
Abstract
Fertilisers, especially nitrogen (N) and phosphorus (P) supplies, are frequently used in agricultural soil management to attain high crop yields. However, the intensive application of these chemical inputs can decrease the quality of agricultural soils and increase the probability of environmental pollution. In this study, the impact of P fertilisation on the diversity of the soil bacterial community was assessed. For this, a culture-independent approach targeting 16 rRNA and phoD genes was used on DNA extracted from pasture soils subjected to three different P fertilisation regimes for a long-term (42 years). As alkaline phosphomonoesterase (ALP) is necessary for mineralisation of organic P, an inverse relationship between the level of potential ALP activity and soil available P was expected. Indeed, a lower ALP activity was observed in soil subjected to higher chemical P fertiliser input. Analysis of the prevalence of three divergent families of ALP (PhoA, PhoD and PhoX) in metagenomic datasets revealed that PhoD is the most frequent ALP in soil samples and was selected as the most representative ALP possessed by the soil bacterial communities. Diversity of the phoD phosphorus mineraliser group, as well as the total bacterial community, was both increased in response to long-term P fertilisation. Specifically, phosphorus fertilisation decreased the relative abundance of certain taxa, including Acidobacteria and Pseudomonas fluorescens. In conclusion, this study shows that P fertilisation affects the microbial diversity of soil ecosystems, which might potentially modulate the soil biogeochemical cycle.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Inorganic fertilisers are frequently used in conventional agriculture to attain high crop yields. However, the intensive application of these chemical inputs can decrease the quality of agricultural soils and increase the risks of environmental pollution (Tilman et al. 2002). Because of concerns regarding economics and environmental protection, viable alternatives to these chemicals are being sought (Goulding et al. 2008). One potential alternative relies on the exploitation of the indigenous soil microbial community (Morrissey et al. 2004). Indeed, microorganisms are essential drivers of nutrient cycles in the soil and can thus influence crop productivity by improving the flow of carbon (C), nitrogen (N), phosphorus (P) and other elements (Nannipieri et al. 2003). Therefore, in reduced-input agro-ecosystems, these microbial activities are likely to be decisive for ecosystem functioning. Understanding the contribution of microbes to plant nutrition is then essential to the development of sustainable agricultural practices.
Many soil bacteria and fungi have the ability to mobilise insoluble soil P into bioavailable forms that can be taken up by plants. Some soil microorganisms (e.g. mycorrhizal fungi and phytohormone-producing bacteria) can enhance the uptake of orthophosphate anions through extension of the root surface area (Karandashov and Bucher 2005; Richardson et al. 2009). Alternatively, other microorganisms can directly dissolve precipitated inorganic phosphate by simply acidifying the soil, or mineralise P from organic compounds via enzymatic hydrolysis. The dissolving of inorganic P is usually attributed to microorganisms which have the ability to acidify the local environment through the release of organic acid (Uroz et al. 2009). For instance, mobilisation of insoluble soil inorganic P by Pseudomonas spp. is mediated through the extracellular production of gluconic acid (Miller et al. 2010; Rice et al. 2012). The mineralisation of insoluble organic P pool by the production of specific enzymes, such as phosphomonoesterases (Rodríguez et al. 2006, Nannipieri et al. 2011), phytases (Yao et al. 2012) and phosphonatases (Kamat et al. 2011), is also a significant aspect of P cycling in the soil. Among the enzymes involved in organic P mineralisation, bacterial alkaline phosphomonoesterases (ALPs) have been most thoroughly investigated in term of their biosynthesis, catalytic properties and genetic controls (Eder et al. 1996; Galperin and Jedrzejas 2001; Monds et al. 2006; Zaheer et al. 2009; Drozd et al. 2011; Sebastian and Ammerman 2011). To date, three distinct ALP families (PhoA, PhoD and PhoX) have been defined based on their sequence similarities and substrate specificities (Luo et al. 2009; Kageyama et al. 2011; Kathuria and Martiny 2011). Enzymes belonging to the PhoD family displayed Ca2+-dependent hydrolysis activity for monoesters but also for diesters and are therefore classified as phosphomonoesterases and phosphodiesterases (Kageyama et al. 2011).
The ecological functions associated with microbial activities in the soil are related with microbial community structures (Nannipieri et al. 2003; Fierer et al. 2012). It is now generally accepted that the composition of these microbial communities is influenced by the plant species and cultivar but also by soil type, climate and land management practises (Berg and Smalla 2009). The consequences of these shifts on microbial community functions related to N cycle, such as nitrification and denitrification, have been investigated in a number of studies (Philippot and Hallin 2005; Chu et al. 2007; Mao et al. 2011). In contrast, our knowledge about the impact of various environmental and anthropogenic factors on the P mineralising microbial communities is still limited and restricted to cultivable bacteria (Browne et al. 2009). Distribution and diversity of functional genes in microbial community could be investigated using the state-of-the-art techniques such as deep sequencing of the soil microbiome (reviewed in Barret et al. (2011)). The recent development of a functional molecular marker targeting the bacterial ALP (Sakurai et al. 2008) provides a good opportunity to study the impact of P fertiliser amendment on the diversity of ALP-producing bacteria.
The aim of this study was to evaluate the impact of long-term P fertilisation regimes on the P mineralising group in bacterial community. To do so, the effect of P fertilisation on the bacterial community diversity was initially compared between three pasture soils managed over 42 years with no, medium and high inputs of chemical P fertiliser. Then, the influence of exogenous P on the P mineralising bacterial group was assessed through the diversity of ALP-producing bacteria using phoD gene sequence as a molecular marker.
