial composition and N-cycling genes, it was not taken into account in the evaluation of the relationships among the soil and microbial communities because of the narrow variation (8.46–7.02). The decrease in plant diversity led to a reduction in bacterial diversity. In addition, alterations in the plant community resulted in concomitant shifts in bacterial and fungal community compositions (Fig. 3). Increases in inorganic N contents were mainly responsible for altering the N-cycling genes. Other variables were not of great significance on their own, but clearly improved the model when incorporated together. Correlations between each of the microbial properties and environmental factors further identified the effects of urea-induced changes in plant and soil properties on microbial community (Table S6). For example, bacterial diversity was positively correlated with plant diversity. MBC and MBN and microbial respiration were positively correlated with OC and aboveground biomass. Exchangeable NH4+-N and NO3−-N contents were positively correlated with the abundances of Acidobacteria, Alphaproteobacteria, Bacteroidete, Nitrosospira, and amoA, amoB, narH, nrfA, nirB, napA, nifD, gdh, ureC, and glnA, where they were negatively correlated with Actinobacteria and Nitrososphaera abundances.

Path analysis diagrams using partial least squares path modeling (PLS-PM) for the effect of urea fertilizer addition on the plant-soil-microbe system. Red arrows indicate a significantly positive effect, while blue indicates a significantly negative effect (P < 0.05). Gray dashed arrows indicate no significant effect (P > 0.05). Numbers are the affecting coefficients. Percentages close to variables refer to the variance accounted for by the model (R2)
Discussion
Effect of urea fertilizer on microbial diversity
As expected, urea fertilization decreased bacterial diversity, which was consistent with a previous study on grassland reporting a decrease in microbial diversity following N enrichment (Zeng et al. 2016). The decline in bacterial diversity may be related to the decrease in plant diversity due to the expansion of nitrophilous species and competitive exclusion by increased N availability (Bobbink et al. 2010; Hong et al. 2017). In our study, graminaceae and compositae species in the upper layer of the community, such as Artemisia capillaris and Phragmites australis, expanded significantly with increasing N amount, whereas species in the lower layer of the community, such as Potentilla bifurca and Oxytropis bicolor, declined or even disappeared (Table S1). This shift caused a shading effect of the upper-layer grasses on the lower-layer grasses, resulting in the loss of some species with weak light competitiveness and thus, reduced aboveground species diversity (DeMalach et al. 2016; Lamb et al. 2009). The positive correlation between plant and bacterial diversity indicated that they may be affected jointly by N enrichment (Table S6) and supported the theory that plant diversity enhances the diversity of soil microbes by increasing the range of food resources available (van der Heijden et al. 2008). Urea addition decreased soil pH by 0.24–1.4 units, a decrease in magnitude similar to findings by Chen et al. (2018) in a nearby region of the semiarid grassland evaluated in this study. The limited response of pH to N addition might be associated with the high buffering capacity of calcareous soil to pH reduction (Yang et al. 2011) and the dry soil conditions.
In contrast to the decrease in plant diversity with increasing levels of N addition, fungal diversity significantly increased with modest N enrichment, but decreased under excess N. This qualitative divergence between plant and fungal diversity along the N addition gradient resulted in the lack of an association between them. Our result contradicted the strong plant–fungal richness relationship observed in a previous study (Yang et al. 2017), but was in agreement with a report by Chen et al. (2018), who found no significant relationship between plant diversity and fungal diversity in semiarid grassland. The uncoupling of plant and fungal diversity following N enrichment indicated that the close association between plant and fungal diversity does not persist in soils under N-enriched conditions. In addition, we found a positive association between fungal diversity and OC content. We observed that while N addition decreased plant diversity, it increased plant productivity, which would have resulted in increases in C pools available to the soil fungi, thus favoring fungal richness (Whittaker 2006). The positive effect of N fertilization on fungal diversity contrasted with studies that found negative (Zhou et al. 2016) or neutral (Leff et al. 2015) impacts of N addition on grassland fungal diversity, but was consistent with the results of a study carried out in N-deficient soils (Chen et al. 2018). Our results highlight the N-limited nature of the semiarid grassland ecosystem, and that responses of fungal diversity to N enrichment can be ecosystem-specific. In contrast to the significant correlation between pH and bacterial diversity, the weak relation between pH and fungal diversity indicated that N fertilization-induced soil acidification has a greater impact on bacteria than on fungi, which have a wider pH range for optimal growth (Rousk et al. 2010).
The similar change patterns in the composition of bacterial and fungal communities and plant communities (Fig. 3) evidenced that above- and belowground communities develop in parallel, confirming the role of plants in determining microbial community composition. Taxonomic analysis indicated consistent responses of microbial groups to N addition (Fig. 1). Acidobacteria and Alphaproteobacteria generally increased in abundance, whereas Actinobacteria decreased with increasing levels of N addition. Shifts in bacterial composition affected by N addition could be explained by the copiotrophic hypothesis, in which copiotrophic groups (e.g., Alphaproteobacteria and Acidobacteria), which have fast growth rates, are more likely to increase in nutrient-rich conditions, whereas oligotrophic groups (e.g., Actinobacteria), which are considered K-strategists that have a slower growth rate, would likely decline (Fierer et al. 2007). However, some copiotrophic organisms, such as Acidimicrobiia, decreased in abundance following N addition. This varied response was also found by Zeng et al. (2016) who reported that N enrichment led to a decrease in the abundance of Acidimicrobiia in temperate grassland. These results suggested that not bacterial taxa within the same taxonomic group do not necessarily shift in a similar manner. Elevated soil N levels select those species genetically better adapted to a particular niche created by N, which outcompete less well-adapted species, including oligotrophic bacteria. Interestingly, we found an increase in the relative abundance of Ascomycota and a decrease in that of Glomeromycota. The phylum Glomeromycota is composed almost entirely of arbuscular mycorrhizal fungi (Redecker and Raab 2006), and their decrease can be explained by the fact they would be less valuable to their hosts and thus, provided with less plant C, under conditions of increased N availability (Wei et al. 2013).
Effect of urea fertilization on microbial biomass and respiration
Three-year urea fertilization addition significantly increased microbial biomass (including C and N) and respiration. The declines in these parameters under high N treatment (N100) were consistent with previous studies in semiarid grasslands (Li et al. 2015; Zhang et al. 2014). Our study site has been proved to be N-saturated under high N treatment, with reductions in aboveground biomass, microbial biomass, and respiration under N100 treatment, indicating that soil available N has exceeded the requirements of plants and microbes and competition for N between communities is less likely to occur (Wang et al. 2018). In semiarid grassland, where aboveground biomass is naturally limited by N, increases in available N often promote plant growth and consequently increase the aboveground C accumulation through enhanced photosynthesis (Zang et al. 2017). Increased C input into the soil through litter decomposition and root exudates provides substrates for microorganism and promotes microbial activity; however, this effect can be reduced or even become negative when the amount of N added exceeds a threshold value, beyond which the allocation of photosynthetic C into the soil is decreased (Nakaji et al. 2001). This hump-shaped relationship between N addition and microbial respiration was also observed by Li et al. (2015), who found that microbial respiration linearly increased in grassland soil in Inner Mongolia (China) when the amount of N added increased from 0 to 8 g N m−2 year−1, and thereafter drastically decreased. Microbial biomass and respiration positively correlated with OC and aboveground biomass, suggesting the regulatory effect of N-fertilization-induced plant changes on microbial activity through driving C input.