Material and methods
Study site and sampling
The experimental site was established on a humic gleysol with a sandy loam texture and managed by Environment Research Centre, Teagasc, Johnstown Castle, Co. Wexford, Ireland. The agricultural managements have been described in detail in previous studies (Culleton et al. 2000, 2002; King-Salter 2008; Griffiths et al. 2012). At the start of the trial, in 1968, the site was ploughed and sown with Lolium perenne. The site was divided into three identical fields with the size of 5.40 ha. Phosphorus fertiliser (calcium superphosphate) was applied annually at 0 (0P), 15 (15P) or 30 (30P) kg ha−1 on each fields. Nitrogen (ammonium nitrate, 240 kg N ha−1) and potassium (potassium chloride, 20 kg K ha−1) were applied annually to all three fields. Phosphorus and K were applied in spring, while N was applied between spring and autumn. The field of each P treatment level was divided into 12 paddocks (0.45 ha each), among which six paddocks were grazed at a low stocking rate (2,200 kg ha−1) and six with a high stocking rate (3,300 kg ha−1). Since the P fertilisation level applied to the low stocking rate paddocks was no longer constant after 1999, only the high stocking rate paddocks were studied (Griffiths et al. 2012). Physicochemical characteristics of 0P, 15P and 30P soils had been determined by Griffiths et al. (2012). In spring 2010, soils of paddock 1#, 2# and 3# of each P treatment were sampled to provide the three field replicates for this study. Soil samples from each paddock were collected from the top 10 cm by taking 20 cores with a gouge auger (1.25 cm diameter) and mixed thoroughly. The soils were handpicked to remove stones and larger soil fauna, sieved to pass through a 3.35-mm mesh. After sampling, the soils were sealed in sterile plastic bags immediately and stored at −20 °C.
Measurement of potential ALP activity
Soil ALP activity was determined as described by Tabatabai and Bremner (1969), using para-nitrophenyl phosphate (pNPP) as a substrate (BioAssay Systems, USA). The soil ALP activity was measured in the three replicate paddocks of each P treatment. One hundred milligrams of wet soil was mixed with 2 ml assay buffer (pH 11) containing 10 mM pNPP. After homogenisation, the mixture was incubated for 4 h at 37 °C. Precipitant was removed by filtration, and absorption was measured at 405 nm. One unit of potential ALP activity was defined as the amount of para-nitrophenol (in milligrams) released by 1 g of soil/h.
DNA extraction
Soil crude DNA was extracted from the three field replicates of each P treatment, using the method described by Carrigg et al. (2007) based on bead beating and incubation with lysozyme at 37 °C for 30 min. However, a modified CTAB extraction buffer (2 % hexadecyltrimethylammonium bromide, 1.0 M sodium chloride and 120 mM sodium phosphate buffer at pH 8.0) was used. The crude DNA was further purified by gel extraction using GELASETM (Epicentre, USA) to remove humic acids prior to PCR.
PCR amplification and DGGE analysis
In order to assess the structure of the bacterial community between the three treatments, a preliminary denaturing gradient gel electrophoresis (DGGE) was performed prior to pyrosequencing. Primers with a 40-bp GC-clamp added to the 5′ end were used to amplify the bacterial 16S rDNA (V1-V3 variable region) and phoD genes (Table S1). PCR was performed in a total volume of 50 μl on a G-STORMTM GS-1 Gradient Cycler (Alpha Metrix Biotech GmbH, Germany). PCR reaction for 16S rDNA consisted of 10 pmol of each primer (Sigma, Germany), 1 U of Platinum High Fidelity Taq Polymerase (Invitrogen, USA), 1× Platinum PCR Buffer (Invitrogen, USA), 0.2 mM of dNTP (Invitrogen, USA), 2 mM of MgSO4 and 200 ng of purified soil DNA as template. The PCR programme consisted of an initial denaturation step of 95 °C (2 min), followed by 30 cycles of 95 °C (30 s), 55 °C (30 s) and 68 °C (45 s) and a final extension step of 68 °C (5 min). In order to amplify phoD, High Fidelity Taq was replaced by Phusion DNA polymerase (NEB Inc, USA). A 50-μl PCR reaction consisted of 50 pmol of each primer (Sigma, Germany), 2 mM dNTP (Invitrogen, USA), 3 % DMSO, 1 U of Phusion Hotstart II DNA polymerase (NEB Inc., USA), 1× Phusion GC Buffer (NEB Inc., USA) and 200 ng of purified soil DNA as template. The PCR programme consisted of an initial denaturation step of 98 °C (2 min), followed by 35 cycles of 98 °C (10 s), 69 °C (23 s) and 72 °C (30 s), then a final extension step of 72 °C (7 min).
DGGE was performed in a D-Code electrophoresis system (BioRad Laboratories, USA). Polyacrylamide gels were prepared with a linear denaturing gradient ranging from 40 to 60 % for 16S rDNA gene and 50 to 75 % for phoD gene. 16S rDNA or phoD fragments amplified from the three field replicates of each P input level were run on a DGGE gel, at 68 V for 15 h. After electrophoresis, gels were stained with SYBR-Gold (Invitrogen, USA) for 30 min, followed by a destaining step in distilled water for 5 min. Gel images were scanned with GelDocItTM Imaging System (UVP, USA), and the intensities of each band were analysed by the gel analysis software QuantityOneTM (BioRad Laboratories, USA). DGGE gels were analysed based on the band presence and intensity of DGGE profiles using MVSP 3.1 software (Kovach Computing Service, Anglesey, Wales, UK). A community dendrogram was constructed based on the Jaccard’s coefficient and clustered by the nearest neighbour-joining method using the unweighted pair group method with arithmetic mean (UPGMA).
Pyrosequencing
Barcode primers with unique combination of 25 bp header and 10 bp linker added to the 5′ end were used to amplify the same region of bacterial 16S rDNA and phoD genes as in the DGGE experiment (Table S1). PCR reactions used the same amplification conditions as stated in the DGGE experiment. All PCR products were purified using GELASETM (Epicentre, USA). The amplicons were pooled and sequenced by Eurofins MWG Operon Inc. with a Roche 454 GS-FLX Titanium system.