Effect of urea fertilization on N-cycling genes and associated microbes
The abundances of amoA and amoB increased with increasing levels of N addition. This finding was consistent with those of previous grassland studies (Di et al. 2009; Xiang et al. 2017) reporting that amo responded positively to N addition. The positive relationship between amoAB abundance and exchangeable NH4+-N and NO3−-N contents suggested that a significant increase in inorganic N might contribute to an increase in the abundance of microbes harboring amoA and amoB genes. Ammonia-oxidizing archaea (AOA) and ammonia-oxidizing bacteria (AOB) are two major groups responsible for converting ammonia to nitrite (Liang et al. 2013). The dominant ammonia-oxidizers found in our grassland were Nitrosospira (AOB) and Nitrososphaera (AOA), which is consistent with other studies that have shown the dominance of these two ammonia-oxidizing microbial groups in terrestrial ecosystems (Pratscher et al. 2011; Zhou et al. 2015). AOA are predominant over AOB in non-fertilized soil because they prefer infertile conditions (Xiang et al. 2017). Accordingly, we found a predominance of Nitrososphaerales in unfertilized soil; however, Nitrosospirales became dominant over Nitrososphaerales after 3-year urea addition (Fig. S2). This finding provides clear evidence of contrasting patterns of AOB and AOA growth under different N conditions. Contents of exchangeable NH4+-N and NO3−-N positively correlated with the abundance of Nitrosospirales and negatively correlated with Nitrososphaerales abundance, suggesting that Nitrososphaera might be less competitive than Nitrosospirales in soils with high substrate concentration (Laverman et al. 2001). Notably, while amoA and amoB were highly abundant, we detected only a few gene copies of hao, which serves as the key gene in transforming NH2OH to NO2− in the nitrification process (Zehr and Kudela 2011). This suggests the potential existence of yet unknown genes with roles similar to that of hao.
The abundance of nirK, involved denitrification, declined following urea addition. Based on taxonomic analysis, we found that most of nirK-harboring denitrifiers were Rhizobiales, similar to findings in a previous study (Pan et al. 2018). The reduction of NO3− to NO2− can be performed by microbes harboring either narI, napA, napB, or all three (Table S3). Only the abundance of napA, encoding periplasmatic nitrate reductase, was significantly affected by N addition. The increased abundance of napA following urea treatment was positively correlated with exchangeable NH4+-N and NO3−-N contents, suggesting that the increased N availability induced by urea addition possibly contributes to the growth of microbes harboring napA. Notably, the non-synergistic effects of urea fertilization on genes involved in denitrification indicated an inconsistent impact increased N availability on denitrifier, which should be well considered in grassland restoration and soil remediation.
Urea fertilization also had a significant effect on the abundance of genes related to NH3 cycling. ureC and gdh, encoding urease and glutamate dehydrogenase, increased in abundance as the amount of N added increased. Moreover, the abundances of gene families involved in dissimilatory nitrate reduction encoding nitrate reductase and nitrite reductase, including napA, nirB, nrfA, and nrfH, were enhanced. ureC, gdh, napA, nirB, nrfA, and nrfH abundances were significantly correlated with soil exchangeable NH4+-N content. Both these findings were consistent with previous findings that the soil ammonification rate and NH4+ content were significantly increased following N fertilizer addition (Wang et al. 2015). The abundances of genes involved in dissimilatory reduction increased, but this did not happen for the abundance of genes involved in assimilatory nitrate reduction, which is another important process for reducing nitrate to ammonium. This inconsistency may be attributed to different roles of the two processes, since dissimilatory nitrate reduction occurs under anaerobic conditions (An and Gardner 2002), whereas the assimilatory process occurs under aerobic conditions. Therefore, the increased N availability may have inhibited the growth of microorganisms with genes involved in the process using nitrate as N resource. High N availability can increase their activities due to the available substrate resources (Mergel et al. 2001). Consistent with the enhanced abundance of nifH following N fertilization observed by Wang et al. (2017), we found an increase in nifH with increasing levels of N addition in grassland. These findings probably suggest a promotive effect of increased N availability on N-fixing microbes. The added N per se, as substrate supply, most likely is a more important factor caused by N addition, for determining nifH abundance. The abundances of gene families that utilize NH3 for glutamine synthesis and anammox decreased under N addition treatment, indicating that microbial communities not only accelerated NH3 production by stimulating corresponding gene families, but also suppressed gene families that consume NH3.
Burkholderiales and Rhizobiales were the dominant groups responsible for the main N processes, especially, nitrification, nitrate reduction (both assimilatory and dissimilatory), glutamine synthesis, and organic N decomposition, indicating the importance of these two taxa in N biogeochemical processes in semiarid grassland. In contrast, Nelson et al. (2015) and Tu et al. (2017) found that Actinomycetales was the main functional group participating in N cycling in grassland. This discrepancy could be due to the varied response of microbial communities to different grassland ecosystems, but also different sampling times. It is worth mentioning that changes in the abundances of certain gene families and their associated microbes cannot actually reveal the variation in corresponding N-cycling pathways, because complex N cycling is a result of collective actions of numerous genes and microbial groups, as evidenced by the distinct taxonomic profiles linked to different N processes in our study.