Sequence analysis
Raw data generated by pyrosequencing were separated into two datasets corresponding to 16S rDNA and phoD reads. Using the pre-proceeding programme of MOTHUR (Schloss et al. 2009), low-quality (quality score <20) and short (<150 bp) reads were removed, and chimera amplicons were filtered. The 10-bp barcodes were used to identify and sort the reads from different phosphorus treatments. Primers, linker and barcode were then trimmed from each sequence (Table S2). Following filtering and removal of noise, a total of 9,650, 8,904 and 8,975 reads of 16S rDNA, as well as 11,685, 12,395 and 13,369 reads of phoD, were obtained from the no-, medium- and high-P input soils, respectively.
The 16S rDNA pyrosequencing reads of each treatment (0P, 15P and 30P) were aligned by MAFFT (Katoh and Toh 2008). Distances matrices were calculated by MOTHUR, and sequences were clustered into operational taxonomic units (OTU) based on a genetic distance of 0.03 using the furthest neighbour method. The phylogenetic affiliation of each OTU was obtained by comparison against the ARB-SILVA taxonomy database using a threshold of 80 % similarity.
The phoD gene sequences were blasted against the NCBI non-redundant protein database using BLASTX with an E value cutoff <1e−10. The filtered phoD sequences were aligned by MAFFT, and distance matrices were calculated by MOTHUR. Assignment of phoD sequences to OTUs was performed by the furthest neighbour method using a genetic distance of 0.25. This genetic distance was defined by the method developed by Palmer et al. (2009) (Fig. S1). Representative sequences of abundant OTUs (≥10 sequences) were selected from each treatment (0P, 15P and 30P) and merged into a single file. These nucleic sequences were aligned using a seed database of 117 phoD sequences derived from draft or complete genomes (see “Recovery of ALP sequences from genomes and metagenome datasets” section). Maximum-likelihood (ML) trees were built with PhyML (Guindon and Gascuel 2003) using the nucleotide substitution model HKY85 (Hasegawa et al. 1985). Branch supports were evaluated using the approximate likelihood-ratio test (Anisimova and Gascuel 2006). Phylogenetic trees were visualised and exported using the web-based tool Interactive Tree Of Life (Letunic and Bork 2011).
Recovery of ALP sequences from genomes and metagenome datasets
Cluster of orthologous group (COG) were associated with representative PhoA, PhoD and PhoX amino acid sequences through the NCBI batch conserved domain search. The following COG identifiers COG1785, COG3540 and COG3211 were used to retrieved 1,358 PhoA, 680 PhoD and 677 PhoX homologs (Berg and Smalla 2009) from 1,392 draft or complete genomes presented in the IMG/M database (Markowitz et al. 2008). Unique protein sequences were aligned using the default parameter of MAFFT. ML phylogenetic trees were built with PhyML using the WAG amino acid substitution model of evolution (Whelan and Goldman 2001) and four categories of substitution rates. The same COG identifiers (COG1785, COG3540 and COG3211) were used to retrieve 2,372 PhoA, 4,990 PhoD and 3,296 PhoX homologs from 236 metagenome datasets. In order to estimate the number of ALP per genome, each ALP was normalised to a single-copy gene (recA). Difference in ALP distribution within each ecosystem was assessed by one-way ANOVA.
Statistic analysis
Soil ALP activity is mean value of three field replicates for each P treatment. Confidence limits of Chao1 and ACE estimators as well as confidence limit of Shannon–Wiener diversity index were calculated by MOTHUR (Schloss et al. 2009). Difference in bacterial composition was compared by 99 % upper and lower confidence intervals for proportions. The relative abundance of PhoA, PhoD and PhoX in different ecosystem categories was compared by one-way ANOVA at a P value <0.05.
Results
Species richness and diversity of total bacterial community altered by P treatment
The long-term impact of P fertilisation on the experimental site that is used to assess the species richness and diversity of total bacterial community in this study has recently been published by Griffiths et al. (2012). This study showed that after four decades of amendment of calcium superphosphate as P fertiliser, the inorganic phosphate levels between the no, medium and high P fertilised soils demonstrated an increasing trend in response to P amendment (8.2 ± 1.7 μg kg−1 in no-P, 31.6 ± 4.0 μg kg−1 in medium-P and 78.9 ± 13.3 μg kg−1 in high-P soil) (Griffiths et al. 2012). The total P content in soils (408 ± 20 mg kg−1 in no-P, 662 ± 54 mg kg−1 in medium-P and 918 ± 47 mg kg−1 in high-P soil) and the biomass P of microbial communities (79 ± 12 μg kg−1 in no-P, 127 ± 25 μg kg−1 in medium-P and 160 ± 26 μg kg−1 in high-P soil) were also increased by P amendment (Griffiths et al. 2012). This demonstrated that the P availability in the soils was indeed affected by the long-term impact of P fertilisation.
Comparison of bacterial community structure between the three different P treatments was initially performed by DGGE analysis of 16S rRNA gene. According to the clustering analysis, no-P soil has a distinct bacterial community structure in comparison to the medium- (15P) and high-input (30P) soils (Fig. 1a). Moreover, the analysis also shows a clear separation between soil samples derived from the medium- (15P) and high-input (30P) soils. The taxonomic information relative to the bacterial community presented in 0P, 15P and 30P soils was further assessed by pyrosequencing of 16S rRNA gene amplicons. Sequences were clustered into OTUs based on a genetic distance of 0.03, a cutoff frequently employed for whole and partial 16S rRNA gene analysis (Schloss and Handelsman 2006). Rarefaction analysis was performed on each soil sample to assess whether further sampling would likely yield additional OTUs. None of the rarefaction curves of observed 16S rDNA OTUs reach the plateau phase (Fig. 2a), which indicates that the soils were not sampled to saturation. Incomplete sampling is a commonly observed feature in studies using high-throughput DNA pyrosequencing to assess the bacterial diversity in soil (Roesch et al. 2007; Uroz et al. 2010; İnceoğlu et al. 2011; Nacke et al. 2011). Therefore, the number of OTUs present in the three soils was predicted with two different non-parametric estimators (Table 1). According to Chao1 estimator, the number of predicted OTU is constant in the three P treatments. However, based on ACE estimator, the number of predicted abundant OTUs (≥10 sequences) is increased in soil with P inputs in comparison to no-P input (Table 1). It suggests that the P fertilisation treatment increased the number of abundant bacterial taxa in the soil.