Conclusions
Our study showed that microbial diversity and N-cycling potential in a semiarid grassland were altered by N fertilization, and these effects appeared to be mediated by plant–soil interactions. Three-year urea addition increased microbial biomass and respiration, fungal diversity, and microbial N-cycling potential, whereas it decreased bacterial diversity. Above the threshold N fertilization rate of 50 kg N ha−1 year−1, microbial biomass and respiration decreased because of inhibitory effects of excess N on aboveground biomass. The changes in plant community strongly impacted microbial biomass, respiration, and diversity, and changes in inorganic N levels were mainly responsible for variation in the microbial N-cycling potential. Our results provide insights into how N enrichment influences above- and belowground communities.
References
Alster CJ, German DP, Lu Y, Allison SD (2013) Microbial enzymatic responses to drought and to nitrogen addition in a southern California grassland. Soil Biol Biochem 64:68–79
An S, Gardner WS (2002) Dissimilatory nitrate reduction to ammonium (DNRA) as a nitrogen link, versus denitrification as a sink in a shallow estuary (Laguna Madre/Baffin Bay, Texas). Mar Ecol Prog Ser 237:41–50
Bardgett RD, Freeman C, Ostle NJ (2008) Microbial contributions to climate change through carbon cycle feedbacks. ISME J 2:805–814
Beck T, Joergensen RG, Kandeler E, Makeschin F, Nuss E, Oberholzer HR, Scheu S (1997) An inter-laboratory comparison of ten different ways of measuring soil microbial biomass C. Soil Biol Biochem 29:1023–1032
Bobbink R, Hicks K, Galloway J, Spranger T, Alkemade R, Ashmore M, Bustamante M, Cinderby S, Davidson E, Dentener F, Emmett B, Erisman JW, Fenn M, Gilliam F, Nordin A, Pardo L, De Vries W (2010) Global assessment of nitrogen deposition effects on terrestrial plant diversity: a synthesis. Ecol Appl 20:30–59
Brooks PC, Landman A, Pruden G, Jenkinson DS (1985) Chloroform fumigation and the release of soil nitrogen: a rapid direct extraction method to measure microbial biomass nitrogen in soil. Soil Biol Biochem 17:837–842
Bremner JM, Mulvaney CS (1982) Nitrogen-total. In: Page AL, Miller RH, Keeney DR (eds) Methods of soil analysis, part 2, chemical and microbial properties. Agronomy Society of America, Madison, pp 595–624
Chen WQ, Xu R, Wu YT, Chen J, Zhang YJ, Hu TM, Yuan XP, Zhou L, Tan TY, Fan JR (2018) Plant diversity is coupled with beta not alpha diversity of soil fungal communities following N enrichment in a semi-arid grassland. Soil Biol Biochem 116:388–398
DeMalach N (2018) Toward a mechanistic understanding of the effects of nitrogen and phosphorus additions on grassland diversity. Perspect Plant Ecol 32:65–72
DeMalach N, Zaady E, Weiner J, Kadmon R (2016) Size asymmetry of resource competition and the structure of plant communities. J Ecol 104:899–910
Di HJ, Cameron KC, Shen JP, Winefield CS, O'Callaghan M, Bowatte S, He JZ (2009) Nitrification driven by bacteria and not archaea in nitrogen-rich grass-land soils. Nat Geosci 2:621–624
Duan CJ, Fang LC, Yang CL, Chen WB, Cui YX, Li SQ (2018) Reveal the response of enzyme activities to heavy metals through in situ zymography. Ecotoxicol Environ Saf 156:106–115
Fierer N, Bradford MA, Jackson RB (2007) Toward an ecological classification of soil bacteria. Ecology 88:1354–1364
Fu L, Niu B, Zhu Z, Wu S, Li W (2012) CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28:3150–3152
Galloway JN, Townsend AR, Erisman JW, Bekunda M, Cai Z, Freney JR, Martinelli LA, Seitzinger SP, Sutton MA (2008) Transformation of the nitrogen cycle: recent trends, questions, and potential solutions. Science 320:889–892
Harpole WS, Ngai JT, Cleland EE, Seabloom EW, Borer ET, Bracken MES, Elser JJ, Gruner DS, Hillebrand H, Shurin JB, Smith JE (2011) Nutrient co-limitation of primary producer communities. Ecol Lett 14:852–862
Hong JT, Ma XX, Zhang XK, Wang XD (2017) Nitrogen uptake pattern of herbaceous plants: coping strategies in altered neighbor species. Biol Fertil Soils 53:729–735
Jung J, Yeom J, Kim J, Han J, Lim HS, Park H, Hyun S, Park W (2011) Change in gene abundance in the nitrogen biogeochemical cycle with temperature and nitrogen addition in Antarctic soils. Res Microbiol 162:1018–1026
Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M (2016) KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res 44:D457–D462
Kou YP, Li JB, Wang YS, Li CN, Tu B, Yao MJ, Li XZ (2017) Scale-dependent key drivers controlling methane oxidation potential in Chinese grassland soils. Soil Biol Biochem 111:104–114
Lamb EG, Kembel SW, Cahill JF Jr (2009) Shoot, but not root, competition reduces community diversity in experimental mesocosms. J Ecol 97:155–163
Laverman AM, Speksnijder AGCL, Braster M, Kowal-chuk GA, Verhoef HA, HWv V (2001) Spatiotemporal stability on an ammonia-oxidizing community in a nitrogen-saturated forest soil. Microb Ecol 42:35–45
LeBauer DS, Treseder KK (2008) Nitrogen limitation of net primary productivity in terrestrial ecosystems is globally distributed. Ecology 89:371–379
Leff JW, Jones SE, Prober SM, Barberán A, Borer ET, Firn JL, Harpole WS, Hobbie SE, Hofmockel KS, Knops JMH, McCulley RL, La Pierre K, Risch AC, Seabloom EW, Schütz M, Steenbock C, Stevens CJ, Fierer N (2015) Consistent responses of soil microbial communities to elevated nutrient inputs in grasslands across the globe. Proc Natl Acad Sci U S A 112:10967–10972
Li H, Durbin R (2009) Fast and accurate short read alignment with burrows–wheeler transform. Bioinformatics 25:1754–1760
Li R, Li Y, Kristiansen K, Wang J (2008) SOAP: short oligonucleotide alignment program. Bioinformatics 24:713–714
Li Y, Liu YH, Wu SM, Niu L, Tian Y (2015) Microbial properties explain temporal variation in soil respiration in a grassland subjected to nitrogen addition. Sci Rep 5:18496–18506
Liang Y, He X, Liang S, Zhang W, Chen X, Feng S, Su Y (2013) Community structure analysis of soil ammonia oxidizers during vegetation restoration in southwest China. J Basic Microbiol 54:180–189
Liu L, Greaver TL (2010) A global perspective on belowground carbon dynamics under nitrogen enrichment. Ecol Lett 13:819–828
Lu M, Yang YH, Luo YQ, Fang CM, Zhou XH, Chen JK, Yang X, Li B (2011) Responses of ecosystem nitrogen cycle to nitrogen addition: a meta-analysis. New Phytol 189:1040–1050
Lu X, Mao Q, Gilliam FS, Luo Y, Mo J (2014) Nitrogen deposition contributes to soil acidification in tropical ecosystems. Glob Chang Biol 20:3790–3801
Mergel C, Kloos A, Bothe KH (2001) Seasonal fluctuations in the population of denitrifying and N2-fixing bacteria in an acid soil of a Norway spruce forest. Plant Soil 230:145–160
Nakaji T, Fukami M, Dokiya Y, Lzuta T (2001) Effects of high nitrogen load on growth, photosynthesis and nutrient status of Cryptomeria japonica and Pinus densiflora seedlings. Trees 15:453–461
Nannipieri P, Paul EA (2009) The chemical and functional characterization of soil N and its biotic components. Soil Biol Biochem 41:2357–2369
Nannipieri P, Trasar-Cepeda C, Dick P (2018) Soil enzyme activity: a brief history and biochemistry as a basis for appropriate interpretations and meta-analysis. Biol Fertil Soils 54:11–19