DGGE analysis of 16S (a) and phoD (b) amplicons to assess the bacterial community. UPGMA clustering analysis was used to calculate the similarities between different P treatments. N no-P soil, M medium-P soil, H high-P soil. Field replicates are numbered 1–3

Rarefaction curves of bacterial 16S (a) and phoD (b) gene sequences. Sequences recovered from the pasture soils with 0P (solid lines), 15P (dashed lines) and 30P inputs (square dots) were grouped as operational taxonomic units (OTUs). OTUs were defined at 97 and 75 % sequence similarities for 16S rRNA (a) and phoD (b), respectively
The diversity of the microbial community within each soil was also estimated by the Shannon–Wiener index. This analysis revealed that the bacterial diversity was potentially enhanced by the high fertilisation treatment (Table 1). These results suggest that the abundance and diversity of the bacterial community are increased by P fertilisation in this grazed pasture. Analysis of the taxonomic composition of the total bacterial community in the different soil samples showed that the dominant bacterial phyla were Acidobacteria, Proteobacteria and to a lesser extends Firmicutes (Table 2). Although all phyla were represented in every treatment, the proportion of each phylum seems to vary slightly in different soils. For instance, the relative abundance of Acidobacteria is higher in the no-P soil in comparison with the soil amended with P fertilisers. In contrast, the relative abundance of Firmicutes and Proteobacteria seems to increase gradually in soil amended with P. Change in the relative abundance of Proteobacteria seems to be mainly mediated by an increase of the Alphaproteobacteria.
phoD representing ALP genes in soil ecosystem
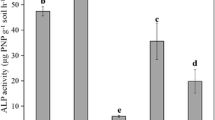
The analysis of 16S rRNA gene amplicons has revealed that P fertilisation treatment may influence the taxonomic composition of the total bacterial community. This change might be explained, at least partially, by a decrease of specific bacterial populations capable of dissolving and mineralising P efficiently in soil. Consequently, it suggests that the inorganic and/or organic P mineralisation capacity of the no-P soil would be higher than the capacities of the amended soils. Interestingly, we observed a lower ALP activity was presented in soil subjected to higher chemical P fertiliser input (37.86 ± 3.74 mg pNP g−1 h−1 in no-P, 22.50 ± 3.41 mg pNP g−1 h−1 in medium-P and 13.68 ± 2.36 mg pNP g−1 h−1 in high-P soil).
Since the difference of bacterial ALP activity could be due to selection of specific ALP genotypes in the low-input soils, the recent development of a molecular marker targeting the bacterial ALPs provides a unique opportunity to test this hypothesis (Sakurai et al. 2008). However, ALP contains three gene families (phoA, phoD and phoX), which are quite divergent at the sequence level (Fig. 3). Moreover, the sequences within each gene family are also very diverse (Figs. S2a, S2b and S2c). As a result, it is practically impossible to design a unique primer set capable of amplifying all three ALP families. In fact, the molecular marker designed by Sakurai et al. (2008) is only representative of the phoD gene family.

Phylogenetic analysis of ALPs. A distance tree (maximum-likelihood) was calculated from 40, 33 and 25 representative sequences of PhoA, PhoD and PhoX, respectively
In order to choose a representative ALP gene family, the abundances of the protein-coding genes phoA, phoD and phoX were assessed in 1,392 genome (Table S3) and 236 metagenome datasets (Table S4) publicly available in the IMG/M database (Markowitz et al. 2008). In order to estimate the number of ALP per genome, each ALP gene was normalised to recA, a single-copy gene. Difference in ALP abundance was assessed by one-way ANOVA (Fig. S3). Overall the relative abundance of phoD was higher than phoX and phoA in the 236 metagenome datasets examined, which is in accordance with previous findings obtained in the ocean (Luo et al. 2009). Moreover, phoD was also highly abundant in plant-associated metagenomes and in terrestrial ecosystem. Therefore, this gene family was chosen for further analysis.
Species richness and diversity of phoD phosphorus mineralising bacterial group
DGGE analysis was performed to estimate the structural diversity associated with phoD in the different soil samples prior to pyrosequencing (Fig. 1b). The most significant separation (18 % similarity) of the bacterial community was observed between the no-P soils (0P) and fertilised (15P and 30P) soils while the bacterial community from the 15P and 30P treatments shared 29 % similarity. These results clearly suggest an impact of the P fertilisation treatments on the phoD phosphorus mineralising community. To get further insights on modifications of the bacterial phosphorus mineralising community following P treatment, pyrosequencing of phoD amplicons was then performed.
According to the rarefaction analysis, the sampling seems to cover all phoD OTUs in the soil (Fig. 2b). Since the P amendment leads to an increased level of soil P and a decreased level of potential ALP activity, we postulate that reduced-P soils (0P and 15P) should select OTUs with particular phoD genotype and consequently decrease phoD diversity. Using different non-parametric estimators, we showed that the fertilisation treatment increased the phoD gene richness (Table 1). Furthermore, the diversity of phoD phosphorus mineraliser group within each soil was also estimated by Shannon–Wiener index. The analysis revealed that the bacterial diversity was increased by both the medium and high fertilisation treatments (Table 1). Altogether these results are in accordance with our initial hypothesis.