Nelson DW, Sommers LE (1982) Total carbon, organic carbon, and organic matter. In: Page AL, Miller RH, Keeney DR (eds) Methods of soil analysis, part 2, chemical and microbial properties. Agronomy Society of America, Madison, pp 539–552
Nelson MB, Berlemont R, Martiny AC, Martiny JBH (2015) Nitrogen cycling potential of a grassland litter microbial community. Appl Environ Microbiol 81:7012–7022
Ning QS, Gu Q, Shen JP, Lv XT, Yang JJ, Zhang XM, He JZ, Huang JH, Wang H, Xu ZH, Han XG (2015) Effects of nitrogen deposition rates and frequencies on the abundance of soil nitrogen-related functional genes in temperate grassland of northern China. J Soils Sediments 15:694–704
Noguchi H, Park J, Takagi T (2006) MetaGene: prokaryotic gene finding from environmental genome shotgun sequences. Nucleic Acids Res 34:5623–5630
Olsen SR, Sommers LE (1982) Phosphorous. In: Page AL, Miller RH, Keeney DR (eds) Methods of soil analysis, part 2, chemical and microbial properties. Agronomy Society of America, Madison, pp 403–430
Pan H, Ying SS, Liu HY, Zeng LZ, Zhang QC, Liu YM, Xu JM, Li Y, Di HJ (2018) Microbial pathways for nitrous oxide emissions from sheep urine and dung in a typical steppe grassland. Biol Fertil Soils 54:717–730
Pratscher J, Dumont MG, Conrad R (2011) Ammonia oxidation coupled to CO2 fixation by archaea and bacteria in an agricultural soil. Proc Natl Acad Sci U S A 108:4170–4175
Ramirez KS, Craine JM, Fierer N (2010) Nitrogen fertilization inhibits soil microbial respiration regardless of the form of nitrogen applied. Soil Biol Biochem 42:2336–2338
Redecker D, Raab P (2006) Phylogeny of the glomeromycota (arbuscular mycorrhizal fungi): recent developments and new gene markers. Mycologia 98:885–895
Rousk J, Bååth E, Brookes PC, Lauber CL, Lozupone C, Caporaso JG, Knight R, Fierer N (2010) Soil bacterial and fungal communities across a pH gradient in an arable soil. ISME J 4:1340–1351
Sanchez G (2013) PLS path modeling with R. Trowchez Editions, Berkeley
Shen XY, Zhang LM, Shen JP, Li LH, Yuan CL, He JZ (2011) Nitrogen loading levels affect abundance and composition of soil ammonia oxidizing prokaryotes in semiarid temperate grassland. J Soils Sediments 11:1243–1252
Stevens CJ, Dise NB, Mountford JO, Gowing DJ (2004) Impact of nitrogen deposition on the species richness of grasslands. Science 303:1876–1879
Tian XF, Hu HW, Ding Q, Song MH, Xu XL, Zheng Y, Guo LD (2014) Influence of nitrogen fertilization on soil ammonia oxidizer and denitrifier abundance, microbial biomass, and enzyme activities in an alpine meadow. Biol Fertil Soils 50:703–713
Tu QC, He ZL, Wu LY, Xue K, Xie G, Chain P, Reich PB, Hobbie SE, Zhou JZ (2017) Metagenomic reconstruction of nitrogen cycling pathways in a CO2-enriched grassland ecosystem. Soil Biol Biochem 106:99–108
van der Heijden MGA, Bardgett RD, Straalen NMV (2008) The unseen majority: soil microbes as drivers of plant diversity and productivity in terrestrial ecosystems. Ecol Lett 11:296–310
Vance ED, Brookes PC, Jenkinson DS (1987) An extraction method for measuring soil microbial biomass C. Soil Biol Biochem 19:703–707
Vestergaard G, Schulz S, Schöler A, Schloter M (2017) Making big data smart—how to use metagenomics to understand soil quality. Biol Fertil Soils 53:479–484
Vitousek PM, Aber JD, Howarth RW, Likens GE, Matson PA, Schindler DW, Schlesinger WH, Tilman DG (1997) Human alteration of the global nitrogen cycle: sources and consequences. Ecol Appl 7:737–750
Wang CH, Butterbach-Bahl K, He NP, Wang QB, Xing X, Han X (2015) Nitrogen addition and mowing affect microbial nitrogen transformations in a C4 grassland in northern China. Eur J Soil Sci 66:485–495