Phylogenetic structure of phoD phosphorus mineralising bacterial group
To assess the taxonomic composition of phoD, abundant OTUs (≥10 sequences) derived from the three P treatments were aligned with 117 reference phoD sequences and subjected to phylogenetic analysis. In the resulting phylogeny, phoD sequences are grouped in four major phylogenetic clusters (Fig. S4 and Table S5). Phylogenetic clusters 1 and 2 are essentially composed of bacteria belonging to the Alphaproteobacteria, cluster 3 is mainly composed of Gammaproteobacteria whereas cluster 4 is a mixture of diverse phyla such as Cyanobacteria, Actinobacteria and Proteobacteria. Although no strong relationship was observed between the four main phylogenetic clusters and the three P treatments (Table 3), some particular OTUs are enriched in specific soil treatment (Fig. S4). For example, OTU#B1_25_408, which is 97 % similar to Pseudomonas fluorescens SBW25, is more frequent in the no-P soil. This could suggest that this OTU can be involved in the increase of ALP activity.
Discussion
Different P availabilities created by long-term P fertiliser amendments
The long-term impacts of P fertiliser amendment on soil physicochemical characteristics of the experimental site were recently published by Griffiths et al. (2012). This showed a relatively lower P availability in 0P soil and a higher P availability in 15P and 30P soils. Since the calcium superphosphate is an acidic fertiliser, which is produced from phosphate rock granules dissolved by sulphuric acid, it is expected that the 15P and 30P soils subjected to more calcium superphosphate amendment should have a more acidic pH. Interestingly, an opposite trend was observed (Griffiths et al. 2012). This might be due to pH variance between different soil paddocks. Another possibility is that in the relatively P-limiting environment of 0P soil, some microorganisms produced organic acids to dissolve the precipitated inorganic phosphate (rock P) and supplement their P nutrition (Kirk 1999; Rodríguez et al. 2006). Indeed, a higher level of dissolved organic carbon, which includes the organic acids, was observed in the no-P soil in comparison to medium-P and high-P soils (Griffiths et al. 2012). It suggests that besides the organic mineralisation activity, the utilisation capacity of inorganic P might also be promoted by the P-limiting condition of no-P soil.
Composite influences of P fertilisation on the diversity and composition of bacterial community
After four decades of P fertilisation, the diversities of the total bacterial community and the phoD phosphorus mineraliser group associated with the P-rich environment resulted from long-term P fertilisation are increased, in comparison to the relatively P-limiting environment in no-P soil. These findings are in accordance with other studies performed on different agricultural and forest soils (Zhong and Cai 2007; Richardson and Simpson 2011; Liu et al. 2012). For example, application of long-term N + P + K fertiliser input to a paddy soil has been shown to enhance the diversity of the soil total microbial community in comparison to soil subjected solely to N + K treatments (Zhong and Cai 2007). However, conflicting results exist, since P fertilisation also decreased the soil microbial diversity in other agro-ecosystems (Ge et al. 2008; Chhabra et al. 2012). For instance, Chhabra et al. (2012) observed that no-P input could decrease the diversity of phosphorus mineraliser bacterial group in the barley rhizosphere. Altogether, it seems that P input may not be the sole factor responsible for alteration in abundance and diversity of the microbial community and that other environmental factor, or a combination of factor, such as soil characteristics and/or climate, might also be involved in this process. Analysis of the taxonomic composition of the total bacterial community in the different P treatments showed that the relative abundance of some bacterial phyla, such as the Acidobacteria, was decreased in P-fertilised soils. Since the abundance of Acidobacteria is positively associated with low soil pH (Jones et al. 2009), the lowest pH of the no-P input soil could explain this shift. Alternatively the decrease in relative abundance of this oligotrophic bacterial phylum along with an increase of copiotrophic phyla such as Proteobacteria in the high-P soil could suggest that the bacterial community become more copiotrophic as P inputs increased, a pattern already observed in soil amended with N fertilisers (Fierer et al. 2012).
Short-term environmental factors such as seasonal cycles (Smalla et al. 2001) and weather events (e.g. acid rain) (Pennanen et al. 1998) could all induce short-term fluctuation of bacterial community. As the diversity and composition of bacterial community were assessed on a single sampling time point, the observation of the bacterial community might be biased. However, these short-term factors could be shaded by the effect of extremely long-term (42 years) fertiliser input, and the bias was minimised since the bacterial communities associated with the three long-term fertilised sites were directly compared.
Decreased soil ALP activity in response to P fertiliser amendment
The shift in bacterial community structure observed across the P gradient could result in a modification of the phosphorous mineralising bacterial community, which might ultimately alter its P mineralisation capacity. Interestingly a lower ALP activity was indeed presented in soil subjected to higher chemical P fertiliser input, while a soil relatively more limited in inorganic P possessed an increased potential ALP activity. The same trend was also observed in other soils subjected to P fertiliser amendments (Van Aarle and Plassard 2010). In addition, P amendment in manure could create a higher ALP activity in soils in comparison to mineral P fertilisers (Nannipieri et al. 2011). It suggests that inorganic P availability might have a negative effect on ALP activity in soils while organic P matters is in contrast.
It was determined that Ca2+ is the co-factor of PhoD and PhoX (Yamane and Maruo 1978, Wu et al. 2007). Since the applied P fertiliser (calcium superphosphate) would introduce extra amount of calcium into the medium-P and high-P soils, the observed lower ALP activity in the medium-P and high-P treatments were not due to limitation of calcium availability in the soil. Moreover, since pH in the medium-P and high-P soils was even more alkaline compared to the no-P soil, the pH will potentially increase the ALP activity in medium-P and high-P soils. However, the total observed ALP activity in no-P was still higher in comparison to medium-P and high-P soils. Therefore, the decreased ALP activity in the P fertilised soils could be explained in part by shift of dominant phosphorus mineralising bacterial group. To test this hypothesis, the diversity of ALP producing bacteria was assessed using phoD as a functional molecular marker. This analysis showed that phoD diversity was decreased in the no-P soil and that some specific phoD phylotypes related to P. fluorescens were enriched in this soil. P. fluorescens is recognised as an important inorganic and organic P mineralising microbe (Hayat et al. 2010; Miller et al. 2010). In consequence, the reduced P fertilisation could increase the abundance of P. fluorescens and improve the release of P from different organic and inorganic sources present in the soil. However, it remains to be demonstrated whether PhoD is responsible for the increase of potential ALP activity since it has been previously reported that PhoD has a minor ALP activity compared to PhoX in P. fluorescens (Monds et al. 2006) and Sinorhizobium meliloti (Zaheer et al. 2009). These lower activities of PhoD in these two bacterial strains could be explained by substrate specificity. Indeed, PhoD of the cyanobacterial strain Aphanothece halophytica displays hydrolysis activities for mono- and di-ester in a Ca2+-dependent manner (Kageyama et al. 2011) and thus could be classified as a phosphomonoesterase and phosphodiesterase.
Potential limit and bias of current adopted ALP functional marker phoD
Compared to the overall ubiquity of phoD within genomic data (Table S3), the distribution of phoD in this study seems to be restricted to few bacterial phyla. For instance, numerous sequences seem to be related to the Alphaproteobacteria. The over-abundance of alphaproteobacterial phoD is likely to be a potential amplification bias caused by the primers specificity rather than the real distribution of phoD. Indeed, the primers used in this study have been designed from phoD sequences derived principally from Alphaproteobacteria (Sakurai et al. 2008). Accumulation of phoD sequences from different genome or metagenome projects will probably help in the near future the design of a more comprehensive set of phoD primers. While this study focused on phoD gene diversity, it would be very interesting for future research to assess the expression level of phoD in the three P treatments, as expression is directly related with the soil ALP activity.
Conclusion
This study has evaluated the ecological impacts of long-term stable P amendment in a pasture system. High level of P input enhances the total microbial biomass in the soil and also seems to increase the soil bacterial diversity. In addition, P amendment seems to decrease soil ALP activity. This trend could be explained at least partially by the selection of specific ALP genotypes encoding enzymes with enhanced activities. Interestingly some specific phoD genotypes were indeed enriched in the no-P soil. However, association between genotype and ALP activity remains to be assessed. Moreover, others studies targeting phoX and phoA could also be interesting complementing approaches. Nonetheless, the changes in the relative abundance of dominant bacterial phyla such as Acidobacteria clearly demonstrate that P fertilisation induces shift in the soil microbial community, which is likely to have implications on ecosystem functioning.
References
Anisimova M, Gascuel O (2006) Approximate likelihood-ratio test for branches: a fast, accurate, and powerful alternative. Syst Biol 55:539–552
Barret M, Morrissey JP, O'Gara F (2011) Functional genomics analysis of plant growth-promoting rhizobacterial traits involved in rhizosphere competence. Biol Fertil Soils 47:729–743
Berg G, Smalla K (2009) Plant species and soil type cooperatively shape the structure and function of microbial communities in the rhizosphere. FEMS Microbiol Ecol 68:1–13
Browne P, Rice O, Miller SH, Burke J, Dowling DN, Morrissey JP, O’Gara F (2009) Superior inorganic phosphate solubilization is linked to phylogeny within the Pseudomonas fluorescens complex. Appl Soil Ecol 43:131–138
Carrigg C, Rice O, Kavanagh S, Collins G, O’Flaherty (2007) DNA extraction method affects microbial community profiles from soils and sediment. Appl Microbiol Biotechnol 77:955–964
Chhabra S, Brazil D, Morrissey JP, Burke J, O'Gara F, Dowling D (2012) Fertilization management affects the alkaline phosphatase bacterial community in barley rhizosphere soil. Biol Fertil Soils (in press)
Chu H, Lin X, Fujii T, Morimoto S, Yagi K, Hu J, Zhang J (2007) Soil microbial biomass, dehydrogenase activity, bacterial community structure in response to long-term fertilizer management. Soil Biol Biochem 39:2971–2976
Culleton N, Coulter B, Liebhardt WC (2002) The fate of phosphatic fertiliser applied to grassland. Ir Geogr 35:175–184
Culleton N, Liebhardt W, Murphy W, Cullen J, Cuddihy A (2000) Thirty years of phosphorus fertiliser on Irish pastures: animal–soil–water relationships. Teagasc, Dublin