Wang CY, Zhou JW, Liu J, Jiang K, Du DL (2017) Responses of soil N-fixing bacteria communities to Amaranthus retroflexus invasion under different forms of N deposition. Agric Ecosyst Environ 247:329–336
Wang C, Lu XK, Moria T, Mao QG, Zhou KJ, Zhou GY, Nie YX, Mo JM (2018) Responses of soil microbial community to continuous experimental nitrogen additions for 13 years in a nitrogen-rich tropical forest. Soil Biol Biochem 121:103–112
Wei C, Yu Q, Bai E, Lu X, Li Q, Xia J, Kardol P, Liang W, Wang Z, Han X (2013) Nitrogen deposition weakens plant-microbe interactions in grassland ecosystems. Glob Chang Biol 19:3688–3697
Whittaker RJ (2006) Island species-energy theory. J Biogeogr 33:11–12
Xiang XJ, He D, He JS, Myrold DD, Chu HY (2017) Ammonia-oxidizing bacteria rather than archaea respond to short-term urea amendment in an alpine grassland. Soil Biol Biochem 107:218–225
Yang HJ, Li Y, Wu MY, Zhang Z, Li LH, Wan SQ (2011) Plant community responses to nitrogen addition and increased precipitation: the importance of water availability and species traits. Glob Chang Biol 17:2936–2944
Yang T, Adams JM, Shi Y, He JS, Jing X, Chen LT, Tedersoo L, Chu HY (2017) Soil fungal diversity in natural grasslands of the Tibetan Plateau: associations with plant diversity and productivity. New Phytol 215:756–765
Zang HD, Blagodatskaya E, Wang JY, Xu XL, Kuzyakov Y (2017) Nitrogen fertilization increases rhizodeposit incorporation into microbial biomass and reduces soil organic matter losses. Biol Fertil Soils 53:419–429
Zehr JP, Kudela RM (2011) Nitrogen cycle of the open ocean: from genes to ecosystems. Annu Rev Mar Sci 3:197–225
Zeng J, Liu XJ, Song L, Lin XJ, Zhang HY, Shen CC, Chu HY (2016) Nitrogen fertilization directly affects soil bacterial diversity and indirectly affects bacterial community composition. Soil Biol Biochem 92:41–49
Zhang CP, Niu DC, Hall SJ, Wen HY, Li XD, Fu H, Wan CG, Elser JJ (2014) Effects of simulated nitrogen deposition on soil respiration components and their temperature sensitivities in a semiarid grassland. Soil Biol Biochem 75:113–123
Zhang C, Liu G, Xue S, Wang G (2016) Soil bacterial community dynamics reflect changes in plant community and soil properties during the secondary succession of abandoned farmland in the Loess Plateau. Soil Biol Biochem 97:40–49
Zhong YQW, Yan WM, Shangguan ZP (2015) Impact of long-term N additions upon coupling between soil microbial community structure and activity, and nutrient-use efficiencies. Soil Biol Biochem 91:151–159
Zhou X, Fornara D, Wasson EA, Wang DM, Ren GD, Christie P, Jia ZJ (2015) Effects of 44 years of chronic nitrogen fertilization on the soil nitrifying community of permanent grassland. Soil Biol Biochem 91:76–83
Zhou J, Jiang X, Zhou B, Zhao B, Ma M, Guan D, Li J, Chen S, Cao F, Shen D, Qin J (2016) Thirty four years of nitrogen fertilization decreases fungal diversity and alters fungal community composition in black soil in northeast China. Soil Biol Biochem 95:135–143
Funding
National Natural Sciences Foundation of China (41771554), National Key Research and Development Program of China (2016YFC0501707), and the Key Technologies R&D Program of China (2015BAC01B03).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 148 kb)
Rights and permissions
About this article

Cite this article
Zhang, C., Song, Z., Zhuang, D. et al. Urea fertilization decreases soil bacterial diversity, but improves microbial biomass, respiration, and N-cycling potential in a semiarid grassland. Biol Fertil Soils 55, 229–242 (2019). https://doi.org/10.1007/s00374-019-01344-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00374-019-01344-z