Drozd M, Gangaiah D, Liu Z, Rajashekara G (2011) Contribution of TAT system translocated PhoX to Campylobacter jejuni phosphate metabolism and resilience to environmental stresses. PLoS One 6:e26336
Eder S, Shi L, Jensen K, Yamane K, Hulett FM (1996) A Bacillus subtilis secreted phosphodiesterase/alkaline phosphatase is the product of a Pho regulon gene, phoD. Microbiology 142:2041–2047
Fierer N, Lauber CL, Ramirez KS, Zaneveld J, Bradford MA, Knight R (2012) Comparative metagenomic, phylogenetic and physiological analyses of soil microbial communities across nitrogen gradients. ISME J 6:1007–1017
Galperin MY, Jedrzejas MJ (2001) Conserved core structure and active site residues in alkaline phosphatase superfamily enzymes. Proteins 45:318–324
Ge Y, Zhang J, Zhang L, Yang M, He J (2008) Long-term fertilization regimes affect bacterial community structure and diversity of an agricultural soil in northern China. J Soils Sed 8:43–50
Goulding K, Jarvis S, Whitmore A (2008) Optimizing nutrient management for farm systems. Philos Trans R Soc Lond B Biol Sci 363:667–680
Griffiths BS, Spilles A, Bonkowski M (2012) C:N:P stoichiometry and nutrient limitation of the soil microbial biomass in a grazed grassland site under experimental P limitation or excess. Ecol Process 1:6
Guindon S, Gascuel O (2003) A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52:696–704
Hasegawa M, Kishino H, Yano TA (1985) Dating of the human ape splitting by a molecular clock of mitochondrial DNA. J Mol Evol 22:160–174
Hayat R, Ali S, Amara U, Khalid R, Ahmed I (2010) Soil beneficial bacteria and their role in plant growth promotion: a review. Ann Microbiol 60:579–598
İnceoğlu Ö, Al-Soud WA, Salles JF, Semenov AV, Van Elsas JD (2011) Comparative analysis of bacterial communities in a potato field as determined by pyrosequencing. PLoS One 6:e23321
Jones RT, Robeson MS, lauber CL, Hamady M, Knight R, Fierer N (2009) A comprehensive survey of soil acidobacterial diversity using pyrosequencing and clone library analyses. ISME J 3:442–453
Kageyama H, Tripathi K, rai AK, Cha-um S, Waditee-Sirisattha R, Takabe T (2011) An alkaline phosphatase/phosphodiesterase, PhoD, induced by salt stress and secreted out of the cells of Aphanothece halophytica, a halotolerant cyanobacterium. Appl Environ Microbiol 77:5178–5183
Kamat SS, Williams HJ, Raushel FM (2011) Intermediates in the transformation of phosphonates to phosphate by bacteria. Nature 480:570–573
Karandashov V, Bucher M (2005) Symbiotic phosphate transport in arbuscular mycorrhizas. Trends Plant Sci 10:22–29
Kathuria S, Martiny AC (2011) Prevalence of a calcium-based alkaline phosphatase associated with the marine cyanobacterium Prochlorococcus and other ocean bacteria. Environ Microbiol 13:74–83
Katoh K, Toh H (2008) Recent developments in the MAFFT multiple sequence alignment program. Brief Bioinform 9:286–298
King-Salter GE (2008) Response of arbuscular mycorrhizal fungi to seasonality and long-term phosphorus fertilisation in an Irish grazed grassland. Ph.D. thesis, University College Dublin, Dublin
Kirk GJD (1999) A model of phosphate solubilization by organic anion excretion from plant roots. Eur J Soil Sci 50:369–378
Letunic I, Bork P (2011) Interactive Tree Of Life v2: online annotation and display of phylogenetic trees made easy. Nucleic Acids Res 39:W475–W478
Liu L, Gundersen P, Zhang T, Mo J (2012) Effects of phosphorus addition on soil microbial biomass and community composition in three forest types in tropical China. Soil Biol Biochem 44:31–38
Luo H, Benner R, Long RA, Hu J (2009) Subcellular localization of marine bacterial alkaline phosphatases. Proc Nat Acad Sci USA 106:21219–21223
Mao Y, Yannarell AC, Mackie RI (2011) Changes in N-transforming archaea and bacteria in soil during the establishment of bioenergy crops. PLoS One 6:e24750
Markowitz VM, Ivanova NN, Szeto E, Palaniappan K, Chu K, Dalevi D, Chen IMA, Grechkin Y, Dubchak I, Anderson I, Lykidis A, Mavromatis K, Hugenholtz P, Kyrpides NC (2008) IMG/M: a data management and analysis system for metagenomes. Nucleic Acids Res 36:D534–D538
Miller SH, Browne P, Prigent-Combaret C, Combes-Meynet E, Morrissey JP, O'Gara F (2010) Biochemical and genomic comparison of inorganic phosphate solubilization in Pseudomonas species. Environ Microbiol Rep 2:403–411
Monds RD, Newell PD, Schwartzman JA, O'Toole GA (2006) Conservation of the Pho regulon in Pseudomonas fluorescens Pf0-1. Appl Environ Microbiol 72:1910–1924
Morrissey JP, Dow JM, Mark GL, O'Gara F (2004) Are microbes at the root of a solution to world food production? EMBO Rep 5:922–926
Nacke H, Thuermer A, Wollherr A, Will C, Hodac L, Herold N, Schoening I, Schrumpf M, Daniel R (2011) Pyrosequencing-based assessment of bacterial community structure along different management types in German forest and grassland soils. PLoS One 6:e17000
Nannipieri P, Ascher J, Ceccherini MT, Landi L, Pietramellara G, Renella G (2003) Microbial diversity and soil functions. Eur J Soil Sci 54:655–670
Nannipieri P, Giagnoni L, Landi L, Renella G (2011) Role of phosphatase enzymes in soil. In: Bünemann EK, Oberson A, Frossard E (eds) Phosphorus in action, vol 100, Soil biology. Springer, Berlin, pp 215–241
Palmer K, Drake HL, Horn MA (2009) Genome-derived criteria for assigning environmental narG and nosZ sequences to operational taxonomic units of nitrate reducers. Appl Environ Microbiol 75:5170–5174
Pennanen T, Perkiömäki J, Kiikkilä O, Vanhala P, Neuvonen S, Fritze H (1998) Prolonged, simulated acid rain and heavy metal deposition: separated and combined effects on forest soil microbial community structure. Microbiol Ecol 27:291–300
Philippot L, Hallin S (2005) Finding the missing link between diversity and activity using denitrifying bacteria as a model functional community. Curr Opin Microbiol 8:234–239
Rice O, Miller S, Morrissey J, O’Gara F (2012) Exploitation of glucose catabolic gene fusions to investigate in situ expression during Pseudomonas–plant interactions. Biol Fertil Soils 48:235–238
Richardson AE, Barea J-M, McNeill AM, Prigent-Combaret C (2009) Acquisition of phosphorus and nitrogen in the rhizosphere and plant growth promotion by microorganisms. Plant Soil 321:305–339
Richardson AE, Simpson RJ (2011) Soil microorganisms mediating phosphorus availability. Plant Physiol 156:989–996
Rodríguez H, Fraga R, Gonzalez T, Bashan Y (2006) Genetics of phosphate solubilization and its potential applications for improving plant growth-promoting bacteria. Plant Soil 287:15–21
Roesch LF, Fulthorpe RR, Riva A, Casella G, Hadwin AKM, Kent AD, Daroub SH, Camargo FAO, Farmerie WG, Triplett EW (2007) Pyrosequencing enumerates and contrasts soil microbial diversity. ISME J 1:283–290
Sakurai M, Wasaki J, Tomizawa Y, Shinano T, Osaki M (2008) Analysis of bacterial communities on alkaline phosphatase genes in soil supplied with organic matter. Soil Sci Plant Nutr 54:62–71
Schloss PD, Handelsman J (2006) Toward a census of bacteria in soil. PLoS Comput Biol 2:e92
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF (2009) Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537–7541
Sebastian M, Ammerman JW (2011) Role of the phosphatase PhoX in the phosphorus metabolism of the marine bacterium Ruegeria pomeroyi DSS-3. Environ Microbiol Rep 3:535–542
Smalla K, Wieland G, Buchner A, Zock A, Parzy J, Kaiser S, Roskot N, Heuer H, Berg G (2001) Bulk and rhizosphere soil bacterial communities studied by denaturing gradient gel electrophoresis: plant-dependent enrichment and seasonal shifts revealed. Appl Environ Microbiol 67:4742–4751
Tabatabai MA, Bremner JM (1969) Use of p-nitrophenyl phosphate for assay of soil phosphatase activity. Soil Biol Biochem 1:301–307
Tilman D, Cassman KG, Matson PA, Naylor R, Polasky S (2002) Agricultural sustainability and intensive production practices. Nature 418:671–677
Uroz S, Buée M, Murat C, Frey-Klett P, Martin F (2010) Pyrosequencing reveals a contrasted bacterial diversity between oak rhizosphere and surrounding soil. Environ Microbiol Rep 2:281–288
Uroz S, Calvaruso C, Turpault M-P, Frey-Klett P (2009) Mineral weathering by bacteria: ecology, actors and mechanisms. Trends Microbiol 17:378–387
Van Aarle IM, Plassard C (2010) Spatial distribution of phosphatase activity associated with ectomycorrhizal plants is related to soil type. Soil Biol Biochem 42:324–330
Whelan S, Goldman N (2001) A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol Biol Evol 18:691–699
Wu JR, Shien JH, Shieh HK, Hu CC, Gong SR, Chen LY, Chang PC (2007) Cloning of the gene and characterization of the enzymatic properties of the monomeric alkaline phosphatase (PhoX) from Pasteurella multocida strain X-73. FEMS Microbiol Lett 267:113–120
Yamane K, Maruo B (1978) Purification and characterization of extracellular soluble and membrane-bound insoluble alkaline phosphatases possessing phosphodiesterase activities in Bacillus subtilis. J Bacteriol 134:100–107
Yao MZ, Zhang YH, Lu WL, Hu MQ, Wang W, Liang AH (2012) Phytases: crystal structures, protein engineering and potential biotechnological applications. J Appl Microbiol 112:1–14
Zaheer R, Morton R, Proudfoot M, Yakunin A, Finan TM (2009) Genetic and biochemical properties of an alkaline phosphatase PhoX family protein found in many bacteria. Environ Microbiol 11:1572–1587
Zhong WH, Cai ZC (2007) Long-term effects of inorganic fertilizers on microbial biomass and community functional diversity in a paddy soil derived from quaternary red clay. Appl Soil Ecol 36:84–91
Acknowledgments
This research was supported in part by grants awarded by the Science Foundation of Ireland (04/BR/B0597, 07/IN.1/B948, 07/SK/B1236b, 08/RFP/GEN1319, 08/RFP/GEN1295, SFI09/RFP/BMT 2350); the Department of Agriculture, Fisheries and Food (DAF RSF 06-321, DAF RSF 06-377, FIRM 08/RDC/629); the Environmental Protection Agency (EPA 2006-PhD-S-21, EPA 2008-PhD-S-2), the Irish Research Council for Science, Engineering and Technology (IRC EMBARK PD/2011/2414), the European Commission (FP6#036314, MTKD-CT2006-042062, TRAMWAYS, MicroB3-287589-OCEAN2012, MACUMBA-CP-TP 311975; PharmaSea-CP-TP 312184, EU 256596); and the Marine Institute (Beaufort award - C&CRA 2007/082) and the HRB (RP/2006/271, RP/2007/290, HRA/2009/146). The authors would like to acknowledge the Boole Centre for Research in Informatics at University College Cork for providing access to computational facilities.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 2561 kb)
ESM 2
(PDF 162 kb)
ESM 3
(PDF 144 kb)
ESM 4
(PDF 144 kb)
ESM 5
(PDF 1408 kb)
ESM 6
(PDF 15302 kb)
ESM 7
(GIF 61 kb)
ESM 8
(GIF 6 kb)
ESM 9
(GIF 89 kb)
ESM 10
(DOCX 13 kb)
ESM 11
(DOCX 14 kb)
ESM 12
(DOC 148 kb)
ESM 13
(DOC 330 kb)
ESM 14
(DOC 996 kb)
Rights and permissions
About this article
Cite this article
Tan, H., Barret, M., Mooij, M.J. et al. Long-term phosphorus fertilisation increased the diversity of the total bacterial community and the phoD phosphorus mineraliser group in pasture soils. Biol Fertil Soils 49, 661–672 (2013). https://doi.org/10.1007/s00374-012-0755-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00374-012-0755-5